
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly stable RNA aptamer probe for the retinoblastoma protein in live cells†
Thao T.
Le
*a,
Andreas
Bruckbauer
b,
Bogachan
Tahirbegi
a,
Alastair J.
Magness
b,
Liming
Ying
 b,
Andrew D.
Ellington
c and
Anthony E. G.
Cass
*a
b,
Andrew D.
Ellington
c and
Anthony E. G.
Cass
*a
aDepartment of Chemistry, Imperial College London, 80 Wood Lane, W12 0BZ, UK. E-mail: t.le@imperial.ac.uk; t.cass@imperial.ac.uk
bNational Heart and Lung Institute, Imperial College London, SW7 2AZ, UK
cMolecular Biosciences, The University of Texas at Austin, Texas 78712, USA
First published on 21st April 2020
Abstract
Although RNA aptamers can show comparable or better specificity and affinity to antibodies and have the advantage of being able to access different live cell compartments, they are often much less stable in vivo. We report here the first aptamer that binds human retinoblastoma protein (RB) and is stable in live cells. RB is both a key protein in cell cycle control and also a tumour suppressor. The aptamer was selected from an RNA library against a unique 12-residue helical peptide derived from RB rather than the whole protein molecule. It binds RB with high affinity (Kd = 5.1 ± 0.1 nM) and is a putative RNA G-quadruplex structure formed by an 18-nucleotide sequence (18E16 - GGA GGG UGG AGG GAA GGG), which may account for its high stability. Confocal fluorescence microscopy of live cells transfected with the aptamer shows it is stable intracellularly and efficient in entering the nucleus where an analogous antibody was inaccessible. The findings demonstrate this aptamer is an advanced probe for RB in live cell applications.
Introduction
Aptamers are short, single stranded DNA or RNA molecules (typically tens of nucleotides in length) selected from random (ribo)oligonucleotide libraries to bind targets of interest.1–3 They can be thought of as nucleic acid-based analogues of antibodies.4–6 Compared to antibodies, aptamers are smaller and hence more efficient in accessing cellular compartments. However, aptamers are susceptible to degradation by a host of nucleases in live cells and may bind other cellular proteins. We present here the first aptamer selected to bind human retinoblastoma protein (RB) that is both highly stable, and specific for RB in cellular environments.RB is a tumour suppressor and plays an important role in cell proliferation through which the RB/E2F pathway regulates initiation of DNA replication.7,8 A number of studies have implicated this protein in other phases of the cell cycle as well as in autophagy.9–11 Given its several roles, altered function of RB either through mutations in the RB gene or changes in expression of RB regulators has been observed in most cancers.12–14 RB is present both in mitochondria15 and in the nucleus (where it is associated with chromatin)16 of human cells. Several anti-RB antibodies are available as reagents for western blotting17,18 and as probes to study its subcellular localization but typically only do so in fixed and permeabilized cells.19 Antibodies are large probes and as such have limitations in live cell applications as transport through the double-layered membrane of the nuclear envelope is difficult.20 Nucleic acid aptamers have been used as an alternative to antibodies in a wide range of applications.5,6,21,22 One advantage of aptamers over antibodies explored here is that they are significantly smaller in size and hence can more effectively access all cell compartments, including the nucleus. However, aptamers, especially RNAs, in their native forms are often prone to degradation by cellular nucleases and those often require chemical modifications to render them suitable as probes for live cell environments.23,24 We report here an RNA G-quadruplex aptamer for RB obtained from an in vitro selection process that shows resistance to degradation without chemical modifications for stabilisation by a host of ribonucleases that exist in human cells growing in culture.25–27 The aptamer is 18-nucleotides in length (corresponding to a molecular weight of less than 6 kDa, which is significantly smaller than an IgG (150 kDa) or even a light chain fragment (23 kDa)) and can efficiently cross the nuclear envelope to probe RB in the nuclei of live cells.
Results and discussion
Aptamer selection and characterisation
The RNA aptamer for RB was selected from a library comprising a 30-nucleotide random region flanked by 5′- and 3′ priming sequences, giving a full length of 80 nucleotides. Instead of using the whole RB molecule, the selection target was a 12 amino acid peptide corresponding to residues 317–328 of the full length protein which forms an accessible alpha helix on its surface. The peptide (LSKR[Y]EEIYLKN) was used as a mix of both phosphorylated and non-phosphorylated forms (represented by [Y]) and in equimolar amounts. Based on the PEP-FOLD6 (ref. 28 and 29) prediction it should retain its helical structure and this was confirmed by circular dichroism spectroscopy30,31 (see Fig. S1 in the ESI†). In using a unique peptide sequence derived from RB instead of the whole protein in the selection it was thought that this would lead to an increase in specificity as previously observed in a selection for the HIV Rev protein.32The selection process using SELEX3 was performed for 12 rounds. Sequence data from the round 12 enriched library showed that one sequence, E16, comprised around 8% (4/48) of the cloned population (see Fig. S2 in the ESI†). E16 also showed the highest affinity for both phosphorylated and non-phosphorylated peptides as well as for the whole RB molecule. The observation that the full length 80-mer aptamer 80E16 binds to the phosphorylated and non-phosphorylated peptides with similar affinities (Kd = 7.4 ± 0.7 nM and 12.6 ± 1.8 nM, respectively) (see Fig. S3A in the ESI†) may result from the use of a mixture as the selection target. When tested on full-length recombinant human RB (expressed in insect cells), the aptamer showed about an order of magnitude higher affinity (Kd = 0.46 nM ± 0.04 nM) (see Fig. S3B in the ESI†). This may be due to the aptamer 80E16 containing regions that also interact with other parts of RB. Truncation of the 80-mer sequence resulted in a much shorter aptamer (18 nucleotides, 18E16 - GGA GGG UGG AGG GAA GGG) with an order of magnitude loss of affinity in binding to RB but with the same affinities for the peptides, consistent with the idea that the full length aptamer makes additional contacts with the whole protein (Table 1). Further removal of nucleotides from 18E16 resulted in significant loss of binding to RB. In a control experiment, an RNA G-quadruplex aptamer that binds the bovine prion protein33 showed no significant binding to RB. Data for binding of the truncated aptamer 18E16 to the peptide derivatives and the whole recombinant RB molecule and the control are in Fig. S4 in ESI.†
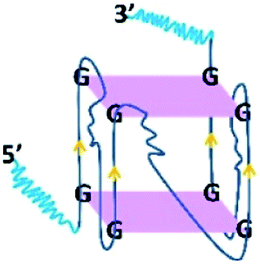
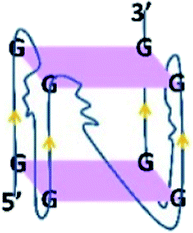
Inspection34 of the 18E16 sequence suggested it could potentially form G-quadruplex structures. Binding of 18E16 to a G-quadruplex specific antibody (BG4) supported this prediction. It was observed that 18E16 binds the anti G-quadruplex antibody BG4 (see Fig. S5 in the ESI†) with an affinity comparable to other known RNA G-quadruplex structures.35
To determine whether a monoclonal antibody (BG4) that recognizes G-quadruplexes and the RB protein have overlapping or independent binding sites on the aptamer, two-site assays were performed as shown in Fig. 1A. The interaction of the BG4 antibody with 18E16 is the same whether or not the latter is also bound to RB, indicating the bound aptamer retained its RNA G-quadruplex structure. This also indicates that the aptamer binds simultaneously to RB and to the BG4 antibody. In addition, the circular dichroism (CD) spectrum of 18E16 shown in Fig. 1B is consistent with the known parallel-stranded RNA G-quadruplex structures.35 Typically DNA G-quadruplexes are more stable in higher concentrations of potassium ions. However, increasing the potassium ion concentration from 5 mM to 150 mM did not lead to a further increase in the 262 nm peak intensity of 18E16 suggesting that it had fully formed a stable G-quadruplex structure even at the lower potassium ion concentrations. It has been reported that RNA G-quadruplex structures are thermodynamically more stable than DNA ones under near-physiological conditions36 due to the involvement of the 2′-hydroxyl group in intramolecular hydrogen bonding within the G-quadruplex.37 Thermal stability of the 18E16 aptamer, assessed by CD spectroscopy (see Fig. S6 in the ESI†) revealed that it has the characteristic high melting temperature reported previously for this type of structure.35 In addition, a dye, N-methylmesoporphyrin IX (NMM), that binds G-quadruplex structures38,39 with parallel stranded orientation40,41 was also used to detect this characteristic structure of 18E16. Fig. 1C shows images of native and denaturing polyacrylamide gels stained with SYBR®Gold for total RNA and NMM for parallel G-quadruplex RNA. The corresponding bands on both native and denaturing gels stained with NMM indicated 18E16 has parallel G-quadruplex and it is highly stable; persisted under the denaturing conditions of the TBE-urea gel.
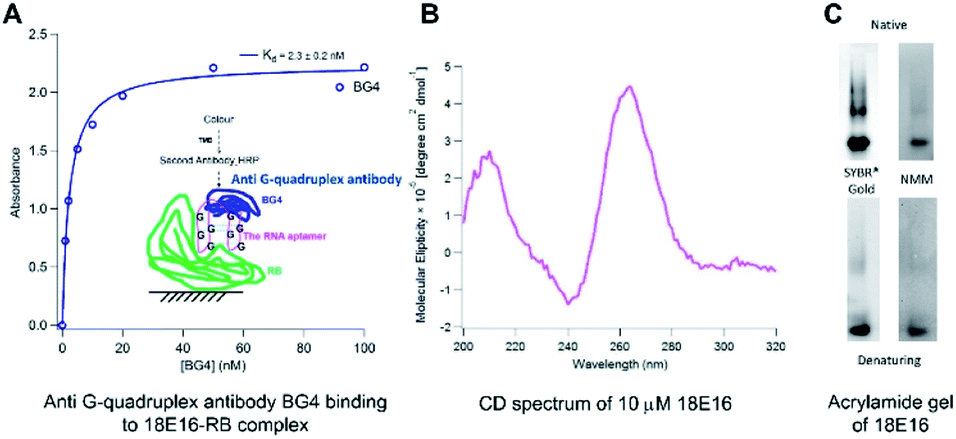 | ||
| Fig. 1 Characterisation of 18E16 's G-quadruplex structure by (A) molecular recognition using an anti G-quadruplex antibody BG4 (inset: a schematic view of the 18E16 -sandwiched binding). The assay was performed in the binding buffer (5 mM MgCl2, 5 mM KCl, 1 mM CaCl2, 150 mM NaCl, 20 mM Hepes, pH 7.4). Changing Hepes to Tris in the binding buffer does not affect binding of the aptamer to RB. The binding curve was obtained by fitting the binding data to Langmuir isotherm model using Igor (WaveMetrics Inc.). (B) CD spectrum of 18E16 showing the characteristic peak for the parallel RNA G-quadruplex structures at 262 nm. The CD measurements were performed using 10 μM 18E16 in the binding buffer using a Chirascan (Applied Photo-Physics). (C) Staining of 18E16 using SYBR® Gold and NMM (a parallel G-quadruplex specific dye) on native (TBE) and denaturing (TBE-urea) gels. | ||
A longer truncated sequence with 24 nucleotides, 24E16 (GGA GGG UGG AGG GAA GGG UUU GGG) shows a comparable affinity for RB as 18E16 (see Fig. S6 and S7 in the ESI†) but has almost double the molar ellipticity at 262 nm in the unbound form, the characteristic peak of parallel-stranded RNA G-quadruplexes (see Fig. S7B in the ESI†). This is probably due to the additional guanines in the longer sequence may add another G-quartet plane resulting in a structure formed by three stacks of G-quartets, which was known to have higher 262 nm peak intensity than that of a structure with two G-quartets.35 The 18E16 core structure is the main determinant of affinity and that additional sequence, even where it forms another G4 layer in the unbound state, makes relatively little contribution. The full sequence (with 80 nucleotides in length) is tighter binding and this may due to the additional sequence making stabilising non G-quadruplex structure contacts with the protein. The use of 24E16 however may be benefited in sensing applications where target-induced conformational changes of the aptamer can provide a valuable detection mode.
Specific extraction of RB from cell lysates
To investigate the specificity of 18E16 in complex cellular environments, a selective extraction method was used. Fig. 2A show a gel electrophoresis analysis of proteins in a HEK293 cell lysate that were captured by the aptamer 18E16. The top intense band of lane 2 has a molecular weight corresponding to that of RB which was absent from lane 1 where 18E16 enrichment was not used. The intensity of the putative RB band captured by 18E16 indicates the aptamer is highly specific for its target in the lysate where thousands of other cellular proteins also present. In addition, a 2-site ELISA where 18E16 was used as the capture molecule for RB in HEK293 lysate and an anti-RB antibody to detect the protein is shown in Fig. 2B.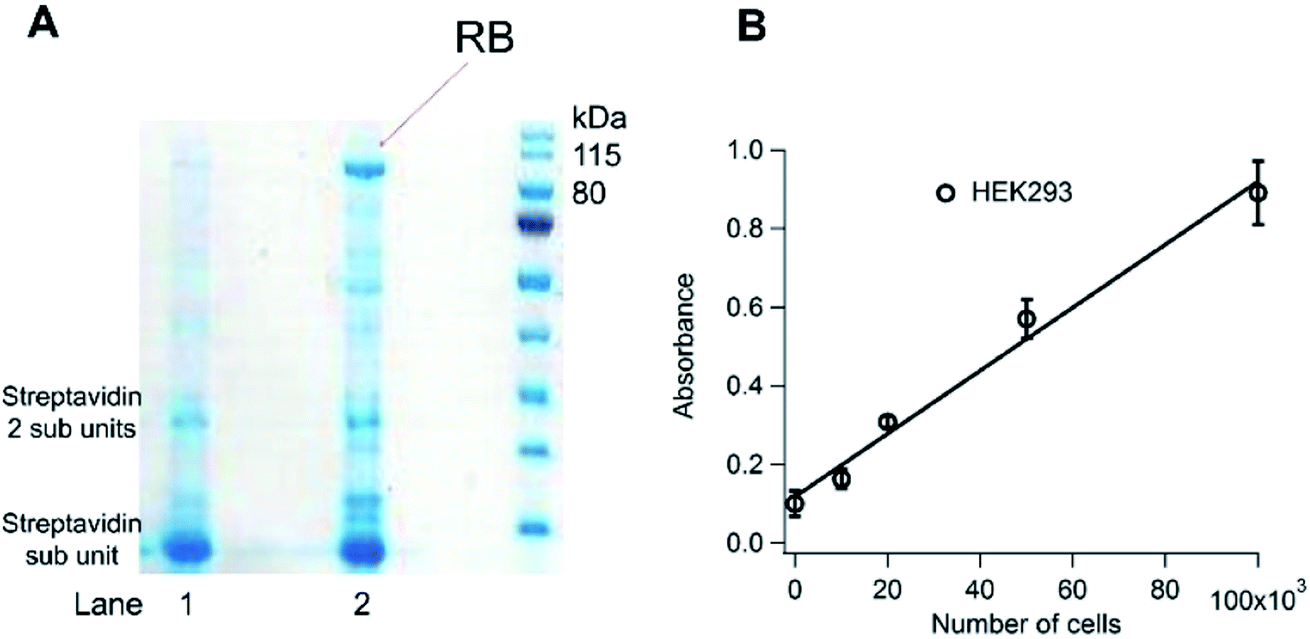 | ||
| Fig. 2 (A) Gel electrophoresis analysis. Lane 1: reference (HEK293 cell lysate with streptavidin magnetic beads in the absence of the aptamer). Lane 2: sample (HEK293 cell lysate captured by the aptamer and separated by streptavidin magnetic beads). A biotin labelled at the 3′-end of the aptamer allowed the complex to be separated from the lysate using streptavidin magnetic beads. The bound molecules were then eluted from beads for gel electrophoresis analysis. The top band of lane 2 showed a molecular weight corresponding to that of RB, where it was not shown from lane 1 of a parallel experiment in the absence of biotinylated 18E16. (B) Direct detection of RB in HEK293 cell lysate by 2-site ELISA using 18E16 as the capture molecule. 3′-end biotin labelled 18E16 was immobilized on a streptavidin-coated microplate to capture RB from the cell lysate. An anti-RB antibody labelled with HRP was used to detect the protein. The assays were run in the binding buffer. | ||
Specific extraction of RB to 18E16 is further confirmed by repeating the selective enrichment procedures with H460 and RB-knockout H460 lysates and western blotting to confirm the putative RB band (see Fig. S8 in the ESI†).
Live cell imaging
RNA aptamers are often considered to be unsuitable as probes in live cell environments as they are readily degraded by intracellular ribonucleases and may also exhibit non-specific interactions with molecules other than the target. Evaluation of the stability of the aptamer was performed in human cells growing in culture, where many cellular and serum ribonucleases are present.25–27 Using the aptamer to probe RB in live cells was investigated using confocal fluorescence microscopy. The aptamer was labelled with Cy3 and transfected into U87MG, MCF7 and HEK293 cells. Confocal images (see Fig. S9 in the ESI†) shows the transfected Cy3-labelled aptamer in both the cytoplasmic and nuclear compartments, where RB is known to be located.15,16 In addition, we used 3D Structured Illumination Microscopy42 for super-resolution imaging of the aptamer to achieve more precise location of RB within the nucleus (see Fig. S10 in the ESI†).To evaluate the background signal, we also transfected U87MG cells with a random 15-mer oligonucleotide labelled with Cy3 and observed no significant fluorescence (see Fig. S11 in the ESI†). The negligible fluorescence from the cells transfected with the random 15-mer oligonucleotide labelled with the dye is due to ‘wash-out’ when it is not bound to its target. It is worth noting that, for the confocal imaging experiments, the transfected cells undergone an extensive course of washing in which after transfection, they were washed 3 times, then detached and reseeded in a microscope chamber for up to 30 hours and washed again before imaging. This washing regime served two purposes; to assess the stability of the aptamer inside living cells and to reduce fluorescent background from unbound aptamer. Moreover, confocal images of RB-knockout H460 and H460 cells transfected with the aptamer (see Fig. S12 in ESI†) show much greater amounts of the aptamer in the latter.
Stability of the aptamer in live cells was investigated by Fluorescence Resonance Energy Transfer (FRET) confocal microscopy using a pair of fluorescent dyes labelled at each end of the molecule (Cy3/donor at the 5′-end and Cy5/acceptor at the 3′-end) as shown in Fig. 3B. High FRET efficiency of this pair in the binding buffer is demonstrated by single molecule FRET (smFRET) in Fig. 3A. Retention of the FRET signal of the doubly labelled aptamer in the human cells growing in culture confirms its intracellular stability as efficient energy transfer will only be observed when the molecule is intact.
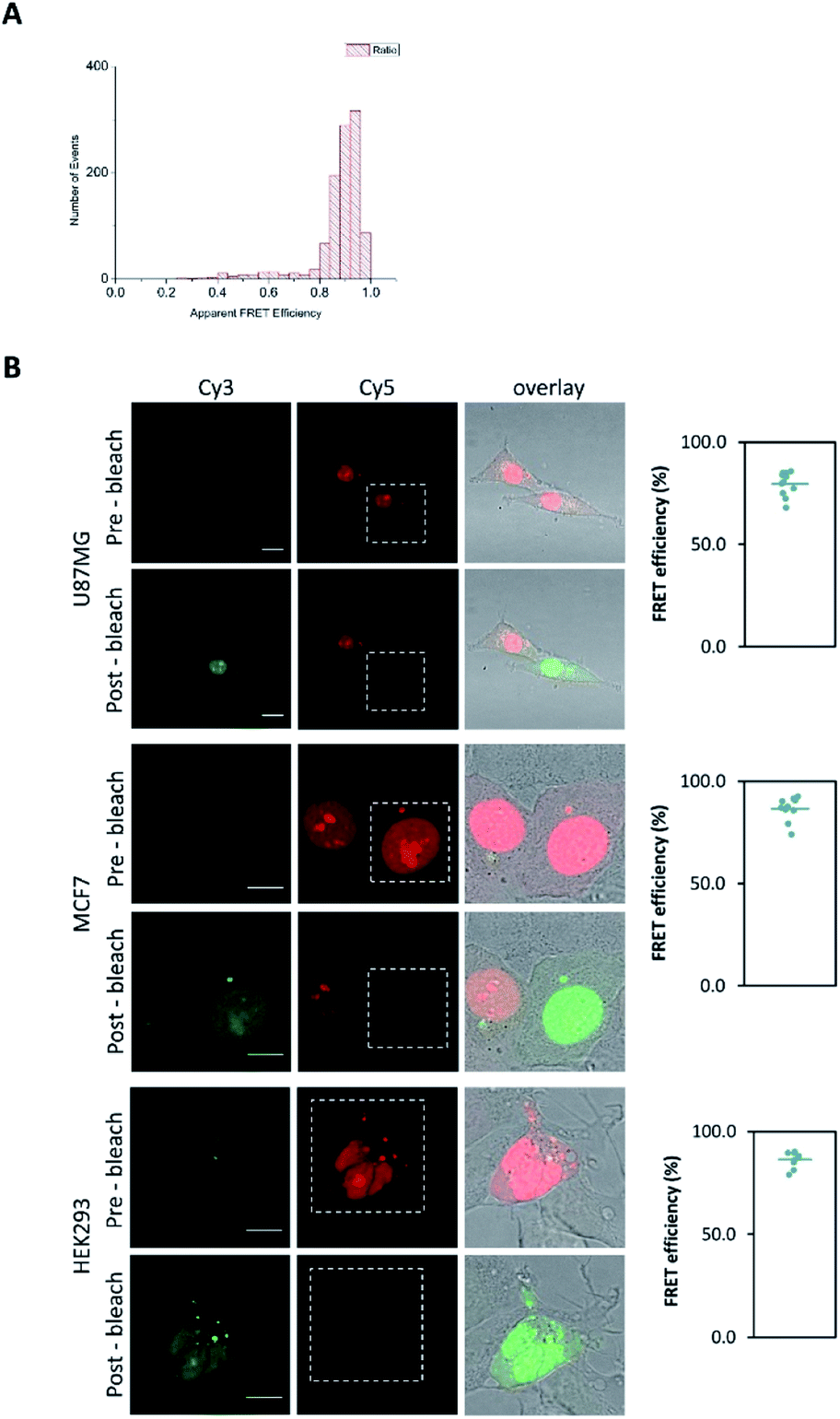 | ||
| Fig. 3 (A) Single molecule FRET (smFRET) histogram of the RNA aptamer in the binding buffer. The aptamer was doubly labelled at the 5′-end with a donor and the 3′-end with an acceptor. It showed high FRET efficiency of the pair when at the 2 ends of the aptamer. (B) Confocal images of Cy3/Cy5 FRET shown by acceptor photobleaching in live cells more than a day after aptamer transfection. U87MG, MCF7, HEK293 cells were incubated with the doubly labelled RNA aptamer (5′-end Cy3 and 3′-end Cy5). For each cell type Cy3 (donor) and Cy5 (acceptor) images are shown before and after photobleaching of the acceptor. Pre-bleach images show very low Cy3 (donor) fluorescence, post-bleach images (bleached region is indicated) demonstrate FRET by Cy3 fluorescence due to the absence of Cy5, quantification as described in methods, scale bar 10 μm. | ||
FRET confocal images shown in Fig. 3B taken more than 1 day after transfection of the doubly labelled aptamer showed strong FRET as demonstrated by acceptor photobleaching.43 The measured FRET efficiencies are very high with 80% ± 2% (SEM, n = 11), 87% ± 2% (SEM, n = 10) and 86% ± 1% (SEM, n = 10) in U87MG, MCF7 and HEK293 cells respectively as shown on the right of Fig. 3B. The FRET efficiencies obtained from confocal microscopy of the transfected aptamer in live cells from two independent experiments are consistent with the FRET efficiency determined by smFRET of the free aptamer in the binding buffer shown in Fig. 3A, confirming the aptamer is intact. In a control experiment, cells transfected with an equimolar mixture of singly labelled aptamers, i.e. 3′-end Cy3-labelled and 3′-end Cy5-labelled showed similar fluorescent intensities signals in both channels confirming negligible FRET when the dyes are on different molecules (see Fig. S13 in the ESI†). This high FRET efficiency indicates that the donor–acceptor dyes are in the same molecule, i.e. the aptamer is not degraded and confirms its intracellular stability, which may be attributed to the structural characteristics of RNA G-quadruplexes.36,37
In live cells, accessibility of imaging agents to different compartments is often a limiting factor in their use. Because this aptamer is significantly smaller than antibodies, it is anticipated that it can gain access to compartments that are inaccessible to the latter. This is demonstrated by comparison of the images of cells transfected with both Alexa Fluor 647-labelled anti-RB antibody IgG1 kappa light chain (shown in red) and the Cy3-labelled RNA aptamer (shown in green) in Fig. 4. This anti-RB antibody light chain fragment is known to bind the RB but at a different site from the aptamer as shown by the results of the sandwich ELISA presented in Fig. 2B, confirming no competitive binding between antibody and aptamer.
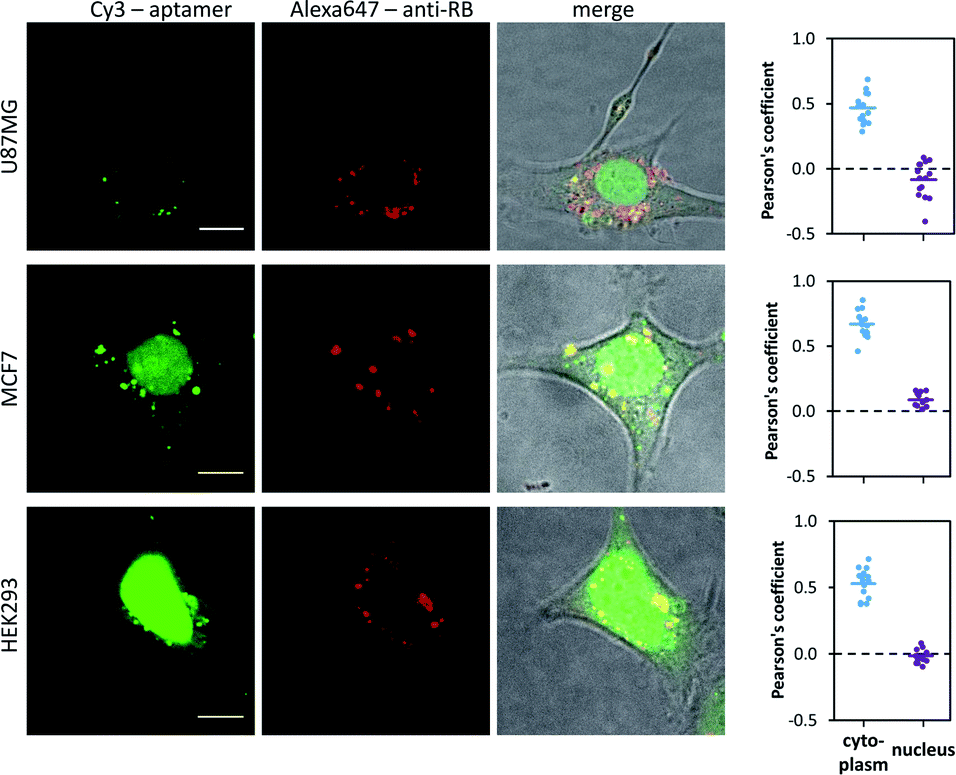 | ||
| Fig. 4 Co-localisation of a Cy3-labelled aptamer and an Alexa Fluor 647-labelled anti-RB antibody IgG1 kappa light chain (sc-73598, Santa Cruz Biotechnology) transfected into live cells, U87MG, MCF7 and HEK293. Quantification of co-localisation in the cytoplasm and nucleus using Pearson's coefficient. The Pearson's coefficients of 0.47 ± 0.03 (SEM, n = 15), 0.67 ± 0.03 (SEM, n = 15) and 0.53 ± 0.01 (SEM, n = 16) were measured for U87MG, MCF7 and HEK293 cells respectively. Data from three independent co-localisation experiments were used for the calculation. Scale bar 10 μm. | ||
The confocal images in Fig. 4 show co-localisation of the aptamer and the antibody fragment fluorescence in the cytoplasmic compartment, Pearson's coefficients of 0.47 ± 0.03 (SEM, n = 15), 0.67 ± 0.03 (SEM, n = 15) and 0.53 ± 0.01 (SEM, n = 16) were measured for U87MG, MCF7 and HEK293 cells respectively (data from three independent co-localisation experiments were used for the calculation). A coefficient of 1 is expected for perfect co-localization, while a score of 0 indicates a random localisation of the fluorescence. Thus, our measurements indicate good co-localization of the aptamer and the antibody in the cytoplasm bind to different sites of the RB. However, only the aptamer fluorescence can be seen in the nucleus consistent with only the aptamer crossing the nuclear membrane, the Pearson's coefficient is close to zero due to random correlation with noise in the anti-RB antibody channel. Lower resolution images of the transfected cells showing panorama view (see Fig. S14 in the ESI†) demonstrate the high delivery efficiency of the aptamer as its fluorescence was observed in both nucleus and cytoplasm of almost all cells.
Experimental
Aptamer selection
A synthetic DNA library with the central region containing 30 randomized nucleotides was used to produce the RNA library (GGG AAU GGA UCC ACA UCU ACG AAU UC-N30-UUC ACU GCA GAC UUG ACG AAG CUU) using T7 RNA polymerase. In the first round, 200 nmol of the RNA library and 1 nmol of an equimolar mixture of phosphorylated tyrosine peptide (LSKRpYEEIYLKN) and non-phosphorylated tyrosine peptide (LSKRYEEIYLKN) both with a biotin at their N-terminus were mixed. Selection buffer comprised 5 mM KCl, 1 mM CaCl2, 5 mM MgCl2, 150 mM NaCl, 20 mM Hepes (pH 7.4). The selection partitioning was done using nitrocellulose membrane filtration. The aptamer and peptide were incubated together for 1 hour prior to addition of streptavidin and filtration through a 0.45 μm nitrocellulose membrane. Counter selection against a biotin/streptavidin complex was performed with the enriched libraries every 2 rounds from the 4th round. After 12 rounds of selection, the enriched pool was cloned and individual colonies were picked for Sanger sequencing. Sequences that had significant consensus regions were then custom synthesized by Eurofins for determination of affinity and specificity using ELISA and ELONA.Binding assays
The assays were performed in 96-well plates and detected with Horseradish Peroxidase (HRP) labelled reagents and 3,3,5,5-tetramethylbenzidine (TMB)/peroxide substrate. The enzymatic reaction was quenched using 2 M H2SO4 solution. Absorbance measurements were taken at 450 nm using a Varioskan Flash (Thermo Scientific). Kd values were calculated from binding data fitted to the Langmuir isotherm using Igor (WaveMetrics Inc.).Detection of parallel G-quadruplex structure by NMM
10 μM of the aptamers in buffer containing 100 mM Tris, 100 mM KCl, 5 mM MgCl2, 1 mM CaCl2, pH 7.2 was heated at 95 °C for 10 minutes and then left at room temperature overnight. The aptamer solutions were then added with 1/5 volume of 20% glycerol in the same buffer and 15 μL volumes of the mix were loaded into a native (20% TBE) gel or denature (10% TBE-urea) gel. The gels were run at 3.5 V cm−1 for 12 hours. The gels was stained with SYBR® Gold (ThermoFisher Scientific, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dilution) or NMM (Santa Cruz Biotechnology, 0.1 mg mL−1) in TBE buffer for 10 minutes before being destained in TBE buffer for 20 minutes and then imaged under a UV lamp.
000 dilution) or NMM (Santa Cruz Biotechnology, 0.1 mg mL−1) in TBE buffer for 10 minutes before being destained in TBE buffer for 20 minutes and then imaged under a UV lamp.
Circular dichroism spectroscopy
10 μM of the RNA aptamer solutions in the binding buffer (20 mM Tris, 5 mM MgCl2, 1 mM CaCl2, 5 mM KCl, pH 7.4) were heated to 84 °C for 5 minutes and left to cool at room temperature for 20 minutes before the CD measurements. Three scans were performed from 200 to 320 nm using a Chirascan (Applied Photo-Physics) spectropolarimeter with the buffer spectrum subtracted and zero correction at 320 nm.Cell culture
H460 and RB-knockout H460 cell lines (from the Sage lab at Stanford University) were cultured in RPMI 1640 medium supplemented with 2 mM L-glutamine and 8% foetal calf serum (FCS). HEK293, MCF7 and U87MG cell lines were cultured in MEM supplemented with 8% FCS. Cells were seeded and grown in 25/75 cm2 corning cell culture flasks in an incubator (37 °C and with 5% CO2). Cell cultures were split when they reached around 80% confluency. Cell counting was done by a Countess II FI (ThermoFisher Scientific).Cell lysis
Cells grown on the 10 cm diameter Petri dishes were washed twice with cold PBS supplemented with 5 mM MgCl2 and 1 mM CaCl2. 1 mL RIPA buffer (Thermo Scientific) was added to each dish at 80–100% confluency to lyse the cells. Cell debris was removed by centrifuging at 16000×g for 30 minutes.
Analysis of aptamer-captured cell lysates by gel electrophoresis and western blotting
Cell lysates were exchanged into the aptamer the binding buffer using an Ultracel-50 centrifugal unit (Millipore) and their protein concentration determined from the absorbance at 280 nm. The lysates were then supplemented with BSA to 0.5% (w/v) concentration and to a volume of 1.5 mL in the binding buffer. The 1.5 mL lysate sample was mixed with 200 μL of 1 μM biotinylated 18E16 in the binding buffer and incubated at room temperature for 1 hour. It was then mixed with 200 μL of 2.5 mg mL−1 streptavidin coated magnetic beads (Promega) which were washed and suspended in 0.5% BSA in the binding buffer. The mixtures were incubated for further 1 hour at room temperature and then washed twice with 1 mL of 0.5% BSA in the binding buffer and 3 times with 0.5 mL of the binding buffer supplemented with 0.05% Tween 20 using a magnet. The beads were then mixed with 26 μL of water and 10 μL of NuPAGE™ LDS Sample Buffer (4×) (Thermo Scientific). Captured proteins were eluted at 99 °C for 4 minutes. The supernatant was separated from the magnetic beads and mixed with 4 μL of 500 mM dithiothreitol and then incubated at 80 °C for another 4 minutes prior to SDS PAGE analysis. Samples were loaded into a 4–10% Tris-bis NuPAGE gel (ThermoFisher Scientific) for electrophoretic separation and visualisation using InstantBlue™ Coomassie stain (Expedeon). For western blotting, after electrophoresis, biomolecules from the polyacrylamide gel was transferred to a 0.45 μm nitrocellulose membrane. The transferred membrane was then blocked with 5% BSA in PBS for 2 hours prior to be detected by an anti-RB antibody labelled with HRP (sc-73598, Santa Cruz Biotechnology). Visualisation of the RB-band was achieved using DAB as the substrate for HRP.Transfection with fluorescently labelled aptamer
Cells grow in a 25 cm2 Corning flasks for 1 day with confluency of around 60%. The cells were treated with the solution of 0.2 nmol singly (Cy3) or doubly fluorescent (Cy3 and Cy5)-labelled aptamer with 25 μL of ‘Lipofectamin™ RNAiMAX reagent in 500 μL optiMEM (Thermo Scientific) and placed in the incubator for 6 hours. The cells were then washed twice with MEM medium supplemented with 8% FCS and then once with PBS buffer. The transfected cells were then detached or lysed for subsequent experiments. For imaging, the detached cells were seeded on poly-L-lysine coated Lab-Tek borosilicate coverglass chambers in MEM medium supplemented with 4% FCS and grown for 15–30 hours prior to confocal microscopy.Transfection of Alexa Fluor 647-labelled anti-RB antibody
Cells were grown in 25 cm2 Corning flasks for 1 day to a confluency of around 60%. The cells were washed 3 times with MEM to thoroughly remove FCS before the solution of 0.2 nmol Alexa Fluor 647-labelled anti-RB antibody IgG1 kappa light chain (sc-73598, Santa Cruz Biotechnology) in 200 μL of PBS and 2.5 reactions of Pierce Protein Transfection Reagent (89850A, ThermoFisher Scientific) was added. Cells transfected with the antibody were placed in an incubator for 6 hours. The cells were then washed twice with the MEM medium supplemented with 8% FCS and then once with PBS buffer and then detached from the flask to be seeded on poly-L-lysine coated Lab-Tek borosilicate coverglass chambers in MEM medium supplemented with 4% FCS for 15–30 hours prior to the confocal microscopy.Transfection of both Cy3-labelled RNA aptamer and Alexa Fluor 647-labelled anti-RB antibody for co-localisation studies
Cells grow in a 25 cm2 Corning flasks for 1 day to confluency of around 50%. The cells were treated with the solution of 0.2 nmol Cy3-labelled aptamer with 25 μL of ‘Lipofectamin™ RNAiMAX reagent in 500 μL optiMEM (ThermoFisher Scientific) and placed in the incubator for 6 hours. The RNA aptamer transfected cells were then washed three times with serum-free MEM to thoroughly remove FCS before the solution of 0.2 nmol Alexa Fluor 647-labelled anti-RB antibody IgG1 kappa light chain (sc-73598, Santa Cruz Biotechnology) in 200 μL of PBS and 2.5 reactions of Pierce Protein Transfection Reagent (89850A, ThermoFisher Scientific) was added and incubated for another 6 hours. The cells were then washed twice with the MEM medium supplemented with 8% FCS and then once with PBS buffer and then detached from the flask to be seeded on poly-L-lysine coated Lab-Tek borosilicate coverglass chambers in MEM medium supplemented with 4% FCS for around 15–30 hours prior to confocal microscopy.Single molecule FRET (smFRET)
A home-built dual-channel confocal fluorescence microscope was used to detect freely diffusing single molecules of dual-labelled RNA aptamers. Apparent FRET efficiencies, Eapp, of each burst were calculated according to Eapp = nA/(nA + nD), where nA and nD are the acceptor and donor counts, respectively.44 The donor, fluorescence, was excited by an argon ion laser (model 35LAP321-240, Melles Griot) with 150 μW at 488 nm. Donor and acceptor fluorescence was collected through an oil-immersion objective (Nikon PlanApo ×60, numerical aperture 1.45) and detected separately by two photon-counting modules (SPCMAQR14, PerkinElmer). The outputs of the two detectors were recorded by two computer-implemented multichannel scalar cards (MCS-PCI, ORTEC). A threshold of 25 counts per millisecond bin for the sum of the donor and acceptor fluorescence signals was used to differentiate single molecule bursts from the background.Confocal microscopy
The cell-seeded microscope slides were washed twice with the MEM medium supplemented with 4% FCS for 2 hours prior to acquiring images using a Leica SP8 Inverted confocal microscope with 63 × 1.4 NA Plan-Apochromat objective. Excitation wavelengths were 561 nm for Cy3 and 633 nm for Cy5 and Alexa Fluor 647. Emission was collected in the bands 566–618 nm for Cy3 and 638–791 nm for Cy5 and Alexa Fluor 647. Cy3 images in Fig. S9 in the ESI† were deconvolved using Huygens software (SVI) with standard settings. The FRET efficiency was calculated in an ROI containing the whole cell as FRETeff = (Ipost − Ipre)/Ipost where Ipre and Ipost are the total, background subtracted fluorescence of the ROI in the Cy3 channel before and after Cy5 bleaching. Co-localisation was quantified as Pearson's coefficients calculated in ROIs containing the cytoplasm or the nucleus using Huygens software.Structured illumination microscopy
Microscopy was performed using a Zeiss Elyra PS.1 microscope using a 63 × 1.4 NA Plan-Apochromat objective with 642 nm excitation, collecting fluorescence through a 650 nm long pass filter. Raw data was acquired with 3 rotations and 5 phases at 144 nm z-spacing and super-resolution images were reconstructed from these using the Zeiss Zen software and auto noise filter settings. Transmitted light bright field images were taken separately and an in-focus transmitted light image was combined with a representative z-slice of the fluorescence.Conclusions
In summary we describe the selection of an RNA aptamer that binds human RB with nanomolar affinity and is both stable and specific in intracellular environments. The selection used as the target a 12-residue peptide corresponding to a surface α-helix on the protein instead of the whole molecule. A truncated version of the aptamer 18 nucleotides in length, 18E16, has a stable RNA G-quadruplex structure in both the free state and complexed with RB. The aptamer's RNA G-quadruplex structure is credited for its resistance against ribonuclease degradation and its stability in live cells. The use of this aptamer as a specific probe for RB in a complex environment was demonstrated both by its selective “pull-down” of the protein from cell lysates. Confocal microscopy showed the aptamer was able to enter nuclei of live cells where the analogous antibody was excluded. In addition, FRET images confirmed the aptamer is not degraded in U87MG, MCF7 and HEK293 cells growing in culture.With the demonstrated properties, this 18E16 aptamer can be used in various applications including for enrichment/purification and for quantification of RB in cell lysates and probing this protein in live cells, in which it can offer more information on the roles of this key protein in normal cells and in diseases. It is also worth noting that this aptamer is small in size and do not required further chemical modifications for stability and it retained its structure even in denaturing conditions (such as of TBE-urea gels).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Scott Knudsen (MIT) for synthesis of the DNA library and valuable advice and Frederic Dick and James MacDonald (University of Western Ontario) and Julien Sage and Andrea Chaikovsky (Stanford University) for generously providing the RB-knockout cells. The authors would like to thank the Facility for Imaging by Light Microscopy (FILM) at Imperial College London for the access to the microscopes and acknowledge funding by BBSRC (grant BB/L015129/1). AEGC and TTL would like to acknowledge BBSRC ZELS Grant BB/L018853/1 and LY, BT and AM to the Leverhulme Trust Grant (RPG-2015-345).References
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- S. D. Jayasena, Clin. Chem., 1999, 45, 1628–1650 CrossRef CAS.
- E. J. Cho, J.-W. Lee and A. D. Ellington, Annu. Rev. Anal. Chem., 2009, 2, 241–264 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- J. W. Harbour and D. C. Dean, Genes Dev., 2000, 14, 2393–2409 CrossRef CAS PubMed.
- C. Giacinti and A. Giordano, Oncogene, 2006, 25, 5220–5227 CrossRef CAS PubMed.
- R. A. Weinberg, Cell, 1995, 81, 323–330 CrossRef CAS.
- F. A. Dick and S. M. Rubin, Nat. Rev. Mol. Cell Biol., 2013, 14, 297–306 CrossRef CAS PubMed.
- N. J. Dyson, Genes Dev., 2016, 30, 1492–1502 CrossRef CAS PubMed.
- J. Ishikawa, H.-J. Xu, S.-X. Hu, D. W. Yandell, S. Maeda, S. Kamidono, W. F. Benedict and R. Takahashi, Cancer Res., 1991, 51, 5736–5743 CAS.
- C. J. Sherr and F. McCormick, Cancer Cell, 2002, 2, 103–112 CrossRef CAS PubMed.
- A. Ertel, J. L. Dean, H. Rui, C. Liu, A. K. Witkiewicz, K. E. Knudsen and E. S. Knudsen, Cell Cycle, 2010, 9, 4153–4163 CrossRef CAS PubMed.
- I. Ferecatu, N. Le Floch, M. Bergeaud, A. Rodríguez-Enfedaque, V. Rincheval, L. Oliver, F. M. Vallette, B. Mignotte and J.-L. Vayssière, BMC Cell Biol., 2009, 10, 50 CrossRef PubMed.
- D. Avni, H. Yang, F. Martelli, F. Hofmann, W. M. ElShamy, S. Ganesan, R. Scully and D. M. Livingston, Mol. Cell, 2003, 12, 735–746 CrossRef CAS PubMed.
- G. Z. Rassidakis, R. Lai, M. Herling, C. Cromwell, A. Schmitt-Graeff and L. J. Medeiros, Am. J. Pathol., 2004, 164, 2259–2267 CrossRef CAS PubMed.
- T. J. Collard, B. C. Urban, H. A. Patsos, A. Hague, P. A. Townsend, C. Paraskeva and A. C. Williams, Cell Death Dis., 2012, 3, e408 CrossRef CAS PubMed.
- L. Szekely, E. Uzvolgyi, W. Jiang, M. Durko, K. Wiman, G. Klein and J. Sumegi, Cell Growth Differ., 1991, 2, 287–295 CAS.
- P. A. Silver, Cell, 1991, 64, 489–497 CrossRef CAS PubMed.
- T. C. Chu, K. Y. Twu, A. D. Ellington and M. Levy, Nucleic Acids Res., 2006, 34, e73 CrossRef PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 CrossRef CAS PubMed.
- M. R. Dunn, R. M. Jimenez and J. C. Chaput, Nat. Rev. Chem., 2017, 1, 0076 CrossRef CAS.
- P. Rothlisberger and M. Hollenstein, Adv. Drug Delivery Rev., 2018, 134, 3–21 CrossRef CAS PubMed.
- S. Sorrentino, FEBS Lett., 2010, 584, 2194–2200 CrossRef CAS PubMed.
- C. M. Arraiano, F. Mauxion, S. C. Viegas, R. G. Matos and B. Séraphin, Biochim. Biophys. Acta, Gene Regul. Mech., 2013, 1829, 491–513 CrossRef CAS PubMed.
- M. von Köckritz-Blickwede, O. A. Chow and V. Nizet, Blood, 2009, 114, 5245–5246 CrossRef PubMed.
- P. Thévenet, Y. Shen, J. Maupetit, F. Guyon, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2012, 40, W288–W293 CrossRef PubMed.
- Y. Shen, J. Maupetit, P. Derreumaux and P. Tufféry, J. Chem. Theory Comput., 2014, 10, 4745–4758 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- G. Holzwarth and P. Doty, J. Am. Chem. Soc., 1965, 87, 218–228 CrossRef CAS PubMed.
- W. Xu and A. D. Ellington, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7475–7480 CrossRef CAS PubMed.
- T. Mashima, A. Matsugami, F. Nishikawa, S. Nishikawa and M. Katahira, Nucleic Acids Res., 2009, 37, 6249–6258 CrossRef CAS PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2005, 33, 2908–2916 CrossRef CAS PubMed.
- M. Wieland and J. S. Hartig, Chem. Biol., 2007, 14, 757–763 CrossRef CAS PubMed.
- B. Saccà, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2005, 33, 1182–1192 CrossRef PubMed.
- G. W. Collie, S. M. Haider, S. Neidle and G. N. Parkinson, Nucleic Acids Res., 2010, 38, 5569–5580 CrossRef CAS PubMed.
- J. Ren and J. B. Chaires, Biochemistry, 1999, 38, 16067–16075 CrossRef CAS PubMed.
- S. M. Lyons, D. Gudanis, S. M. Coyne, Z. Gdaniec and P. Ivanov, Nat. Commun., 2017, 8, 1127 CrossRef PubMed.
- E. O. Ariyo, E. P. Booy, T. R. Patel, E. Dzananovic, E. K. McRae, M. Meier, K. McEleney, J. Stetefeld and S. A. McKenna, PLoS One, 2015, 10, e0144510 CrossRef PubMed.
- N. C. Sabharwal, V. Savikhin, J. R. Turek-Herman, J. M. Nicoludis, V. A. Szalai and L. A. Yatsunyk, FEBS J., 2014, 281, 1726–1737 CrossRef CAS PubMed.
- M. G. L. Gustafsson, L. Shao, P. M. Carlton, C. J. R. Wang, I. N. Golubovskaya, W. Z. Cande, D. A. Agard and J. W. Sedat, Biophys. J., 2008, 94, 4957–4970 CrossRef CAS PubMed.
- Y.-B. Zheng, Y.-Y. Xiao, P. Tan, Q. Zhang and P. Xu, PLoS One, 2012, 7, e51849 CrossRef CAS PubMed.
- M. I. Wallace, L. Ying, S. Balasubramanian and D. Klenerman, J. Phys. Chem. B, 2000, 104, 11551–11555 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01613f |
| This journal is © The Royal Society of Chemistry 2020 |