
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical radical reactions of alkyl iodides: a highly efficient, clean, green alternative to tin reagents†
Diyuan
Li
 ,
Tsz-Kan
Ma
,
Reuben J.
Scott
and
Jonathan D.
Wilden
*
,
Tsz-Kan
Ma
,
Reuben J.
Scott
and
Jonathan D.
Wilden
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: j.wilden@ucl.ac.uk
First published on 8th May 2020
Abstract
An electrochemical ‘redox-relay’ system has been developed which allows the generation of C-centered radicals. Intermolecular ‘tin-like’ radical reactions can subsequently be conducted under the most benign of conditions. The yields and efficiency of the processes are competitive and even superior in most cases to comparable conditions with tributyltin hydride. The use of air and electricity as the promotor (instead of a tin or other reagent) combined with the aqueous reaction media make this a clean and ‘green’ alternative to these classic C–C bond forming processes.
Introduction
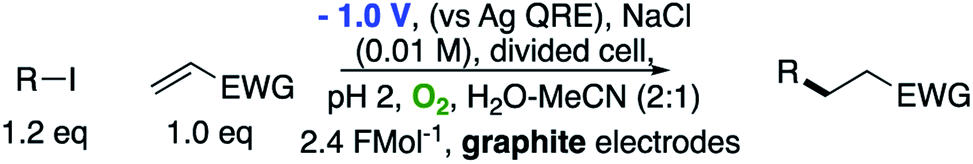
The use of radical reactions for the construction of complex organic molecules are now almost ubiquitous in synthetic chemistry.1 Historically however, organic chemistry has relied on tributytin hydride (and similar reagents) to mediate such processes.2 Organotin reagents have proved remarkably efficient for the generation and mediation of carbon-centered radicals. Their drawbacks however are significant; the reagents are extremely toxic, expensive and the side-products are often difficult to remove from the reaction medium post reaction. As such, recent years have seen intense interest in alternatives to these extremely useful reagents.3 Almost without exception however, none have matched the efficiency, generality and functional group tolerance of tin-based reagents. The result of this is that many chemists, particularly those in industry have shunned tin reagents, however, in doing so they necessarily preclude the advantages that this class of radical reactions can offer in terms of the construction of complex organic architectures. Two methods that are becoming more popular for the generation of radical species are electrochemistry and photoredox catalysis.4,5 Photoredox catalysis has already, in a short amount of time, proved to be a powerful tool for the activation of organic molecules.6 Similarly, synthetic organic electrochemistry is becoming an increasingly popular alternative way to activate organic molecules via direct addition or removal of electrons to generate reactive radical species.7 Unfortunately, the direct electrochemical reduction of alkyl halides requires highly reducing potentials and as such has not found general application because of the incompatibility of these extreme potentials with various functional groups.8,9 Herein, we outline an operationally simple, mild and molecular oxygen mediated indirect electrochemical approach to the formation of carbon centered radicals from alkyl iodides using the ‘standard’ feedstocks used in tributyltin hydride mediated reactions, namely an alkyl halide and an alkene acceptor (i.e. the classic Giese reaction).10 We also demonstrate its synthetic utility in intermolecular C–C bond formation with a range of alkene and alkyne acceptors.We here demonstrate that ‘tin-like’ radical reactions can be achieved using only mildly reductive electrochemical conditions combined with aerial oxygen in semi-aqueous conditions. The mildly reducing conditions (−1.0 V vs. Ag QRE) are selective for the activation of molecular oxygen and the desired radical reaction only and does not interfere with other functional groups in the molecule. Similarly, although aqueous conditions are employed, the applied potential is not sufficient to effect the reduction of water (ca. −1.5 V vs. Ag QRE under our conditions). Furthermore, no tin, silicon, phosphorous or any other mediator apart from air is required, and the construction of the electrochemical cell employs only environmentally benign and cheap materials. In particular, inexpensive graphite electrodes are employed. Fig. 1 compares the previous conditions with the work described here.11–14
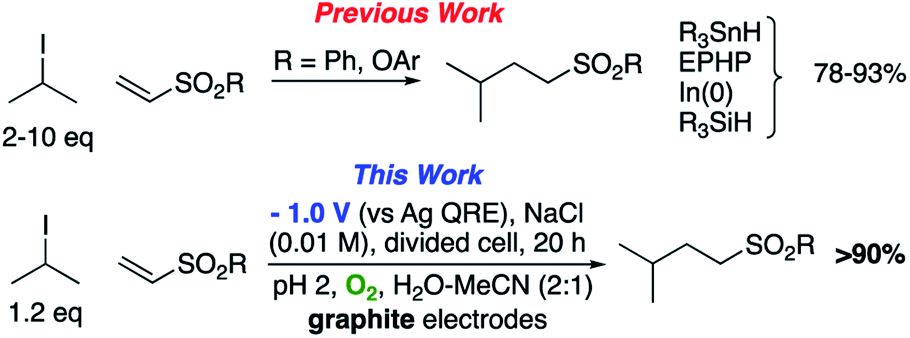 | ||
| Fig. 1 Comparison of classic intermolecular Giese reactions with the electrochemical method. | ||
Results & discussion
We first made the observation that a methanol–water solution of phenyl vinyl sulfone and 2-iodopropane when exposed to a mild reducing potential (constant potential, 1.0 V vs. Ag wire quasi-reference electrode, graphite rod working electrode, divided cell) undergo efficient C–C bond formation to yield the sulfone 1 in good yield as well as some β-hydration product 2 (Scheme 1). The reactants were contained in the cathodic chamber and no mixing of organic species were observed with the anodic compartment. Furthermore, only the electrochemical oxidation of a small amount of water was observed in the anodic chamber, meaning no toxic or hazardous materials are required to be removed from the apparatus after the reaction.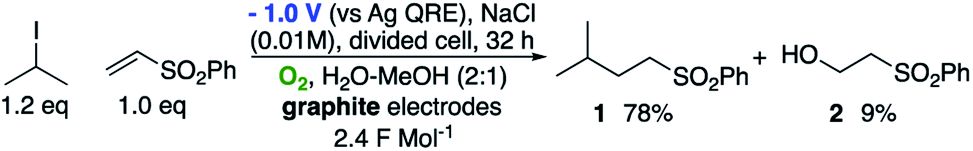 | ||
| Scheme 1 Initial observations. | ||
Intrigued by this reaction and suspecting that exposure to air was a critical facet, we set about exploring the optimal conditions for this process as shown in Table 1. It seems likely that the hydroxy product results from a simple base-mediated addition reaction of water to the electron deficient alkene. As such, this undesired product was relatively easily eliminated by lowering the pH and employing acetonitrile as the organic co-solvent.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Atmos. | Time, h | pH | 1 , % | 2 , % |
| a Isolated yield. b pH adjusted with NaOH. c pH adjusted with HCl. d PBS 7.4 buffer. | ||||||
| 1 | MeOH | Air | 32 | 7 | 0 | 70 |
| 2 | H2O–MeOH (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
Air | 32 | 7 | 78 | 9 |
| 3 | MeCN | Air | 32 | 7 | Trace | 0 |
| 4 | H2O–MeCN (1:3) |
Air | 32 | 7 | Trace | Trace |
| 5 | H2O–MeCN (1:1) |
Air | 32 | 7 | 68 | Trace |
| 6 | H2O–MeCN (2:1) |
Air | 32 | 7 | 90 | Trace |
| 7 | H2O–MeCN (2:1) |
Ar degassed | 32 | 7 | 0 | 0 |
| 8 | H2O–MeCN (2:1) |
Stream of O2 | 32 | 7 | Trace | Trace |
| 9 | H2O–MeCN (2:1) |
Air | 20 | 9b | 47 | 53 |
| 10 | H2O–DMF (2:1) |
Air | 20 | 2c | 74 | 0 |
| 11 | H2O–MeCN (2:1) |
Air | 20 | 7.4d | 86 | 14 |
| 12 | H2O–MeCN (2:1) |
Air | 20 | 5c | 82 | 18 |
| 13 | H2O–MeCN (2:1) |
Air | 20 | 2c | 100 | 0 |

Table 1 shows the optimisation for the reaction outlined in Scheme 1. A number of observations are noteworthy. Firstly, water is essential for the success of the reaction. Secondly, it was noted that the pH of the solution slowly rises as the reaction proceeds, and as such, the formation of the undesired hydroxy addition product accelerates. Lowering the pH from the outset, therefore, minimises this undesired side-reaction. Finally, as demonstrated in entry 7, oxygen is required for the reaction to proceed however entry 8 demonstrates that there is no advantage to performing the reaction in an oxygen atmosphere. In fact, such an approach is actually detrimental and almost completely attenuates the reaction with only traces of products detected and the almost quantitative recovery of starting material. This gave us the first inclination that oxygen may be required only in sub-stoichiometric quantities for the putative radical reaction that ensues.
Having established a robust set of conditions that resulted in high yields of the desired product 1, we were then keen to establish the scope of the reaction with a more diverse set of alkyl halides and acceptors. The results are outlined in Table 2.

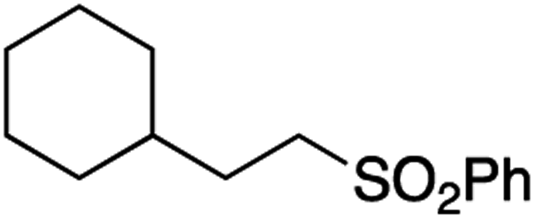
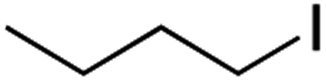

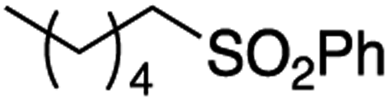
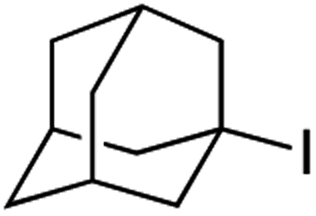

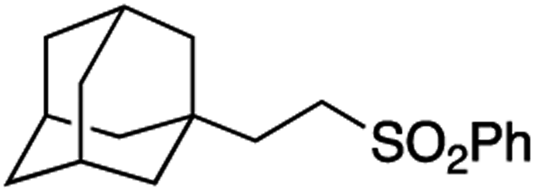
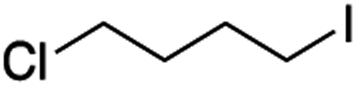

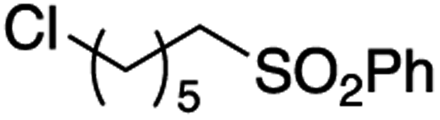
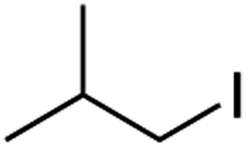

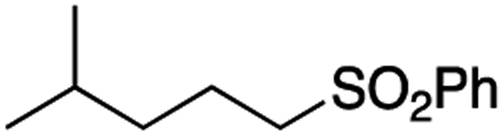
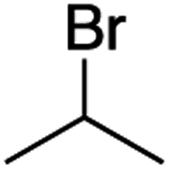

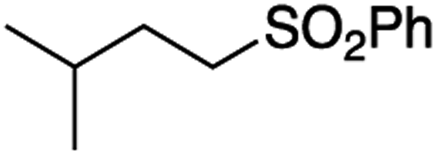
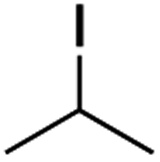
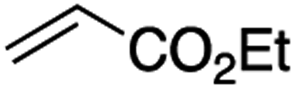
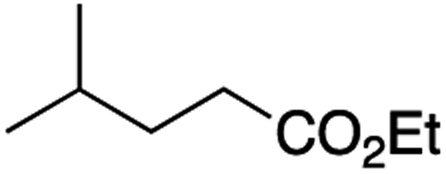
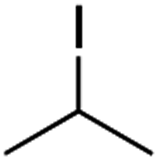
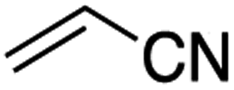
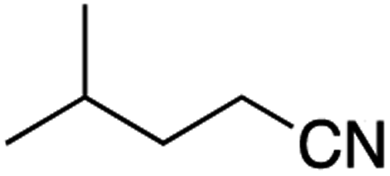
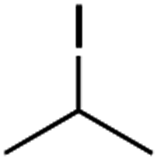
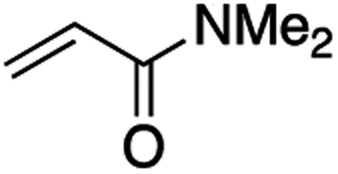
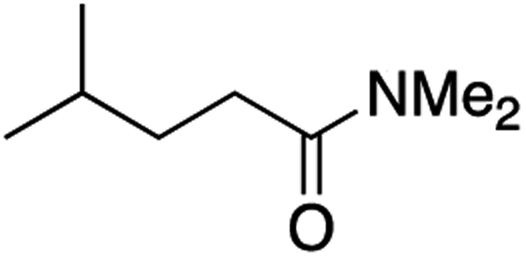
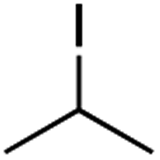
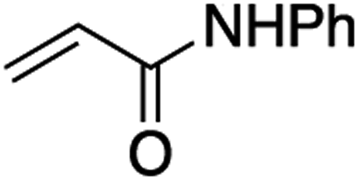
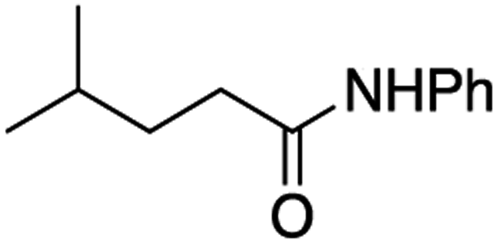
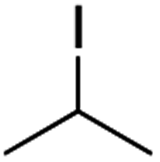



It is noteworthy that, in terms of the amount of the alkyl iodide employed, the efficiency of the reactions outlined in Table 2 is generally higher than with other radical chain carriers such as tributyltin hydride where, in order to circumvent competing reduction by Bu3SnH, multiple equivalents of the alkyl halide are typically employed. Tertiary iodides are also applicable (e.g. entry 3) however the high reactivity and poor solubility of accessible tertiary-iodides prevented a more diverse screen at this point. Surprisingly, even some alkyl bromides proved reasonably effective (entry 6) although admittedly, this is far from optimised. Table 2 demonstrates that a wide range of alkyl iodides could be coupled with various electron-deficient acceptors in generally excellent yields. We were also encouraged by entry 10 where an amide with a free N–H can be employed in excellent yields. The conditions employed are more amenable to our future ambitions to manipulate biological molecules than classical tin-mediated radical methods and illustrate a further advantage of the mild electrochemical technique.
With these results in hand, we were keen to undertake some preliminary mechanistic investigations into this reaction. We first turned to cyclic voltammetry (plots available in the ESI†) of the various reactants present in the solution. We discovered that the only species that was redox active at the potentials employed was molecular oxygen which was reduced between −0.7 and −0.8 V (vs. QRE in our system). We then examined the total amount of charge passed for the reaction of isopropyliodide with phenylvinylsulfone under the optimal conditions in Table 1 (entry 13). The charge–time graph is shown in Fig. 2 and shows a relatively smooth transfer of charge from the start of the reaction to the point where no more sulfone was observed.
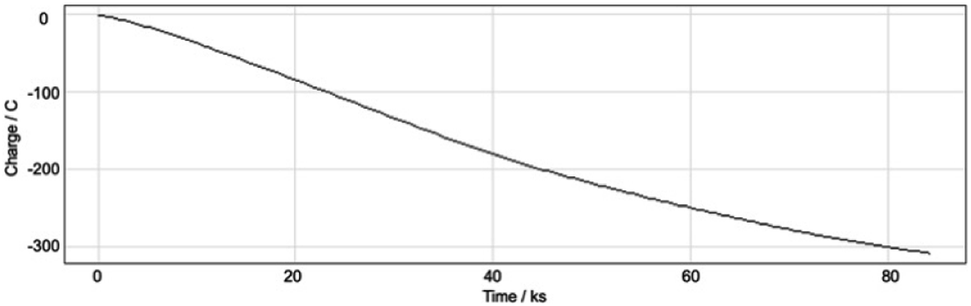 | ||
| Fig. 2 Charge transferred over the course of the reaction. | ||
We have calculated that for a 1.2 mmol reaction scale (1.44 mmol of alkyl iodide), around 300 C of charge was passed which corresponds to no more than two moles of electrons per mole of alkyl iodide. This is extraordinarily efficient in terms of the amount of electricity used. Given that we know that a small quantity of oxygen is required for a successful reaction, two general pathways are envisaged to explain the observed reactivity. The first and simplest of these is outlined in Scheme 2.
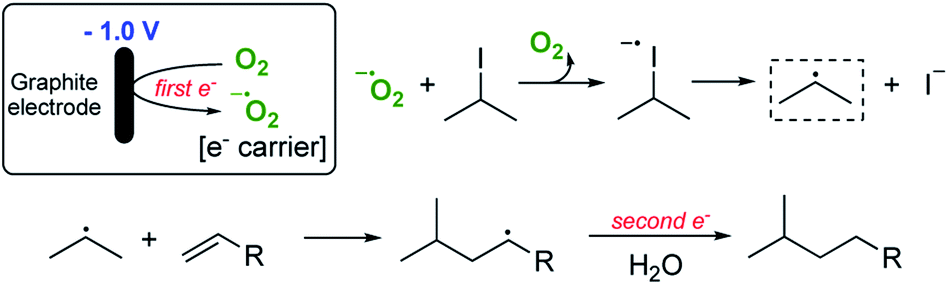 | ||
| Scheme 2 Proposed electron-transfer pathway ‘pathway 1’. | ||
Pathway 1 involves initial reduction of O2 to generate superoxide which then acts as an electron carrier, transferring the electron to σ* of the C–I bond and liberating O2 which is therefore, formally catalytic. This leads to collapse of the subsequent radical anion and the release of the required carbon-centred radical. Pathway 2 (shown in Scheme 3) on the other hand involves initial reduction of O2 to give a reactive species capable of activating an alkyl iodide, presumably by oxidation. Fragmentation of the unstable I(II) species then occurs to yield the alkyl radical.
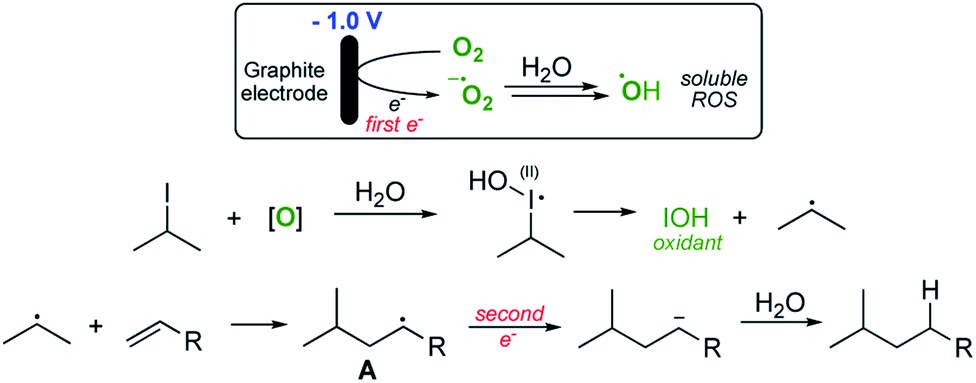 | ||
| Scheme 3 Proposed ‘redox-relay’ ‘pathway 2’. | ||
With regard to pathway 1, we concluded that this pathway was unlikely to be in operation. Two observations led us to this conclusion; firstly, as mentioned above, cyclic voltammetry of isopropyl iodide (see ESI†) demonstrated that the alkyl halide is not reduced within the redox window of the solvent so it is unlikely that superoxide (which is relatively easily generated) will be a sufficiently powerful reducing agent to deliver an electron to σ* of the C–I bond to effect homolysis. Secondly, entry 3 in Table 1 shows that water is an essential component for successful reaction, again suggesting that more reactive oxygen species are formed as depicted in pathway 2 (Scheme 3). We also considered the possibility that under the acidic conditions employed, the reduction of molecular oxygen to give hydrogen peroxide could also be occurring and therefore this reactive oxygen species could be implicated in the process. In order to ascertain if this was the case, we also performed the reaction in the absence of oxygen but with hydrogen peroxide present. No terminal product was observed in this case. Furthermore, we also examined the addition of iron(II) sulfate to the reaction medium in order to catalyse the formation of hydroxyl radicals from any putative hydrogen peroxide in the solution (the Fenton reaction)15 in the hope that this might accelerate the rate of these reactions. No effect was observed on the reaction and consequently we concluded that hydrogen peroxide was unlikely to be a major player in the main reaction process, although it is possible that traces of H2O2 generated via initial reduction of aerial O2 are responsible for initiation of the process.
Given the observations outlined above, we believe that pathway 2 is more likely, with the generation of the highly reactive hydroxyl radical as the species responsible for the reaction turnover. Initially we assumed that the iodides were simply being oxidised to give the I(III) iodanes which then undergo reduction to unstable I(II) species that fragment to yield IOH and the alkyl radical. However, given that the reaction is also applicable to some alkyl bromides (entry 7, Table 2), this seems unlikely, since hypervalent bromine reagents are extremely difficult to access under mild oxidative conditions and consequently are not commonly employed in organic synthesis.16–18 As such, we needed to consider an alternative reactive species that might be promoting the reaction pathway. As the only likely reactive species capable of activating both bromides and iodides in our solution, we suspected that the hydroxyl radical might be fulfilling this function.
Such a pathway is appealing because the hydroxyl radical is known to be sufficiently reactive to activate organic halides19,20 and the differing efficiencies between alkyl bromides and iodides can be easily explained by the accessibility of the lone pairs of the relevant halogen. Furthermore, it is known (particularly in biological systems) that the superoxide radical (or, since these reactions are generally performed under acidic conditions, the hydroperoxyl radical – pKa 4.88)21 reacts with hypohalous acids and hydrogen peroxide to yield hydroxyl radicals.22 A system therefore exists where the alkyl halide is continually activated to form alkyl radicals via the two interlocking cycles outlined in Scheme 4.
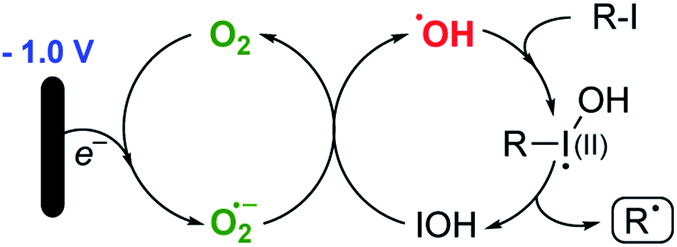 | ||
| Scheme 4 Proposed reactive species in the redox relay pathway. | ||
It is noteworthy that when pure methanol (a known hydroxyl radical scavenger)23 is used as the reaction solvent (Table 1, entry 1), no reaction is observed until a significant amount of water is added as a co-solvent (Table 1, entry 2). Even then, the reaction rate is significantly attenuated, and the yield falls far short of the optimised conditions. More detailed mechanistic studies will follow.
An obvious question if such a redox relay pathway involving a mutually cooperative interaction between hypohalous acid and superoxide is the fact that hypohalous acid is not present at the beginning of the reaction to initiate the process, how is the reaction initiated? Given that only a trace of the hydroxyl radical is needed to be generated before the redox relay pathway outlined in Scheme 4 can then take over, it is possible that trace amounts of hydrogen peroxide could be formed and then react with superoxide in the uncatalyzed (and slow) Haber–Weiss reaction.24 Alternatively, trace amounts of iodide present in the alkyl halide starting materials could also be responsible since H2O2 and iodide under acidic conditions has been suggested as a source of hydroxyl radicals in iodine-based chemical oscillators.25
In order to further support the mechanistic proposal outlined in pathway 2 and the involvement of a transient hypervalent iodine species, we attempted to emulate the initial oxidation of the alkyl iodide to the iodane species with a classical oxidant in the absence of O2 followed by exposure of that species to the reducing potential that would allow the radical reaction to occur. Knowing that alkyl iodides can be oxidised to I(III) iodanes by mCPBA we accordingly stirred the peracid with isopropyl iodide in an inert atmosphere for 1 h before applying a reducing potential and the addition of phenyl vinyl sulfone. After 18 h, only 9% of the required product was obtained (Scheme 5). The fact that the yield is so poor given that mCPBA would be expected to completely oxidise this alkyl iodide to the iodane suggests that the I(III) species is not a major intermediate in the reactive pathway. Interestingly however, others have noted that aryl peracids are sources of the hydroxyl radical26,27 and this may explain the low level of conversion observed here.
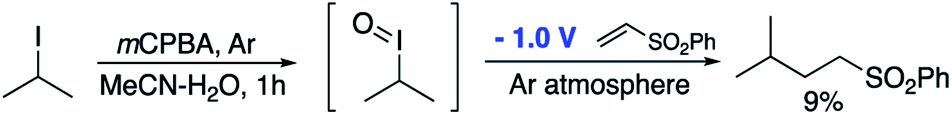 | ||
| Scheme 5 An intermediate I(III) iodane is unlikely to be a major intermediate. | ||
We also wished to demonstrate that the intermediate radical (A in Scheme 3) is reduced by an electron transfer step resulting in an anion which is then quenched by a proton from the aqueous reaction medium rather than a radical hydrogen atom transfer step (most likely from acetonitrile). This was easily achieved by performing the reaction in a D2O–MeCN mixture as shown in Scheme 6. Performing the reaction in this way led to deuterium incorporation adjacent to the sulfonyl group. To check that the alkyl sulfone was not simply undergoing exchange after the reaction had occurred, we also subjected the undeuterated alkyl sulfone to the reaction conditions for 72 h. No deuterium incorporation was observed.
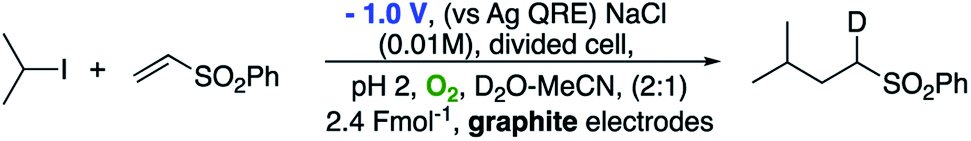 | ||
| Scheme 6 Demonstration that protonation of a sulfonyl anion occurs from the aqueous medium. | ||
Now reasonably convinced by our proposed mechanism, we wished to gain more insight into the key electron transfer steps occurring within the pathway. Given that we have already established that only two electrons are ‘consumed’ during the reaction (i.e. 2.4 F mol−1) per mole of iodide, and noting that excess O2 did not improve (and in fact attenuated) the reaction, we suspected that only a sub-stoichiometric amount of O2 was required to catalyse the reaction. This was easily demonstrated by again performing the reaction in an inert atmosphere (Ar) and observing that no reaction occurred until a small amount of air (1 mL, ca. 0.2 mL O2) was injected into the system. This corresponds to approximately 0.0083 mmol of O2 or 0.57 mol%. Once this had occurred the reaction proceeded smoothly to completion in 20 h, demonstrating that oxygen is only required in extremely low quantities and suggesting that it is likely to be the mediator in the process.
With some understanding of the reaction mechanism and some excellent results (both inter and intramolecular examples) employing alkenes, we were keen to test our methodology with alkynes to discover if they would be suitable partners for intermolecular radical reactions. Consequently, we employed our optimised conditions from Table 1, replacing the alkene acceptor with ethyl propiolate (Scheme 7).
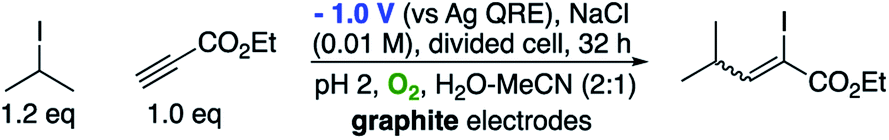 | ||
| Scheme 7 Intermolecular atom-transfer process. | ||
To our surprise, we isolated not the expected reduced addition product, but the addition product as the α-iodo alkene as a 1.2:1 mixture of E and Z isomers. The electrochemical conditions appear to have promoted the atom transfer radical addition (ATRA) reaction in this case. The results are consistent with the experiments of Curran, who achieved the same transformation employing heat and bistributyl tin.28 Presumably the mechanism outlined in Scheme 8 is in operation.
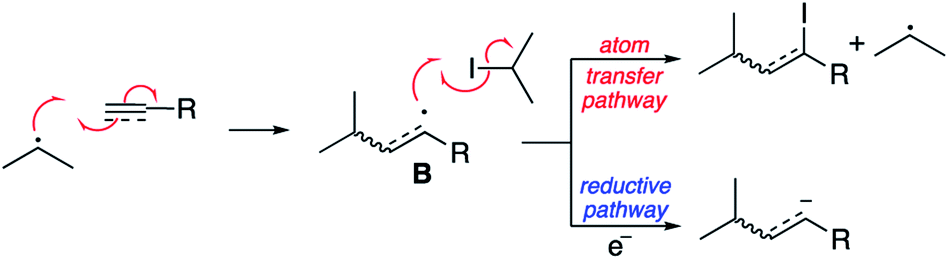 | ||
| Scheme 8 Proposed mechanistic pathways. | ||
We have attributed this observation to the stability of the intermediate radical B. In our previous reactions with alkenes the intermediate radical is directly resonance stabilised by the adjacent electron withdrawing group. In the example outlined in Scheme 8 however, the intermediate will be a highly reactive vinyl radical not directly stabilised by the adjacent ester. As such, presumably the rate of iodine atom abstraction is significantly faster than reduction to give a similarly unstabilised vinyl anion. Support for this hypothesis is observed when 4-methylphenyl ethynyl sulfone is employed as the reacting partner (Scheme 9). The product of this reaction is a mixture of the atom transfer reaction and reduction. This is explained by the fact that although still highly reactive, both the vinyl radical and anion obtain some degree of stabilisation via n–σ* interactions between the radical or anion and the C–S σ* orbital.29,30 As such in this case, both pathways are competitive in terms of rate.
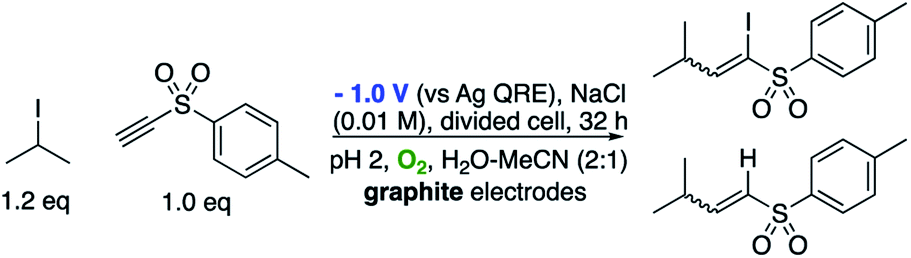 | ||
| Scheme 9 Competitive atom-transfer vs. reduction pathways. | ||
Finally, with such encouraging intermolecular radical processes in hand we also wanted to examine if our conditions could be applied to redox-neutral intramolecular cyclisations to aromatic systems. Accordingly, we prepared 1-(4-iodobutyl)-1H-indole-3-carbaldehyde. Pleasingly, exposure of this molecule to our conditions led to the cyclised, oxidised system in excellent yield (Scheme 10).
 | ||
| Scheme 10 Redox-neutral cyclisation. | ||
Although it is tempting to assume that the oxidative cyclisation would require an excess of molecular oxygen to deliver the observed product, we did not perform this reaction under such conditions but rather using the optimised procedure outlined in Table 1. The rate and efficiency also seemed to be comparable (or better) than the other examples in Table 2 suggesting that adventitious aerial oxygen was not responsible for this observation. In 1991 Bowman suggested a pseudo SRN1 process to explain this phenomenon for tin mediated cyclisations to aryl systems in the absence of any other oxidising agent, where the radical resulting from cyclisation is a powerful single electron donor capable of activating another molecule of iodide.31 This explanation was also favoured by Moody for the Bu3SnH mediated cyclisation of the same frameworks as those outlined in Scheme 10.32 For our system following electrochemical initiation, this would lead to a self-perpetuating combination of three different cycles leading to the oxidised product as illustrated in Scheme 11.
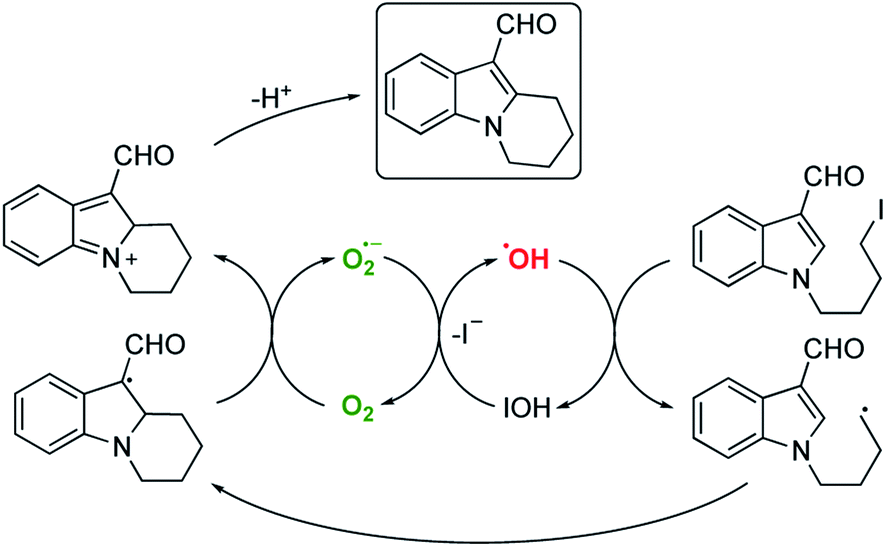 | ||
| Scheme 11 Likely pathway for redox-neutral cyclisation. | ||
Conclusions
In conclusion, we have described a new electrochemical approach to performing radical reactions, (particularly intermolecular radical reactions) between alkyl iodides and a variety of alkene and alkyne acceptors. The conditions employed are environmentally benign and the yield and efficiency of the reaction often exceeds the classical method employing tin reagents. We have described the reaction mechanism in terms of a ‘redox relay’ sequence initiated by molecular oxygen. Our work here will provide a framework for the further development of this chemistry to reach other examples where tin reagents were considered the ‘gold-standard’ for radical reactions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We wish to thank the Leverhulme Trust (RPG-2019-183) for generous financial support of our programme. The authors also gratefully acknowledge Dr D. Macmillan and Dr K. Karu for mass spectrometry and Dr A. Aliev for NMR support.Notes and references
- G. J. Rowlands, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2011, 107, 19–33 RSC.
- P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2001, vol. 1 and 2 Search PubMed.
- P. A. Baguley and J. C. Walton, Angew. Chem., Int. Ed., 1998, 37, 3072–3082 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- For a review see: M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed.
- C. A. Paddon, F. L. Bhatti, T. J. Donohoe and R. G. Compton, J. Phys. Org. Chem., 2007, 20, 115–121 CrossRef CAS.
- J. A. Cleary, M. S. Mubarak, K. L. Vieira, M. R. Anderson and D. G. Peters, J. Electroanal. Chem. Interfacial Electrochem., 1986, 198, 107–124 CrossRef CAS.
- B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef.
- A. Studer, S. Amerin, F. Schleth and T. Schulte, J. Am. Chem. Soc., 2003, 125, 5726–5733 CrossRef CAS PubMed.
- S. Caddick, J. D. Wilden, H. D. Bush, S. J. Wadman and D. B. Judd, Org. Lett., 2002, 4, 2549–2551 CrossRef CAS PubMed.
- H. Miyabe, M. Ueda, A. Nishimura and T. Naito, Org. Lett., 2002, 4, 131–134 CrossRef CAS PubMed.
- O. Edetanlen-Elliot, R. J. Fitzmaurice, J. D. Wilden and S. Caddick, Tetrahedron Lett., 2007, 48, 8926–8929 CrossRef CAS.
- W. H. Koppenol, Free Radical Biol. Med., 1993, 15, 645–651 CrossRef CAS PubMed.
- M. Ochiai, Y. Nishi, S. Goto, M. Shiro and H. J. Frohn, J. Am. Chem. Soc., 2003, 125, 15304–15305 CrossRef CAS PubMed.
- M. Ochiai, Y. Nishi, S. Goto, M. Shiro and H. J. Frohn, Angew. Chem., Int. Ed., 2005, 44, 406–409 CrossRef CAS PubMed.
- M. Ochiai, K. Miyamoto, T. Kaneaki, S. Hayashi and W. Nakanishi, Science, 2011, 332, 448–451 CrossRef CAS PubMed.
- U. Brühlmann, H. Büchler, F. Marchetti and R. E. Bühler, Chem. Phys. Lett., 1973, 21, 412–414 CrossRef.
- H. Mohan and K.-D. Asmus, J. Chem. Soc., Perkin Trans. 1, 1987, 1795–1800 RSC.
- M. Hayyan, M. A. Hashim and I. M. Alnashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- L. P. Candeias, K. B. Patel, M. R. L. Stratford and P. Wardman, FEBS Lett., 1993, 333, 151–153 CrossRef CAS PubMed.
- V. Múčka, P. Bláha, V. Čuba and J. Červenák, Int. J. Radiat. Biol., 2013, 89, 1045–1052 CrossRef PubMed.
- W. H. H. Koppenol, Redox Rep., 2001, 6, 229–234 CrossRef CAS PubMed.
- D. R. Stanisavljev, M. C. Milenković, M. D. Mojović and A. D. Popović-Bijelić, J. Phys. Chem. A, 2011, 115, 2247–2249 CrossRef CAS PubMed.
- T. Katsumi and S. Osamu, Bull. Chem. Soc. Jpn., 1962, 35, 1678–1683 CrossRef.
- A. Bravo, H.-R. Bjorsvik, F. Fontana, F. Minisci and A. Serri, J. Org. Chem., 1996, 61, 9409–9416 CrossRef CAS.
- D. P. Curran and D. Kim, Tetrahedron, 1991, 47, 6171–6188 CrossRef CAS.
- S. Wolfe, A. Stolow and L. A. LaJohn, Tetrahedron Lett., 1983, 24, 4071–4074 CrossRef CAS.
- D. A. Bors and A. J. Streitwieser, J. Am. Chem. Soc., 1986, 108, 1397–1404 CrossRef CAS.
- W. R. Bowman, H. Heaney and B. M. Jordan, Tetrahedron, 1991, 47, 10119–10128 CrossRef CAS.
- C. J. Moody and C. L. Norton, Tetrahedron Lett., 1995, 36, 9051–9052 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and details of the electrochemical apparatus employed. See DOI: 10.1039/d0sc01694b |
| This journal is © The Royal Society of Chemistry 2020 |