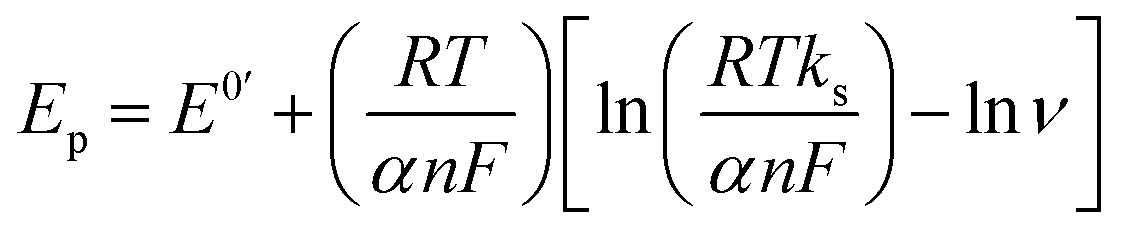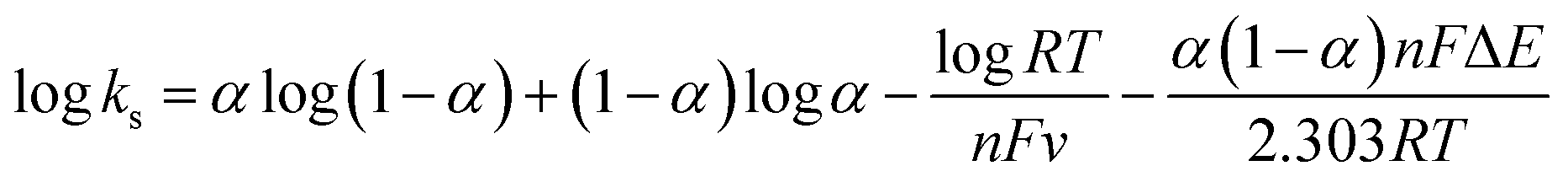DOI:
10.1039/D1AN00537E
(Paper)
Analyst, 2021,
146, 4066-4079
Label-free electrochemical immunosensor based on gold nanoparticle/polyethyleneimine/reduced graphene oxide nanocomposites for the ultrasensitive detection of cancer biomarker matrix metalloproteinase-1
Received
28th March 2021
, Accepted 27th April 2021
First published on 29th April 2021
Abstract
Matrix metalloproteinase-1 (MMP-1) is associated with many types of cancers, including oral, colorectal, and brain cancers. This paper describes the fabrication of an MMP-1 immunosensor based on a gold nanoparticle/polyethyleneimine/reduced graphene oxide (AuNP/PEI/rGO)-modified disposable screen-printed electrode (SPE). A microwave-assisted single-step method was employed for the simultaneous reduction of gold and graphene oxide in a PEI environment to avoid AuNP agglomeration. The crystal structure, chemical composition, optical properties, and interior morphology of the materials were probed by X-ray diffraction, Raman spectroscopy, UV-visible spectrometry, and transmission electron microscopy techniques. To assemble a label-free MMP-1 immunosensor layer-by-layer, 3-mercaptopropionic acid was utilized due to its strong sulfur-gold bonding ability, and its tail end was attached to a carboxyl group, allowing the MMP-1 antibody (anti-MMP-1) to be subsequently cross-linked using the traditional N-(3-dimethylaminopropyl) and N′ ethylcarbodiimide hydrochloride method. Differential pulse voltammetry analysis showed a linear relationship with MMP-1 concentration in the range of 1–50 ng ml−1 with an R2 value of ∼0.996 (n = 5, RSD < 5%). This immunosensor was successfully applied for MMP-1 detection in urine, saliva, bovine serum, and cell culture media (HSC-3 & C6) of oral and brain cancers showing results comparable to those of the credible ELISA method.
1 Introduction
Matrix metalloproteinases (MMPs), or matrixins, are a group of enzymes that degrade the extracellular matrix (ECM) proteins. The MMP structure consists of a group of zinc-containing endopeptidases that vary in size and substrate specificity. MMPs have a three-domain structure consisting of a C-terminal domain (Hpx) of a propeptide, a catalytic domain, and a heme-binding protein.1 Humans have 24 matrixin genes, including the duplicate MMP-23 gene, making 23 unique MMPs in the human body. These MMPs play important roles in normal and abnormal cells during the process of tumorigenesis.2 The breakdown of the extracellular matrix and basement membranes by MMPs allows for easy penetration of cancer cells into blood vessels and subsequently, tissues.3 Among them, matrix metalloproteinase-1 (MMP-1) is an interstitial collagenase that acts on fibrillar collagen. MMP-1 plays a predominant role in the breakdown of the extracellular matrix during ordinary physiological processes including reproduction, embryonic development, tissue remodeling, and disease processes, for example, arthritis and metastasis.4 Specifically, MMP-1 degrades fibrous collagen of types I, II, and III. Numerous studies have demonstrated the association of MMP-1 with metastasis or expression of various cancers, including oral,5,6 colorectal,7,8 breast,9 thyroid,10 lung,11 prostate,12 and bladder cancers.13 Biosensors have been used for the determination of MMP-1 predominantly as point-of-care devices, which should also enable further examination of a diverse spectrum of diseases in clinical studies. In recent years, there have been many studies on electrochemical cancer detection due to the specificity, speed, portability, and low cost of electrochemical detection methods providing excellent utility in clinical care.14 Thus, electrochemical detection provides a label-free method for immunosensing, in which MMP-1 antibodies (anti-MMP-1) can be modified by layer-by-layer assembly, endowing it with the advantages of rapid detection and high sensitivity. This method is superior to traditional detection methods as it allows for a detection limit to be established, is convenient and accurate,15,16 and holds promise for trampoline detection, which involves a detection platform utilizing various body fluids for detecting the biomarker MMP-1. Previously, various nanomaterials were applied for the electrochemical immunosensing of different kinds of MMPs such as MMP-2, MMP-3, and MMP-9.17–22 Hitherto, no one reported the electrochemical immunosensing of MMP-1. For the first time, our research group reports an electrochemical immunosensor for the detection of MMP-1 using AuNP/PEI/rGO nanocomposites.
Metal nanoparticles have received much attention in electrochemical and biosensor applications. Precious metal particles with nanometer sizes, in particular gold nanoparticles (AuNPs), have received much attention in various fields such as chemistry, medicine, biology, and materials science due to their excellent electrical conductivity and optical, thermal, and catalytic properties. In the field of electrochemistry, AuNPs have enabled signal amplification in various types of sensors due to their biocompatibility and excellent electrical conductivity, as well as their specificity and ease of simple functionalization.23,24 AuNPs are catalytically active because of their large surface-area-to-volume ratio and dominant interfacial properties and can reduce the overpotential of many electroanalytical reactions and maintain the reversibility of redox reactions. These NPs have also been reported for the detection of various substances such as small molecules, toxic chemicals, and drugs.25 In this study, an electrocatalytic method was used for detection, involving modifying electrochemical cells for the detection of tumor cells. AuNPs functionalized with antibodies were selectively bound to molecules on tumor cells to catalyze hydrogen evolution in an acidic environment, generating an electrochemical response. Besides, 3-mercaptopropionic acid (3-MPA) acts as a building block for complex arrays of AuNPs and the carboxyl (–COOH) group of 3-MPA was activated by EDC/NHS. The MMP-1 antigens were easily immobilized after activating the –COOH group of 3-MPA with EDC/NHS.26
Polymer-assisted synthesis of metal nanoparticles has attracted great attention as it allows for the effective stabilization of the nanoparticles by acting as both a stabilizer and a reducing agent to effectively combine monodispersed particles. Examples include NH2-containing,27 OH-containing,28 and polychloride29 polymers. Polymeric layers have also been used to overcome the problem of selectivity.30 Polyethyleneimine (PEI) is an amine-containing cationic PE and is also water-soluble. Nowadays, linear PEI is utilized as both a reducing and protecting/stabilizing agent for the preparation of AuNPs.31 PEI is mixed with a variety of inorganic NPs to improve the therapeutic potential of nanosystems for biological activities via their physical properties.32 Formerly, Khetani et al. reported PEI modified graphene screen-printed electrodes for the highly selective and sensitive determination of glial fibrillary acidic protein in different biofluids. Also, PEI displayed some specific properties such as stability at ambient temperature, quickly rendered reduction to a Schiff's base reaction using glutaraldehyde, and certainly adsorbed onto graphene surfaces, thus making it a promising candidate for the rapid functionalization of graphene.33 Hence, PEI was utilized in this work to simultaneously reduce GO to rGO and Au3+ to Au0 in AuNPs, and as a stabilizing agent for preventing gold nanoparticles from agglomeration. Graphene is the fundamental structural element of graphite and carbon nanotubes. In graphite, adjacent graphene layers are bound through weak van der Waals forces, allowing for easy mechanical separation into single graphene layers. Graphene is a two-dimensional material in which carbon atoms form an sp2 honeycomb lattice34 and it exhibits many unique properties such as the quantum Hall effect, electron mobility of more than 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1,35 large specific surface area, excellent mechanical properties, and excellent thermal conductivity (3000–5000 W m−1 K−1).36 Therefore, graphene has been used widely in sensors, supercapacitors, fuel cells, conductive films, and other fields.37 There are many graphene composites currently used in the field of sensors; however, graphene is a hydrophobic material, is largely insoluble in most solvents, and also lacks functional groups. Oxidation of graphene to graphene oxide (GO) provides an abundance of functional groups on the edges and surfaces of the sheet, thus solving the problem of poor hydrophilicity and allowing for further modification of the side chains.38 Although graphene oxidation improves the hydrophilicity of graphene, oxidation introduces defects into the C
000 cm2 V−1 s−1,35 large specific surface area, excellent mechanical properties, and excellent thermal conductivity (3000–5000 W m−1 K−1).36 Therefore, graphene has been used widely in sensors, supercapacitors, fuel cells, conductive films, and other fields.37 There are many graphene composites currently used in the field of sensors; however, graphene is a hydrophobic material, is largely insoluble in most solvents, and also lacks functional groups. Oxidation of graphene to graphene oxide (GO) provides an abundance of functional groups on the edges and surfaces of the sheet, thus solving the problem of poor hydrophilicity and allowing for further modification of the side chains.38 Although graphene oxidation improves the hydrophilicity of graphene, oxidation introduces defects into the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C structure, resulting in a decrease in conductivity. To restore the electrical, mechanical, and thermal properties and high surface area and conductivity,39 reduced graphene oxide (rGO) can be prepared from GO. rGO has been prepared using various methods, including electrochemical reduction, high temperature and high-pressure processes, microwave reduction,40,41 and ultraviolet (UV) reduction.42 None of these methods require a reducing agent or other chemical agents. Amongst them, microwave-aided synthesis techniques have numerous advantages, including lower processing cost, rapid and uniform heating, higher yield and shorter preparation time, energy saving process, higher purity, no selective heating of the surface, and smaller narrow particle size distribution, over other conventional techniques.43 As a consequence, the microwave method for chemical reduction is used to rapidly heat and simultaneously reduce graphene oxide and gold ions and is the method employed in the work described herein, which involves the preparation of gold nanoparticle/polyethyleneimine/reduced graphene oxide (AuNP/PEI/rGO), followed by the self-assembly of an immunosensor using grafted antibodies for the modification of the surface of an electrode.
C structure, resulting in a decrease in conductivity. To restore the electrical, mechanical, and thermal properties and high surface area and conductivity,39 reduced graphene oxide (rGO) can be prepared from GO. rGO has been prepared using various methods, including electrochemical reduction, high temperature and high-pressure processes, microwave reduction,40,41 and ultraviolet (UV) reduction.42 None of these methods require a reducing agent or other chemical agents. Amongst them, microwave-aided synthesis techniques have numerous advantages, including lower processing cost, rapid and uniform heating, higher yield and shorter preparation time, energy saving process, higher purity, no selective heating of the surface, and smaller narrow particle size distribution, over other conventional techniques.43 As a consequence, the microwave method for chemical reduction is used to rapidly heat and simultaneously reduce graphene oxide and gold ions and is the method employed in the work described herein, which involves the preparation of gold nanoparticle/polyethyleneimine/reduced graphene oxide (AuNP/PEI/rGO), followed by the self-assembly of an immunosensor using grafted antibodies for the modification of the surface of an electrode.
2 Experimental section
2.1 Materials
Graphite powder was obtained from Bay Carbon, USA. Sulfuric acid, potassium permanganate, sodium nitrate, hydrogen peroxide, polyethyleneimine, potassium hexacyanoferrate(II), potassium hexacyanoferrate(III), bovine serum albumin, and 3-mercaptopropionic acid were obtained from Sigma, USA. Monosodium hydrogen phosphate, potassium dihydrogen phosphate, and potassium chloride were obtained from J.T Baker, USA. Gold(III) chloride and the MAB-901 MMP-1 antibody were obtained from Acros, Belgium, and R&D, USA, respectively. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, N-hydroxysulfosuccinimide sodium salt, and fetal bovine serum were obtained from Thermo Fisher Scientific, USA. Screen printed three-pole electrodes (model TE-100) were obtained from Zensor, USA. All chemicals were utilized without purification.
2.2 Methods
The optical properties of the materials were investigated by UV-visible spectrometry (Cary 5000 UV-vis NIR (Agilent, USA)), and the chemical compositions of GO and rGO were confirmed by Raman spectroscopy (Ramboss 500i Micro (DINGWOO/USA)). The crystal structures of the materials were confirmed using X-ray diffraction (XRD) (X'Pert3 Powder (PANalytical, the Netherlands)), and the microstructure was studied by field emission scanning electron microscopy (FESEM, Hitachi Regulus 8100) and transmission electron microscopy (TEM) (JEOL 2100 F). A pH tester [pH510 (Eutech Instrument/UK)] was employed to maintain the pH throughout the experimental processes. The electrochemical characterization of various modified electrodes was performed using a CHI6114E instrument (CH Instruments, USA). The three-pole screen printing electrode (SPE) acted as the counter, working, and reference electrodes. The electrolyte solution contained a mixture of 5 mM Fe(CN)63−/4− dissolved in 0.1 M phosphate buffered saline (PBS) and 0.1 M KCl (pH 7.4). The employed electrochemical potential window ranges from −0.6 V to +0.6 V and the scan sweep rate was 0.05 V s−1.
2.3 AuNP/PEI/rGO preparation process
GO was prepared by a procedure based on modified Hummers and Offeman's methods.44 To prepare the composite (AuNP/PEI/rGO), a mixture of 5.88 ml of GO and 0.12 ml of gold trichloride solution was subjected to shock treatment for 20 min. 12 μl of PEI was then added to the mixture and ultrasonicated for 10 min. The mixed sample was then placed in a microwave oven (CEM Explorer-48) with the temperature and wattage set to 150 °C and 150 W, respectively, for 5 min. The obtained AuNP/PEI/rGO solution was centrifuged three times with DI water.
2.4 Preparation of chlorinated electrodes
The electrode was first cleaned 3–5 times with DI water before being placed in an oven at 50 °C for 3 min. The silver electrode in the SPE was used as the working electrode, and platinum wire and Ag/AgCl were used as the counter and reference electrodes, respectively. A 3 M KCl solution was used as the electrolytic solution; a CHI641E electrochemical instrument was used to chlorinate the electrode by amperometry. The chlorinated electrode was rinsed with DD water, then dried and stored in a dry box in the dark.
2.5 Fabrication of AuNP/PEI/rGO and immobilization of MMP-1
The AuNP/PEI/rGO composite dispersed in solution was mixed with ethanol in a ratio of 8:2, and approximately 6 μl of this solution was then dropped onto the working surface of the electrode, which was dried in an oven at 50 °C for 40 min. 1 μl of the 3-MPA solution was drop-cast on the SPE surface and left at room temperature for 6 h. A 0.2 M EDC/0.05 M sulfo-NHS solution was mixed at a volume ratio of 1:1 for 30 min, followed by the addition of 6 μl of the solution to the SPE for 30 min at ambient temperature. The SPE surface was then washed with PBS and placed in a fume hood to air dry. 4 μl of a 0.1 mg ml−1 solution of anti-MMP-1 was dropped onto the electrode, which was then stored in a refrigerator for 6 h, then washed with PBS and dried in an oven. A 1% BSA solution was prepared and 3 μl was drop cast on the modified SPE and allowed to react at room temperature for 30 min. Different concentrations of the MMP-1 antigen were prepared and 2 μl of each solution was drop cast on an electrode and kept at 4 °C in a refrigerator for 2 h. The electrode surface was washed with PBS and kept in a fume hood to air dry. Detection of the MMP-1 antigen was performed by differential pulse voltammetry (DVP). The fabrication process of the modified electrode is shown in Fig. 1.
 |
| | Fig. 1 Fabrication of AuNP/PEI/rGO/SPE and immobilization of MMP-1 antibodies for the immunoelectrochemical sensing of MMP-1. | |
2.6 Collection of human samples
Urine and saliva samples were collected from a normal healthy adult person and centrifuged at 9000 rpm for 15 min. The acquired supernatant was then separated and stored at −20 °C. These solutions were used in a real sample test. Ethical statement: The research protocols of the urine sample were performed in compliance with the relevant laws and institutional guidelines for care and use of laboratory human subjects of Chang-Gung Memorial Hospital. Informed consent was obtained from all the human subjects for experimentation and approved by the Institutional Review Board of Chang-Gung Memorial Hospital (IRB no. 201801660B).
3 Characterization
3.1 XRD, Raman, and UV-vis spectroscopy
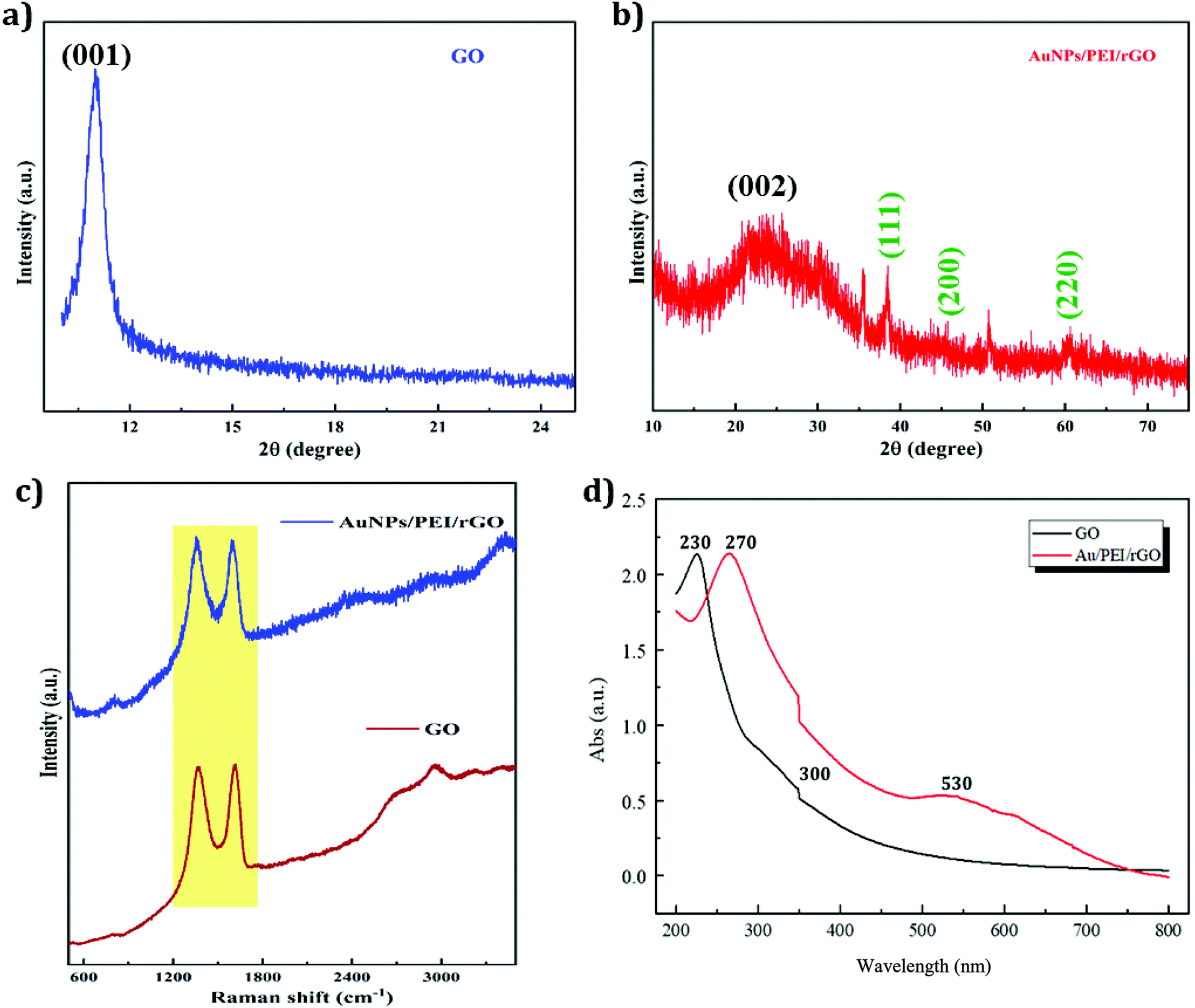
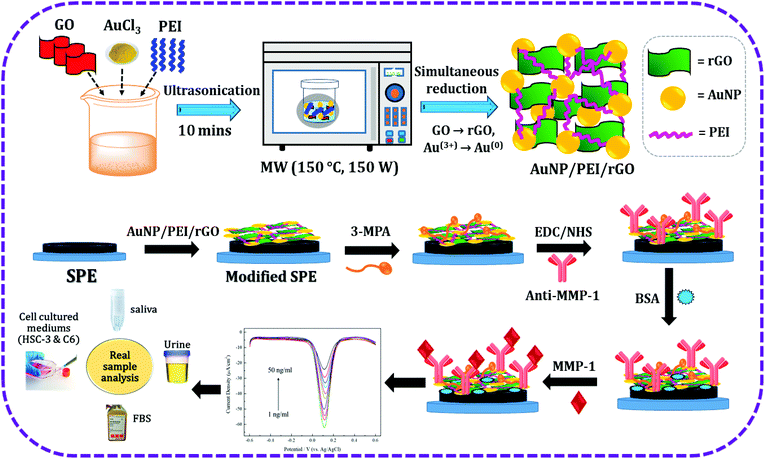
The structural features of the resultant materials were examined by various spectrometric techniques including XRD, Raman, and UV-vis spectroscopy. The XRD pattern of GO is shown in Fig. 2a. The characteristic carbon peak of GO apparent at 12° represents the (001) plane.45 The XRD pattern of AuNP/PEI/rGO is displayed in Fig. 2b. It can be seen that rGO exhibits a characteristic peak at 25.6° which is attributed to the (002) plane and AuNPs show peaks at 38.4, 45.4, and 60.3° corresponding to the (111), (200), and (220) planes, respectively [JCPDS no.: 04-0787].46 GO and AuNP/PEI/rGO were characterized by Raman spectroscopy, which showed two unambiguous peaks at 1350 cm−1 and 1595 cm−1 corresponding to the D and G peaks, respectively (Fig. 2c). These peaks arise from the lattice defects in the atomic crystal structure of the carbon material. GO also exhibited a weak characteristic peak in the Raman spectrum at around 2800 cm−1, which was not present in the spectrum of rGO, due to the reduction of the functional groups in rGO. UV-vis spectrophotometry was used to check the absorbance variation in GO and rGO, and the results are shown in Fig. 2d. The spectrum of GO showed absorption peaks at 230 and 300 nm, corresponding to the C–C π → π* and CO n → π* electron transfer transitions, respectively. However, the spectrum of AuNP/PEI/rGO synthesized in this experiment showed a peak at ∼270 nm, due to a redshift of the π → π* transitions following the reduction. Due to the reduction of the oxygen functional groups, the peak at 300 nm disappeared, and a characteristic peak at 530 nm confirmed the presence of AuNPs.
 |
| | Fig. 2 XRD pattern of GO (a) and AuNP/PEI/rGO (b). Raman spectra of GO and AuNP/PEI/rGO (c). UV-vis spectra of GO and AuNP/PEI/rGO (d). | |
3.2 FESEM and TEM analyses
The structural morphologies of the GO and AuNP/PEI/rGO nanocomposites were examined using FESEM and TEM analyses. Fig. 3(a–f) shows the FESEM images of GO and AuNP/PEI/rGO at various magnifications. GO shows a randomly aggregated, thin, crumpled layer structure in Fig. 3(a–c) at different magnifications. Besides, the AuNPs were decorated on the sheet like structures of rGO in the composites (Fig. 3(d–f)). Besides, the TEM images of the AuNP/PEI/rGO nanocomposites at lower to higher magnifications are presented in Fig. 4(a–e). The AuNPs are decorated on the surface of the rGO sheets via the microwave synthesis approach in a PEI environment. Fig. 4(f and g) shows the HRTEM image with crystal lattice fringes and the SAED pattern of the AuNPs decorated on the rGO. This result indicates the crystalline nature of the AuNPs present in the prepared composite. The elemental mapping results of the composites are depicted in Fig. 4h–l. Furthermore, the weight percentages of the elements such as Au = 39.19, C = 47.05, O = 7.58, and N = 6.17% present in the AuNP/PEI/rGO composite were obtained by EDX analysis (Fig. 4m and n). These results confirmed the successful formation of AuNP/PEI/rGO nanocomposites.
 |
| | Fig. 3 FESEM images of GO (a–c) and AuNP/PEI/rGO (d–f) at various magnifications. | |
 |
| | Fig. 4 TEM images of AuNP/PEI/rGO at various magnifications (a–e). HRTEM (f) and SAED (g) patterns of AuNPs in the nanocomposite. Elemental mapping results of the AuNP/PEI/rGO nanocomposite (mixture) in (h) the presence of different elements including Au (i), C (j), O (k), and N (l). EDX result (m) and the corresponding weight percentage plot (n). | |
3.3 Electrochemical impedance spectroscopy (EIS)
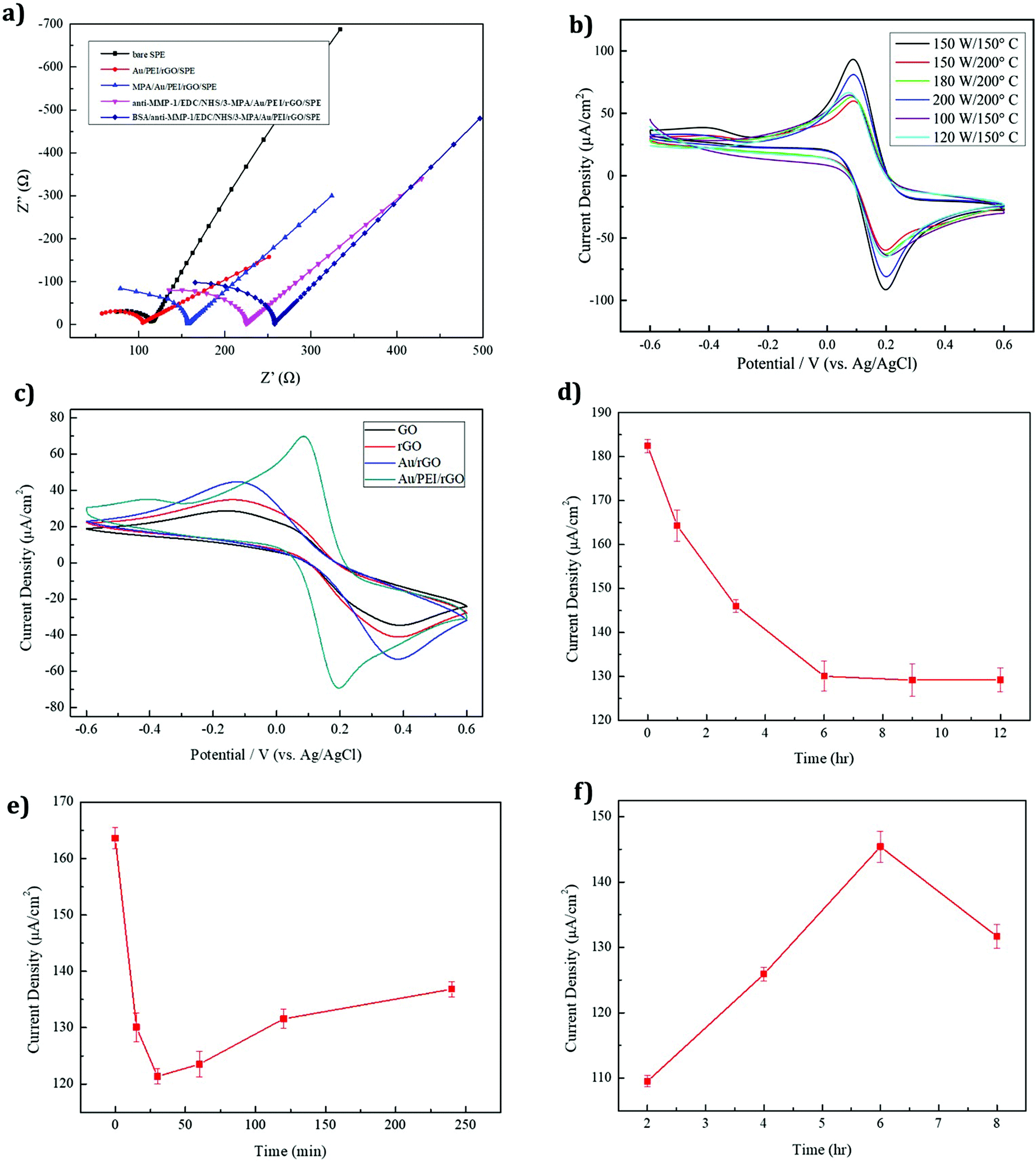
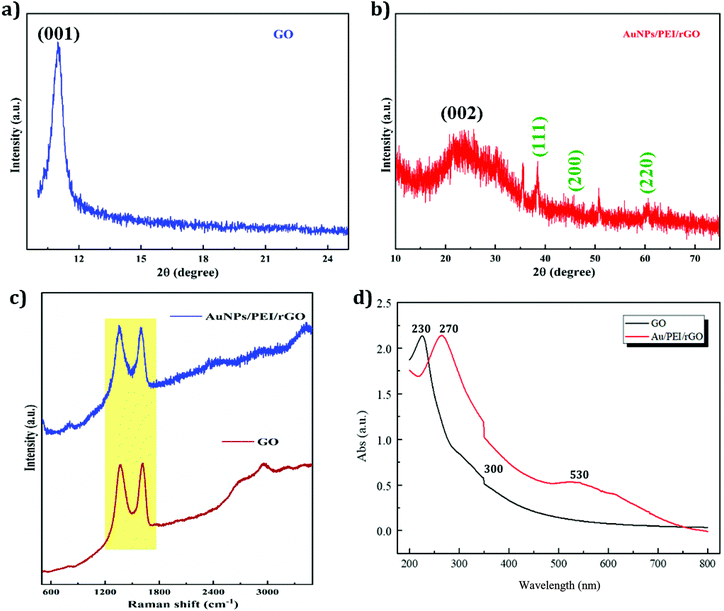
EIS is an efficient method for evaluating the electron transport properties. The MMP-1 assembly on an immunosensor was examined by EIS with 5 mM Fe(CN)63−/4− being dissolved in a 0.1 M PBS/0.1 M KCl (pH 7.4) electrolyte solution at a scan rate of 50 mV s−1, and the obtained spectra are shown in Fig. 5a. The EIS layers of each modified electrode, namely bare SPE, Au/PEI/rGO/SPE, 3-MPA/Au/PEI/rGO/SPE, anti-MMP-1/EDC/NHS/3-MPA/Au/PEI/rGO/SPE and BSA/anti-MMP 1/EDC/NHS/3-MPA/Au/PEI/rGO/SPE, are shown in Fig. 5a. The large semicircular regions correspond to the areas of high electron transfer resistance (Rct). The Au/PEI/rGO/SPE shows noteworthy electron transfer kinetics between redox peaks which means lower Rct than that of the bare SPE. Because of their high surface area and highly conductive nature, rGO and AuNPs were coated on the SPE surface. Moreover, the Rct value was considerably enhanced after the immobilization of the antibodies on the SPE surface. This indicated the successful formation of the anti-MMP-1 layer on the modified electrode surface, which demonstrates the blocking effect on the electron transfer kinetics. After the layer-by-layer assembly, the fabricated electrode resistance was gradually increased. Hence, the Rct value of the modified electrode was increased. The Rct values of the various modified electrodes are given in Table 1.
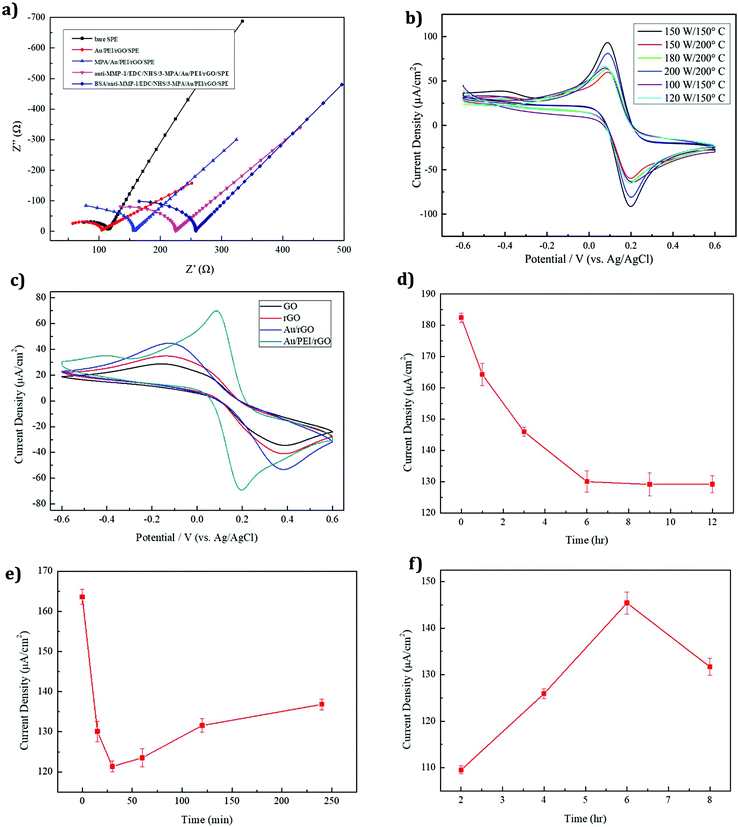 |
| | Fig. 5 (a) EIS spectra of different modified electrodes (conditions: 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH 7.4), scan rate 50 mV s−1). (b) CVs of the AuNP/PEI/rGO-modified SPE (conditions: 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH 7.4), scan rate 50 mV s−1, n = 3) prepared under different microwave synthesis conditions. (c) CV responses of four different modified electrodes, namely GO, rGO, AuNP/rGO and AuNP/PEI/rGO-modified SPE. (d) Plot of the 3-MPA modification time versus current change. (e) Plot of the EDC/sulfo-NHS modification time (0, 15, 30, 60, 120, and 240 min) versus current change. (f) Plot of the MMP-1 antibody modification time versus current change. | |
Table 1 Electrochemical impedance values for the different modified electrodes
| Modified material |
Impedance radius (Ohm) |
| Bare SPE |
116.35 |
| Au/PEI/rGO/SPE |
104.49 |
| 3-MPA/Au/PEI/rGO/SPE |
156.82 |
| anti-MMP-1/EDC/NHS/Au/PEI/rGO/SPE |
225.87 |
| BSA/anti-MMP-1/EDC/NHS/Au/PEI/rGO/SPE |
256.93 |
3.4 Optimization study
3.4.1 Current response under different microwave synthesis conditions.
Cyclic voltammetry (CV) was used to investigate the AuNP/PEI/rGO nanocomposites prepared under different synthesis conditions. An optimum current response was obtained under the conditions of 150 W and 150 °C, as shown in Fig. 5b. These parameters produced a maximum peak current and led to the optimum performance of the AuNP/PEI/rGO nanocomposite. Upon reduction of the synthesis temperature below 150 °C, no apparent change in the structure was observed, due to the reduction reaction state giving a sufficient number of functional groups for modifying the AuNPs.47 Compared to the other modified electrodes, the AuNP/PEI/rGO/SPE (150 W and 150 °C) shows the lowest peak to peak separation and higher peak current ratio, due to the higher electron transfer rate.
3.4.2 Different compositions of the modified electrodes.
The CV responses of four different modified electrodes, namely GO, rGO, AuNP/rGO, and the AuNP/PEI/rGO-modified SPE, are shown in Fig. 5c. The rGO-modified electrode showed a higher peak current response than GO due to the reduction of GO creating defects and disorders that enhance the electrochemical performance. AuNP-decorated rGO showed a higher peak current than the GO and rGO-modified electrodes. Owing to the high conductivity and catalytic activity of the AuNPs, the modified electrode exhibited good performance. The AuNP/PEI/rGO-modified SPE exhibited an even larger peak current and well-defined redox peak. The addition of PEI prevented the AuNPs from agglomeration by the stabilizing effect of the amine groups, accounting for the excellent electrochemical performance of the AuNP/PEI/rGO-modified SPE.
3.4.3 Testing the optimal conditions for Au–S bond modification.
The immunosensor developed in this study required modification with antibodies for specific analyses. AuNPs are commonly used for modifying the properties of other biological materials, and thus functionalized AuNPs were also used in this study. 3-Mercaptopropionic acid (3-MPA) was used as a source of S, as it contains –SH and –COOH functional groups. Natural sulfur–gold bonding, along with the tail carboxyl groups, was used to facilitate antibody modification. As can be seen in Fig. 5d, the Au–S reaction is rapid, with a modification time of just an hour producing substantial current changes. The peak current was found to gradually decrease over time, due to an increase in the number of functional groups on the modified SPE surface. This was seen as an increase in the peak current resistance during the first 6 h, after which the electrode was stabilized. Thus, an optimum modification time of 6 h was selected.
3.4.4 EDC/NHS cross-linking modification time.
EDC/NHS offers a traditional cross-linking method that has been widely used for many years. In this method, EDC (1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride) is used to couple a primary amine to a carboxyl or phosphate group. This is often also used in combination with NHS (N-hydroxysuccinimide) or sulfo-NHS (N-hydroxythiosuccinimide). Since EDC is water-soluble and highly reactive, the main function of NHS is to serve as an intermediate for the reaction to increase the stability of the active ester and improve the efficiency of crosslinking. As can be seen in Fig. 5e, the cross-linking reaction is quite rapid. As a result of the cross-linking reaction, the surface of the electrode increased by a single layer, resulting in a significant decrease in the current. The reaction was completed in 30 min. Increasing the cross-linking time resulted in poor electrode activity and a higher current. Thus, 30 min was chosen as the optimum cross-linking reaction time.
3.4.5 Optimal anti-MMP-1 modification time.
The antibody modification time affects the amount of antibody grafted, and subsequently the current, as the antibody contains an amine group. Fig. 5f shows a current density plot, with the current gradually increasing with the reaction time, reaching a maximum value at a reaction time of 6 h. After 8 h, a drop in current was observed due to the modification reaching saturation, with too many antibodies adhering to the surface of the electrode causing biomolecular blockage of the working electrode, with the current drop hindering electron conduction. Thus, an optimal antibody modification time of 6 h was selected.
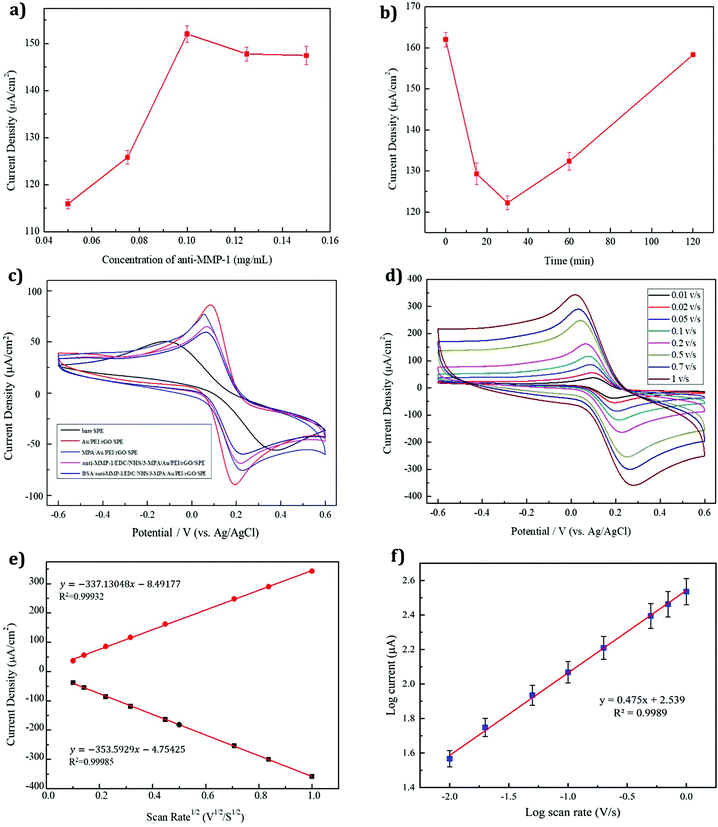
3.4.6 The optimal concentration of the anti-MMP-1 modifier.
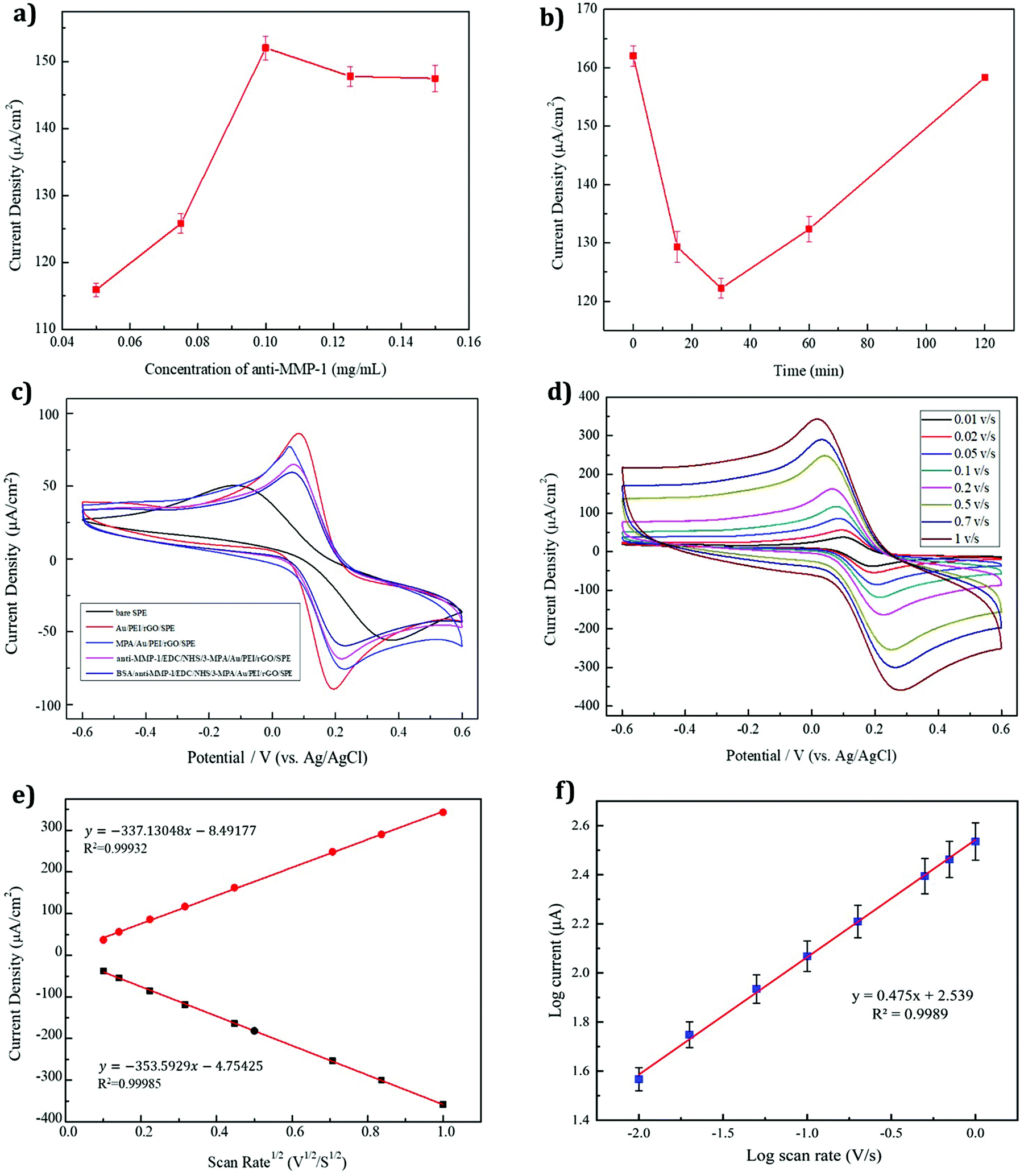
In an immunosensor, the concentration of the antibody modifier also affects the current response. Fig. 6a shows that as the concentration of the MMP-1 antibody increases, the number of active sites of the modified electrode also increases, as shown by an increase in the current density. At an antibody concentration of 0.1 mg ml−1, the current density reached a maximum, exhibiting a plateau as the antibody concentration was increased further due to the saturation of the electrode surface. Thus, an optimal antibody concentration of 0.1 mg ml−1 was selected for the experiments.
 |
| | Fig. 6 (a) Plots of the MMP-1 antibody modification concentration (0.05, 0.075, 0.1, 0.125, and 0.15 mg ml−1) versus current change and (b) BSA modification time (0, 15, 30, 60, and 120 min) versus current change (conditions: 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH 7.4), scan rate 50 mV s−1, n = 3). (c) Electrochemical CV responses of the various modified electrodes for MMP-1 sensing in 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH 7.4), scan rate 50 mV s−1. (d) CVs of different scan sweeps ranging from 0.01 to 1 V s−1 with 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH: 7.4). (e) Calibration plot for the square root of the scan sweeps versus anodic and cathodic peak currents. (f) Calibration plot for the log of the scan sweeps versus the log of peak current. | |
3.4.7 Optimal bovine serum albumin (BSA) modification time.
Detection by using an immunosensor involves specific binding between the antigen and antibody. Following the modification of the antibody on the working electrode, non-specific sites on the surface of the electrode must be blocked to prevent them from affecting antigen detection. Errors, as well as the modification time, can also affect the results of such blockage. Fig. 6b shows a plot of the BSA modification time versus the current response. The blocking response was shown to be rapid, with a modification time of 30 min giving optimum performance, and an increase in the modification time leading to poor activity.
3.5 Layer-by-layer analysis of the electrochemical sensors
Electrochemical changes in the various modified electrodes were evaluated by CV. The CV responses of the differently modified electrodes (bare SPE, AuNP/PEI/rGO/SPE, 3-MPA/AuNP/PEI/rGO/SPE, anti MMP-1/EDC/NHS/3-MPA/AuNP/PEI/rGO/SPE and BSA/anti-MMP-1/EDC/NHS/3-MPA/AuNP/PEI/rGO/SPE) for MMP-1 sensing in 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH 7.4) are evaluated at a scan rate of 50 mV s−1 (Fig. 6c). The bare SPE exhibited poor catalytic activity and electron transport properties compared to the other modified electrodes. The AuNP/PEI/rGO-modified nanocomposite on the SPE showed a better response than the bare SPE, due to the electron transfer-promoting properties of the Au nanocomposite and the PEI amine base being beneficial for the red blood salt K3[Fe(CN)6]. Furthermore, the Au/PEI/rGO graph displays a weak peak at around −0.4 V due to the combination of gold ions with the polymer material in the potassium chloride/ferro-ferricyanide solution system.48 The yellow blood salt K4[Fe(CN)6] is close to each other, so the overall resistance decreases, and the current density increases. Subsequently, following the 3-MPA modification, the current density decreased and the resistance increased due to an increase in the thickness of the film layer, confirming the 3-MPA modification. The AuNP/PEI/rGO/SPE has Au attached to the S atom of 3-MPA, generating a thiol-functionalized gold (Au–S) bond. Subsequently, the terminal carboxylic acid group (–COOH) of 3-MPA was activated by the modification of the EDC/NHS solution. Using EDC/NHS as a cross-linking agent for anti-MMP-1 modification resulted in an increase in the current-reducing resistance, with the antibody crosslinking hindering the electron transfer. Herein, the 3-MPA has a terminal –COOH group which is covalently bound to the terminal –NH2 group of anti-MMP-1 through the EDC/NHS cross-linking chemistry.49 The binding of anti-MMP-1 to MMP-1 was highly specific, with the non-specific sites likely being occupied by BSA as a barrier. This further reduced the electron conduction, increasing the current resistance due to the decrease in the current density. Thus, the successful modification of BSA/anti-MMP-1/EDC/NHS/3-MPA/AuNP/PEI/rGO/SPE was evident.
3.6 Analysis of different scan rates
Analysis of the electrochemical kinetics, including the identification of the rate-determining step, was performed using CV with different scan rates. The reaction rate at the electrode surface was rapid, allowing for the effective transfer between the electrode and the solution interface. CV is performed with scan rates in the range of 0.01–1 V using 5 mM Fe(CN)63−/4− dissolved in a 0.1 M PBS/0.1 M KCl solution (pH 7.4) (Fig. 6d). The CV results showed that changing the scan rate produced changes in the peak current and potential. Linear regression of the redox peak current values obtained at different scan rates showed a linear relationship with the square root of the rate, allowing the rate-determining step of diffusion to be defined for this sensor using the Randles–Sevcik equation:50| | | ip = 2.69 × 105An3/2Dυ1/2 | (1) |
where ip is the cathodic or anodic peak current in the CV curve, A is the electrochemical reaction area, n is the number of electrons transported in the redox reaction, C is the concentration of the reactant in the solution, D is the diffusion coefficient of the reactant, and υ is the scan rate. From Fig. 6d, it is apparent that the relative redox peak current value increases with the reaction rate, and the current density and υ1/2 are linearly derived from Fig. 6e. The correlation coefficients of the oxidation and reduction peaks were both R2 = 0.999. Thus, the rate-determining step of the immunosensor was found to be related to the diffusion, and the electrons were stable during the transfer process. Besides, Fig. 6f shows the calibration plot for the log of peak current response versus the log of scan rates.
The electron transfer rate constant was calculated using the following equations:51
| | 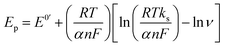 | (2) |
| | 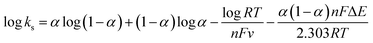 | (3) |
where
ks is the surface electron transfer rate constant,
α is the electron transfer coefficient and Δ
E is the peak potential difference.
α was calculated using the following equation (equation
(4)):
| | 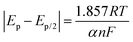 | (4) |
where
Ep/2 is the potential corresponding to half of the peak current and
Ep is the peak potential. The values were determined to be as follows:
α = 0.663,
v = 0.001 V s
−1,
F = 96
485 C mol
−1,
n = 1,
E° = −0.6 V,
Ep/2 = 0.12 V,
R = 8.314 J mol
−1 K
−1,
Ep = 0.192 V, and
T = 298.15 K, respectively. Then the electron transfer rate (
ks) was calculated to be 0.174 s
−1.
3.7 DPV analysis of MMP-1
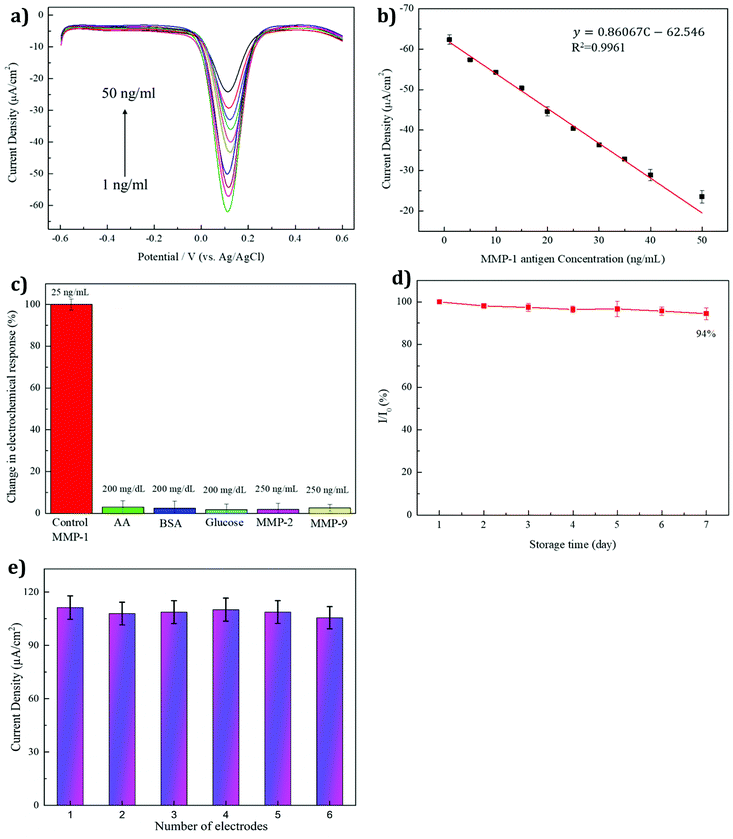
Differential pulse voltammetry is one of the important useful techniques for measuring the sensitivity, detection limit, and dynamic linear range of a chemical reaction. As shown in Fig. 7a, the differential pulse voltammetry (DPV) response decreases when the antigen concentration is higher, due to the increased antigen concentration hindering the electrode surface. Hence, the corresponding electron transfer rate also decreased. Owing to the formation of antigen–antibody immune complexes, they create an insulating layer and evolved repulsive electrostatic interaction between antigen and [Fe(CN)6]3−/4−. As shown in Fig. 7b, the MMP-1 concentration shows a near-linear relationship with the current density in the range of 1–50 ng ml−1. The corresponding linear regression equation is as follows: current density (μA cm−2) = 0.860 concentration (ng ml−1) – 62.546 with an R2 value of 0.996 (n = 5, RSD < 5%). The lowest detection limit (LOD: 3 × SD/b) was calculated to be ∼0.219 ng ml−1.
 |
| | Fig. 7 (a) DPV responses at MMP-1 concentrations of 1, 5, 10, 15, 20, 25, 30, 35, 40 and 50 ng ml−1 in 5 mM Fe(CN)63−/4− dissolved in 0.1 M PBS/0.1 M KCl (pH 7.4), scan rate 50 mV s−1. (b) Linear plot of the MMP-1 concentrations versus peak current density. (c) Plot depicting the different interfering substances versus relative error percentage. (d) Storage stability over 7 days at a temperature of 4 °C versus I/I0 (%). Reproducibility analysis (e). | |
3.8 Selectivity, stability, and reproducibility analyses
Effectively avoiding interference is a basic requirement of an immunosensor. To investigate the interference effects of the MMP-1 sensor in this study, several interference substances (vitamin C (AA), protein (BSA), and glucose) and the MMP family (MMP-2 and MMP-9) were tested. In the human body, the normal blood glucose concentration is 70–110 mg dl−1. The concentrations of the interference substances were tested at a glucose concentration of 200 mg dl−1, higher than that in the body, while the concentrations of the MMP family interference substances were found to be ten times higher than the concentration of MMP-1. The concentrations used were as follows: 200 mg dl−1 for the interfering substances BSA, glucose, and vitamin C, and 250 ng ml−1 for the interfering substances MMP-2 and MMP-9, with an MMP-1 antigen concentration of 25 ng ml−1. The antigen was used without adding interference substances as a control, and the test results are shown in Fig. 7c. Upon the addition of AA, BSA, glucose, MMP-2, and MMP-9 to the MMP-1 antigen, the relative error percentage was calculated to be less than 5% (n = 5). This result suggested that the MMP-1 immunosensor had good specificity for MMP-1 binding. The stability of the sensor is also important to retain the specificity. The stability of the sensor was measured while storing the electrode at 4 °C for one week, with the results shown in Fig. 7d. The obtained relative standard deviation (RSD) was approximately 94%, which was only 6% lower than that obtained on the first day. This result indicated that the sensor remained stable while stored at 4 °C, retaining the excellent anti-MMP-1 activity. The reproducibility was evaluated using six different MMP-1/BSA/anti-MMP-1/EDC/NHS/3-MPA/AuNP/PEI/rGO fabricated working SPEs via the single measurement. The RSD was assessed to be 3.68% based on the electroanalytical signal of the six replicates (Fig. 7e), demonstrating the good reproducibility of the developed immunosensor.
3.9 Real sample analysis
3.9.1 Enzyme immunoassay.
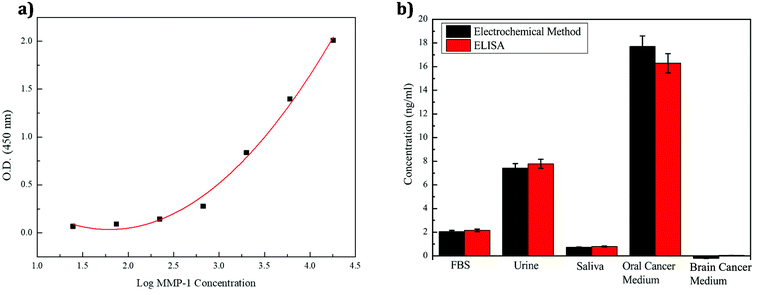
To verify the accuracy and practicability of the electrochemical method for MMP-1 detection in different environments, enzyme immunoassays were used to detect the presence of MMP-1 in real samples. A commercial human MMP-1 ELISA kit was also purchased for an authentic sample content analysis of MMP-1 for comparison. Using standard concentrations of 18000, 6000, 2000, 666.7, 222.2, 74.07, and 24.69 pg ml−1, the absorbance of each sample was measured using a full-wavelength absorption light-fluorescence composite analysis system at a wavelength of 450 nm (Table 2), and logarithmic regression curves were obtained using the equation y = 0.331x2 − 1.187x + 1.098, R2 = 0.9904 (Fig. 8a). As shown in Fig. 8a, the absorbance and concentration are determined from the regression curve.
 |
| | Fig. 8 (a) Graph of the log of the MMP-1 concentration versus absorbance. (b) Comparison of ELISA and electrochemical methods used for the measurements in real samples. | |
Table 2 Light absorbance values for each standard concentration
| Standard (pg mL−1) |
Optical density (450 nm) |
| Standard A 18000 |
2.00845 |
| Standard B 6000 |
1.39651 |
| Standard C 2000 |
0.8366 |
| Standard D 666.7 |
0.27699 |
| Standard E 222.2 |
0.14259 |
| Standard F 74.07 |
0.09131 |
| Standard G 24.69 |
0.06721 |
3.9.2 Determination of MMP-1 concentrations in various body fluids.
In these experiments, the concentrations of MMP-1 in different body fluid environments, including cell culture media of oral cancer cells with MMP-1 and brain tumor cells without MMP-1, were determined. Since the human serum is difficult to obtain, it was instead simulated using bovine serum, saliva, and urine samples. The three samples were first centrifuged, followed by the removal of the supernatant and addition of the MMP-1 antigen to each sample. Cell culture media for detecting human oral squamous carcinoma (HSC-3 and JCRB) and brain tumor (C6 and ATCC) cell lines were prepared similarly. Table 3 shows the MMP-1 concentrations measured by the electrochemical method, and Table 4 shows the MMP-1 concentrations determined by the ELISA method. The concentrations of MMP-1 and cell culture fluids in various environments were thus estimated by both the enzyme immunoassay and the electrochemical method. Interestingly, the concentration value detected in the brain tumor cell culture solution was negative, indicating that no MMP-1 was detected by the electrochemical method, with only a low concentration detected by the ELISA method. Thus, the results of this study did not indicate the presence of MMP-1 in brain tumors. As shown in Fig. 8b, the error (the difference between the results of the two methods) is less than 10%. Thus, the similarity of the results obtained by the electrochemical measurements and enzyme immunoassay indicated that the proposed sensor is highly practicable.
Table 3 MMP-1 concentrations in the real samples measured by the electrochemical method (n = 3)
| Electrochemical detection |
Average measured electrochemical response (A cm−2) |
RSD % |
Calculated amount detected (ng mL−1) |
| FBS |
−6.08 × 10−5 |
1.69% |
2.04 |
| Urine |
−5.65 × 10−5 |
3.07% |
7.43 |
| Saliva |
−6.19 × 10−5 |
6.19% |
0.72 |
| Oral cancer medium |
−4.73 × 10−5 |
5.84% |
17.71 |
| Brain cancer medium |
−6.27 × 10−5 |
1.92% |
−0.21 |
Table 4 Measurements for the enzyme immunoassays (MMP-1, n = 3) of authentic samples
| ELISA |
Average measured response (O.D.) |
RSD % |
Calculated amount detected (ng mL−1) |
| FBS – fetal bovine serum; O.D. – optical density; and RSD – relative standard deviation. |
| FBS |
0.824 |
2.2% |
2.157 |
| Urine |
1.488 |
3.7% |
7.78 |
| Saliva |
0.657 |
2.9% |
0.791 |
| Oral cancer medium |
1.97 |
1.6% |
16.293 |
| Brain cancer medium |
0.046 |
0.5% |
0.037 |
4 Conclusion
In this study, an AuNP/PEI/rGO composite was prepared using a microwave reduction technique involving the simultaneous reduction of GO and gold chloride in a polyethyleneimine environment. The as-prepared material was examined by suitable spectrophotometric and voltammetric techniques, and an optimization study revealed the following optimum conditions for synthesis: (i) a microwave temperature of 150 °C and a power of 150 W, (ii) a modification time (for 3-mercaptopropionic acid) of 6 h, (iii) an EDC/NHS optimum crosslinking time of 30 min, (iv) an anti-MMP-1 modification time of 6 h, (v) a optimal concentration of the anti-MMP-1 modifier of 0.1 mg ml−1 and (vi) a time of 30 min for the BSA blocking reaction. The EIS and CV results were used to compare the differently modified electrodes for the detection of the MMP-1 antigen. DPV showed a dynamic linear range of 1–50 ng ml−1 with a LOD of ∼0.219 ng ml−1 and a sensitivity of 0.086 mA cm−2. The proposed modified electrode showed excellent selectivity and stability, and a comparison of ELISA and electrochemical methods showed that the modified electrode is highly practicable for the determination of MMP-1 concentrations in body fluids.
Author contributions
Xinke Liu: writing – review and editing, conceptualization, methodology, project administration, and funding acquisition. Lu-Yin Lin: writing – review and editing, methodology, and funding acquisition. Fu-Yen Tseng: writing – review and editing, methodology, formal analysis, data curation, and visualization. Yu-Cheng Tan: investigation, formal analysis, data curation, and validation. Jian Li: investigation, formal analysis, and data curation. Li Feng: investigation, formal analysis, and data curation. Lijun Song: resources, formal analysis, and data curation. Chih-Fang Lai: methodology and funding acquisition. Xiaohua Li: methodology, project administration, funding acquisition, and writing – review and editing. Jr-Hau He: methodology, funding acquisition, and writing – review and editing. Rajalakshmi Sakthivel: writing – original draft, writing – review and editing, methodology, formal analysis, data curation, and visualization. Ren-Jei Chung: conceptualization, methodology, supervision, project administration, funding acquisition, and writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support of this research from the Ministry of Science and Technology of Taiwan (MOST 109-2222-E-027-004) and the National Taipei University of Technology-Shenzhen University Joint Research Program (NTUT-SZU-109-02 (2020009); NTUT-SZU-110-09 (2021009)); in part from the Guangdong Province Key Research and Development Plan (2019B010138002; 2020B010174003); and the International Distinguished Visiting Professor support for Prof. Jr-Hau He from the National Taipei University of Technology. The first and second authors contributed equally to this work.
References
- H. Nagase, R. Visse and G. Murphy, Cardiovasc. Res., 2006, 69, 562–573 CrossRef CAS PubMed.
- A. Winer, S. Adams and P. Mignatti, Mol. Cancer Ther., 2018, 17, 1147–1155 CrossRef CAS PubMed.
-
M. Ferrantini and F. Belardelli, Gene therapy of cancer with interferon: lessons from tumor models and perspectives for clinical applications, 2000, vol. 10, pp. 145–157. Academic Press Search PubMed.
- S. D. Shapiro, Curr. Opin. Cell Biol., 1998, 10, 602–608 CrossRef CAS PubMed.
- S. C. Lin, M. Y. Chung, J. W. Huang, T. M. Shieh, C. J. Liu and K. W. Chang, J. Oral Pathol. Med., 2004, 33, 323–326 CrossRef CAS PubMed.
- Y. Feng, Q. Li, J. Chen, P. Yi, X. Xu, Y. Fan, B. Cui, Y. Yu, X. Li, Y. Du and Q. Chen, Int. J. Oral Sci., 2019, 11, 7 CrossRef CAS PubMed.
- E. Sunami, N. Tsuno, T. Osada, S. Saito, J. Kitayama, S. Tomozawa, T. Tsuruo, Y. Shibata, T. Muto and H. Nagawa, Oncologist, 2000, 5, 108–114 CrossRef CAS PubMed.
- E. A. Baker, F. G. Bergin and D. J. Leaper, Br. J. Surg., 2000, 87, 1215–1221 CrossRef CAS PubMed.
- P. Boström, M. Söderström, T. Vahlberg, K. O. Söderström, P. J. Roberts, O. Carpén and P. Hirsimäki, BMC Cancer, 2011, 11, 348 CrossRef PubMed.
- K. Kameyama, Pathol., Res. Pract., 1996, 192, 20–26 CrossRef CAS.
- M. Li, T. Xiao, Y. Zhang, L. Feng, D. Lin, Y. Liu, Y. Mao, S. Guo, N. Han, X. Di and K. Zhang, Lung Cancer, 2010, 69, 341–347 CrossRef PubMed.
- F. Ozden, C. Saygin, D. Uzunaslan, B. Onal, H. Durak and H. Aki, J. Cancer Res. Clin. Oncol., 2013, 139, 1373–1382 CrossRef CAS PubMed.
- D. H. Shin, U. Dier, J. A. Melendez and N. Hempel, Biochim. Biophys. Acta, 2015, 1852, 2593–2602 CrossRef CAS PubMed.
- J. Wang, Biosens. Bioelectron., 2006, 21, 1887–1892 CrossRef CAS PubMed.
- X. Chen, X. Jia, J. Han, J. Ma and Z. Ma, Biosens. Bioelectron., 2013, 50, 356–361 CrossRef CAS PubMed.
- D. Wang, Y. Yuan, Y. Zheng, Y. Chai and R. Yuan, Chem. Commun., 2016, 52, 5943–5945 RSC.
- G. Yang, L. Li, R. K. Rana and J. J. Zhu, Carbon, 2013, 61, 357–366 CrossRef CAS.
- Z. Yin, Y. Wang, T. T. Zheng, R. Zhang, X. Li, J. J. Zhu and X. Chen, Sci. Adv. Mater., 2015, 7, 1581–1588 CrossRef CAS.
- B. S. Munge, J. Fisher, L. N. Millord, C. E. Krause, R. S. Dowd and J. F. Rusling, Analyst, 2010, 135, 1345–1350 RSC.
- E. Shabani, M. J. Abdekhodaie, S. A. Mousavi and F. Taghipour, Biochem. Eng. J., 2020, 164, 107772 CrossRef CAS.
- J. J. Shi, T. T. He, F. Jiang, E. S. Abdel-Halim and J. J. Zhu, Biosens. Bioelectron., 2014, 55, 51–56 CrossRef CAS PubMed.
- B. Arévalo, A. ben Hassine, A. Valverde, V. Serafín, A. Montero-Calle, N. Raouafi, J. Camps, M. Arenas, R. Barderas, P. Yáñez-Sedeño and S. Campuzano, Talanta, 2021, 225, 122054 CrossRef PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- A. de la Escosura-Muñiz, C. Sánchez-Espinel, B. Díaz-Freitas, A. González-Fernández, M. Maltez-da Costa and A. Merkoçi, Anal. Chem., 2009, 81, 10268–10274 CrossRef PubMed.
- C. S. Santos, R. Mossanha, K. Wohnrath, J. Inaba and C. A. Pessôa, J. Electrochem. Soc., 2016, 163, B158–B162 CrossRef CAS.
- X. Sun, S. Dong and E. Wang, Polymer, 2004, 45, 2181–2184 CrossRef CAS.
- H. Chen, Y. Wang, Y. Wang, S. Dong and E. Wang, Polymer, 2006, 47, 763–766 CrossRef CAS.
- T. Ishii, H. Otsuka, K. Kataoka and Y. Nagasaki, Langmuir, 2004, 20, 561–564 CrossRef CAS PubMed.
- R. Sakthivel, M. Annalakshmi, S. M. Chen and S. Kubendhiran, J. Electrochem. Soc., 2019, 166, B680–B689 CrossRef CAS.
- V. K. Ponnusamy, V. Mani, S. M. Chen, W. T. Huang and J. F. Jen, Talanta, 2014, 120, 148–157 CrossRef CAS PubMed.
- V. Mulens-Arias, A. Nicolás-Boluda, A. Gehanno, A. Balfourier, F. Carn and F. Gazeau, Nanoscale, 2019, 11, 3344–3359 RSC.
- S. Khetani, V. Ozhukil Kollath, V. Kundra, M. D. Nguyen, C. Debert, A. Sen, K. Karan and A. Sanati-Nezhad, ACS Sens., 2018, 3, 844–851 CrossRef CAS PubMed.
- J. Basu, J. K. Basu and T. K. Bhattacharyya, Int. J. Smart Nano Mater., 2010, 1, 201–223 CrossRef CAS.
- K. Ojha, O. Anjaneyulu and A. K. Ganguli, Curr. Sci., 2014, 107, 397–418 CAS.
- S. K. Tiwari, S. Sahoo, N. Wang and A. Huczko, J. Sci.: Adv. Mater. Devices, 2020, 5, 10–29 Search PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- R. Sakthivel, M. Annalakshmi, S. M. Chen, S. Kubendhiran, R. Anbazhagan and H. C. Tsai, J. Taiwan Inst. Chem. Eng., 2019, 96, 549–558 CrossRef CAS.
- K. S. Divya, A. Chandran, V. N. Reethu and S. Mathew, Appl. Surf. Sci., 2018, 444, 811–818 CrossRef CAS.
- X. Liu, L. Pan, T. Lv, G. Zhu, Z. Sun and C. Sun, Chem. Commun., 2011, 47, 11984–11986 RSC.
- Y. H. Ding, P. Zhang, Q. Zhuo, H. M. Ren, Z. M. Yang and Y. Jiang, Nanotechnology, 2011, 22, 215601 CrossRef CAS PubMed.
-
D. Gupta, D. Jamwal, D. Rana and A. Katoch, Applications of Nanocomposite Materials in Drug Delivery, Woodhead Publishing, 2018, pp. 619–632 Search PubMed.
- W. S. Hummers Jr. and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef.
- G. Yasin, M. Arif, M. Shakeel, Y. Dun, Y. Zuo, W. Q. Khan, Y. Tang, A. Khan and M. Nadeem, Adv. Eng. Mater., 2018, 20, 1701166 CrossRef.
- J. Borowiec, K. Yan, C. C. Tin and J. Zhang, J. Electrochem. Soc., 2015, 162, H164 CrossRef CAS.
- C. Jun-Gang, P. Tong-Jiang, S. Hong-Juan, L. Bo and Z. Er-Zheng, Chin. J. Inorg. Chem., 2014, 30, 779–785 Search PubMed.
- N. B. Li, J. H. Park, K. Park, S. J. Kwon, H. Shin and J. Kwak, Biosens. Bioelectron., 2008, 23, 1519–1526 CrossRef CAS PubMed.
- M. Braiek, K. B. Rokbani, A. Chrouda, B. Mrabet, A. Bakhrouf, A. Maaref and N. Jaffrezic-Renault, Biosensors, 2012, 2, 417–426 CrossRef CAS PubMed.
- K. B. Oldham, J. Electroanal. Chem., 1979, 105, 373–375 CrossRef CAS.
- J. Hong, Y. X. Zhao, B. L. Xiao, A. Moosavi-Movahedi, H. Ghourchian and N. Sheibani, Sensors, 2013, 13, 8595–8611 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |

 *c
*c