
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe oxazolomycin family: a review of current knowledge
Patrik Oleksak ab,
Jozef Gondab,
Eugenie Nepovimovaa,
Kamil Kucaa and
Kamil Musilek*a
ab,
Jozef Gondab,
Eugenie Nepovimovaa,
Kamil Kucaa and
Kamil Musilek*a
aUniversity of Hradec Kralove, Faculty of Science, Department of Chemistry, Rokitanskeho 62, 500 03, Hradec Kralove, Czech Republic. E-mail: kamil.musilek@uhk.cz
bPavol Jozef Safarik University, Faculty of Science, Department of Organic Chemistry, Moyzesova 11, 040 01, Kosice, Slovak Republic
First published on 9th November 2020
Abstract
Oxazolomycin A and neooxazolomycin were firstly isolated in 1985 by the group of Uemura et al. from the Streptomyces sp. bacteria. To date, there have been reported 15 different natural compounds commonly classified as part of the oxazolomycin family. All oxazolomycin compounds possess extraordinary structures and they represent a synthetic challenge. Such molecules are additionally known for their wide range of biological activity including antibacterial, antiviral and cytotoxic effects. The present review summarizes the structural elucidation and classification of oxazolomycin compounds, their biosynthesis and biological activity. It is further focused on the total syntheses of oxazolomycins and one formal synthesis reported to date.
1 Introduction
1.1 Discovery of oxazolomycins
The oxazolomycin family is a commonly used term for a group of molecules which includes 15 derivatives isolated from natural sources to date. As the first members of this group oxazolomycin A (1; Fig. 1) and neooxazolomycin (2) were isolated and recognized in 1985 by Uemura et al. from Streptomyces sp.1,2 In the same year (1985), curromycin A (6) and curromycin B (7) were described as the products of a genetically modified strain of Streptomyces hygroscopicus.3 Curromycin A and curromycin B were formerly termed as triedimycin A and triedimycin B.4 In 1997 16-methyloxazolomycin (5) was isolated as a product from Streptomyces sp. as another member of the oxazolomycin family.5 Two novel oxazolomycins, specifically oxazolomycin B (3) and oxazolomycin C (4) were reported in 1998 by Kanzaki et al. as the products of Streptomyces albus JA3453.6 Otani et al. in 2000 reported isolation of KSM-2690 B (8) and KSM-2690 C (9) from the broth filtrate of Streptomyces strain KSM-2690 and they were classified as novel oxazolomycins.7 In 2005 Potts et al. reported discovery of lajollamycin (10) as the product of the strain Streptomyces nodosus (NPS007994) and was named for the area of collection in Scripps Canyon, La Jolla in California, USA.8 Other lajollamycins were isolated in 2014 by Oh et al. from Streptomyces strain SMC72 and they were identified as lajollamycin B (11), lajollamycin C (12) and lajollamycin D (13).9 The latest two oxazolomycins reported in 2017 by Koomsiri et al. were identified from a culture broth of Streptomyces subflavus subsp. irumaensis AM-3603 and called oxazolomycin A2 (14) and bisoxazolomycin (15).10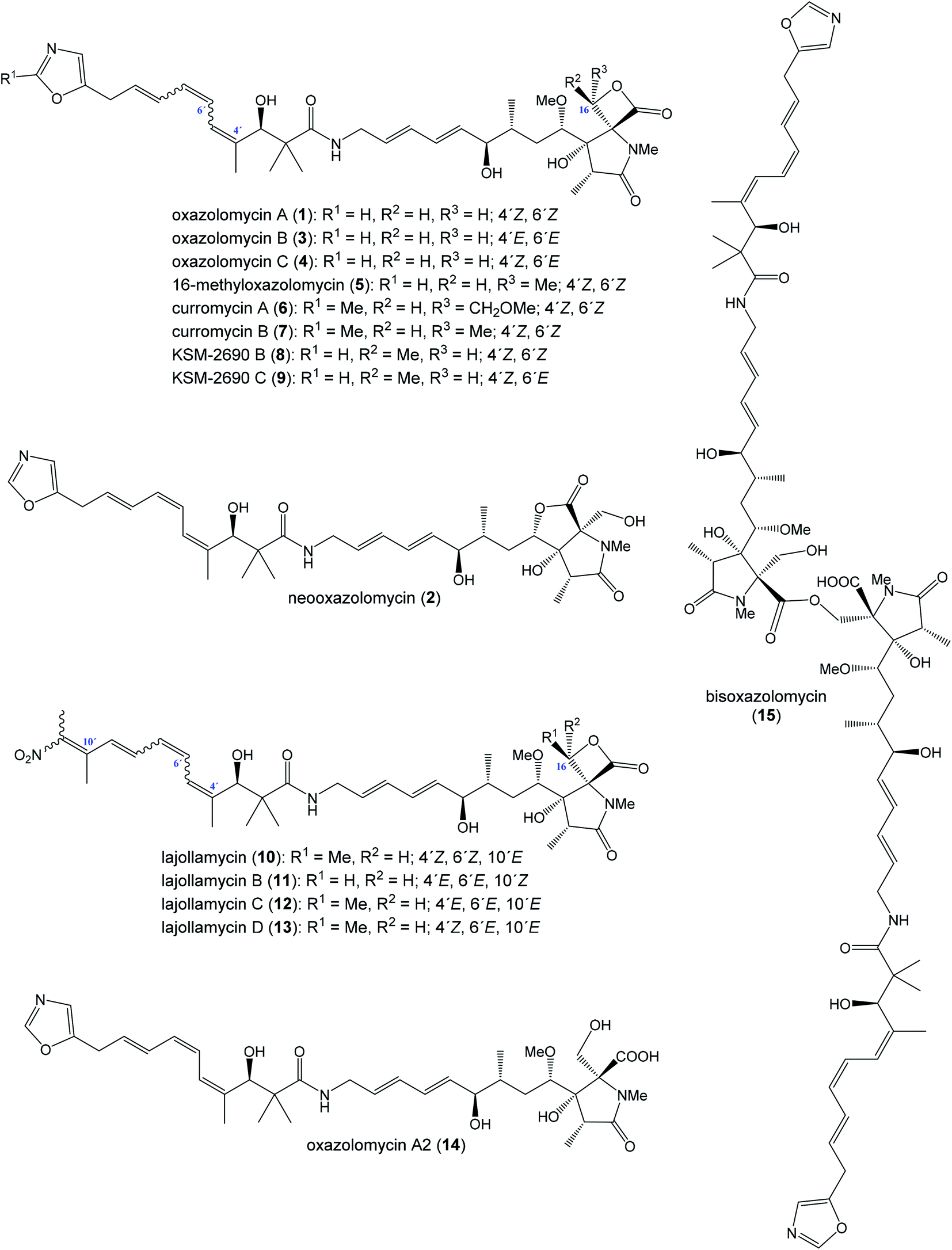 | ||
| Fig. 1 Oxazolomycin family members. | ||
1.2 Structural elucidation of oxazolomycins
Uemura et al. reported as the first structure of oxazolomycin A (1), involving spiro-β-lactone-γ-lactam moiety.1 They also identified presence of 5-substituted oxazole ring in the structure as well as (Z)-configurated trisubstituted double bond considering nuclear Overhauser effect (NOE) enhancements between methyl (C4′) and H5′. Uemura et al. also reported a structure of neooxazolomycin (2), which in contrast to 1 contains bicyclic γ-lactone-γ-lactam moiety.2 Neooxazolomycin is the only member of oxazolomycin family which contains bicyclic core. Structural elucidation of both 1 and 2 was achieved by application of a combination of X-ray crystallography and chemical correlation of the degradation products.11 A completely geometric and stereochemical alignment of 1 and 2 was reported still in 1985.1,2 Moreover, the structure of 1 was unambiguously provided in 2011 in the first total synthesis reported by Hatakeyama et al.12 The first total synthesis of 2 was reported earlier in 1990 by Kende et al. and confirmed the assigned configuration.13Further in 1985, structures of curromycin A (6) and curromycin B (7) were reported with double bond geometries, however without the stereochemical alignment.3 Structure of both curromycins is similar to 1, but these curromycins involve 2-methyl substituted oxazole ring. Additionally, compound 6 contains –CH2OMe residue at C16-position and compound 7 possesses the methyl group at the same position C16.3 Kim et al. reported in 1997 isolation, structural elucidation and biological properties of 16-methyloxazolomycin (5), with partial stereochemical determination.5 Structural recognition of 5 revealed considerable similarities with the structure of 1. Geometry of diene and triene system as well as presence of 5-substituted oxazole moiety are identical to 1. In contrast to oxazolomycin A, spiro-β-lactone-γ-lactam core of 16-methyloxazolomycin incorporates methyl group at C16-position, alike in the structure of curromycin B. The stereochemistry of β-lactone-γ-lactam moiety of 5 was deduced to be identical to 1 by NMR (NOESY) experiments, which also proved (S)-configuration of C16. Moreover, a stereochemical determination of β-lactone-γ-lactam core of 5 was determined as (2R,3S,15S)-configuration using NOESY experiments.5 Completely stereochemical determination of 5 was reported in 1999 by Kim et al., which confirmed identical configurations at all common, backbone stereocenters with 1.14
Kanzaki et al. reported in 1998 structures of two novel oxazolomycins: oxazolomycin B (3) and oxazolomycin C (4), both are geometrical isomers of oxazolomycin A.6 Olefinic proton signals were assigned by 1H–1H COSY and the coupling constants values of 1H NMR, the geometry was assigned by an analysis of NOE difference spectra and NOESY. The obtained data showed that the geometries of diene system in 3 and 4 are identical to 1, but configuration of triene system is (4′E,6′E,8′E)- for 3 and (4′Z,6′E,8′E)- for 4 in contrast to (4′Z,6′Z,8′E)-configurated triene system of 1. Kanzaki et al. reported also biological activities of 3 and 4, but stereochemical aspects were not determined.6
Otani et al. reported in 2000 isolation and structures of KSM-2690 B (8) and KSM-2690 C (9), the two novel members of oxazolomycin family.7 Structure of both 8 and 9 were elucidated using FAB-MS, UV and IR spectra, 1D and 2D NMR methods. Acquired spectral data of 8 were compared with those for 16-methyloxazolomycin (5) reported by Kim et al., which suggested that 8 is a stereochemical isomer of 5.14 To clarify stereochemical differences Otani et al. analyzed ROESY and NOE correlations of 8, when β-lactone-γ-lactam moiety of 8 was assigned with (2R,3S,15S,16R)-configurations in contrast to that of 16-methyloxazolomycin (5) with (2R,3S,15S,16S)-configurated stereocenters.7 But some stereocenters (C4, C6, C7 and C3′) were not examined, 1H NMR data indicated close similarities of both compounds except for the C16 center and consequently compound 8 was determined as a novel stereoisomer of 5.
The spectral data indicated that 9 is an isomer of 8 but differences were observed in the chemical shifts for the triene moiety of 9. And the chemical shifts of triene moiety in 9 were closely similar to that reported by Kanzaki et al. for oxazolomycin C.6 Otani et al. determined (4′Z,6′E,8′E)-configurations of triene moiety in 9 when NOE correlations of β-lactone-γ-lactam core were identical to these of 8.7 In addition, KSM-2690 C (9) was determined as geometric isomer of 8, however configuration of four stereocenters (C4, C6, C7 and C3′) were not examined.7
Isolation and structural characterization of lajollamycin (10) was reported by Potts et al. in 2005.8 NMR data of 10 exhibited similarities with other oxazolomycins such as presence of spiro-β-lactone-γ-lactam core, amide linkage and conjugated olefinic system. Surprisingly, structure of 10 exhibited absence of oxazole ring system and further analysis revealed presence of nitro group in the structure. Analysis of NOESY and HMBC correlations indicated elongation of the triene system typical for oxazolomycins by an additional double bond that was substituted with two methyl moieties. The double bond geometries of the tetraene system were determined by NOESY and NOEDS analysis, which revealed (4′Z,6′Z,8′E,10′E)-configurated tetraene in the structure of 10. Absolute configuration of 10 was determined by Oh et al. in 2014 by analysis of the ROESY NMR correlations, applied a J-based configuration analysis and the modified Mosher's method.9
In the same work Oh et al. reported isolation and structural elucidation of another lajollamycins as the members of the oxazolomycin family.9 Structures of lajollamycin B (11), lajollamycin C (12) and lajollamycin D (13) were determined by FAB-HRMS, 1D and 2D NMR analysis. The double bond geometries of these lajollamycins were determined by analysis of coupling constants and ROESY correlations. The configurations of tetraene moiety were elucidated as (4′E,6′E,8′E,10′E)-configurations for 11 and 12 and as (4′Z,6′E,8′E,10′E)-configurations for 13. Oh et al. reported proposition of the absolute configurations for 11, 12 and 13 that were identical to those determined for 10 based on the similarities in acquired data and a shared biogenesis.9 However, in 2019 Hatakeyama et al. reported first total synthesis of lajollamycin B and its C10′ geometric isomer.15 Surprisingly, a spectroscopic comparison with data for 11 reported by Oh et al.9 found to be identical with unexpected isomer. Thus, Hatakeyama et al. carefully reinvestigated Oh's 2D NMR spectroscopic data of 11 and unambiguously assigned NOESY spectra to both prepared isomers. 1H and 13C NMR spectra of both isomers exhibited close similarities with (E)- and (Z)-configurated terminal nitrotetraene derivatives prepared by Hatakeyama and co-workers. According to these findings Hatakeyama et al. revised the initially assigned geometry of lajollamycin B from (10′E)- to (10′Z)-configurated double bond.15
The last two members of oxazolomycin family were isolated, characterized and reported in 2017 by Koomsiri et al.10 Both structures were elucidated using HR-ESI-MS, UV and IR spectra, optical rotation, 1D and 2D NMR spectroscopy. Structure of oxazolomycin A2 (14) indicated close similarity to oxazolomycin A (1), but difference in chemical shifts of the oxymethylene H2-16 was observed. These observations suggested, that 14 is the analog of 1 with hydrolyzed β-lactone. Indeed, 14 could be prepared by basic hydrolysis of 1.10 Moreover, oxazolomycin A2 has been synthetized as an intermediate in total synthesis of 1 reported by Hatakeyama et al. in 2011, however crude 14 was directly used to further reaction without isolation.12 Spectral data of bisoxazolomycin (15) suggested that structure is an asymmetric dimer of 1 consisting of N-methyl-γ-lactam segments I and II.10 Both segments were confirmed by the 1H–13C long-range correlations. The chemical shifts of the oxymethylene of H2-16 in segment I were similar to those of 14, but the chemical shifts of the oxymethylene of H2-16′′ in segment II were moved to more shaded area (higher ppm). Moreover, 15 could be transformed by basic hydrolysis to 14. Thus, bisoxazolomycin was revealed to be a new dimer of 14, with an ester bond between C17 and C16′′.
1.3 Structural classification of oxazolomycins
Relatively complicated structures of oxazolomycins can be divided into three fragments, usually termed as left-hand, middle and right-hand fragment. The structure of dimeric bisoxazolomycin (15) consist of two segments, named as segment I and segment II by Koomsiri et al. (Fig. 2).10 The both segments can be divided into three fragments. These fragments and their analogues are crucial intermediates for total syntheses of all members of oxazolomycin family reported to date.12,13,15–17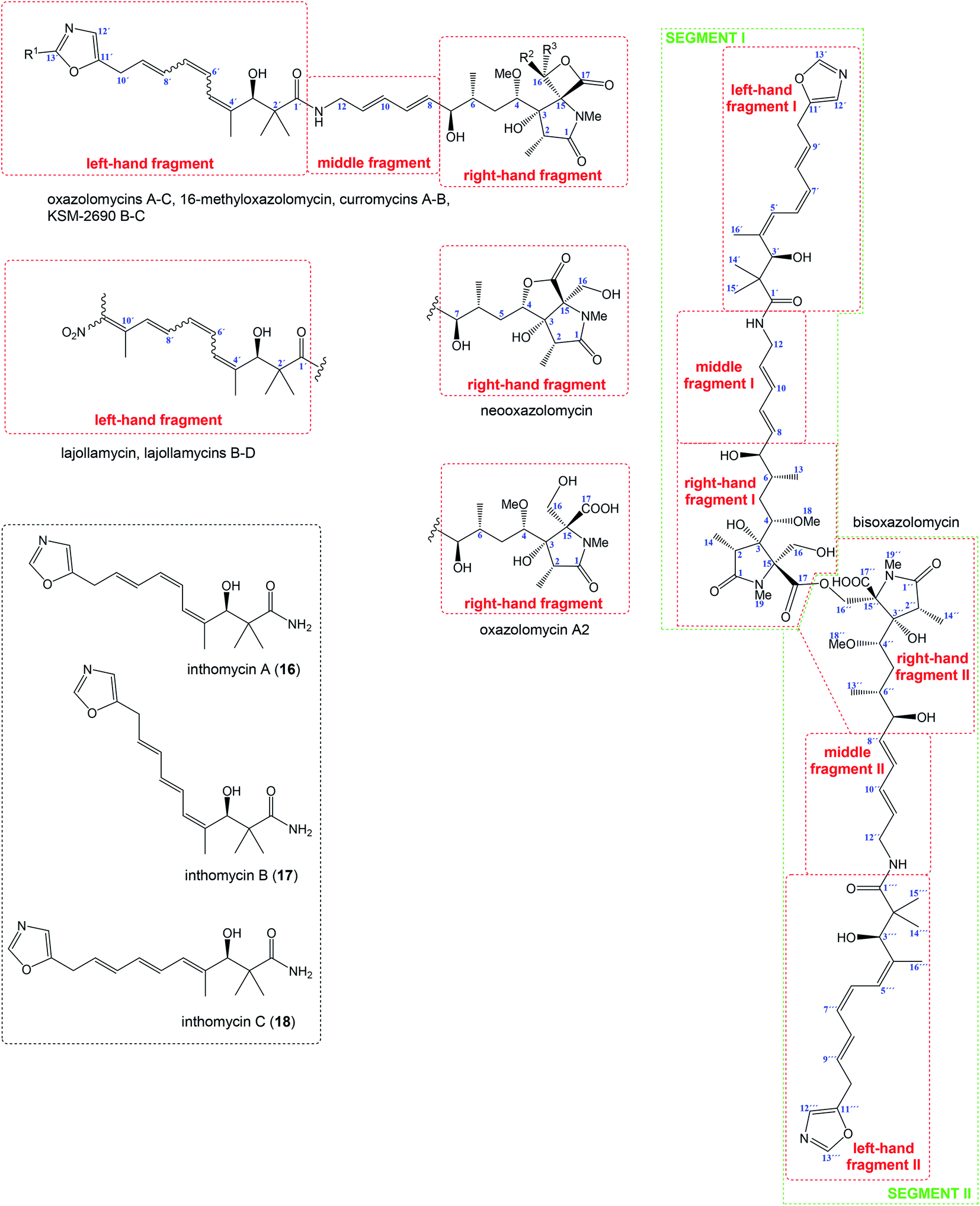 | ||
| Fig. 2 Structural division of oxazolomycin family members and structure of inthomycins. | ||
The left-hand fragment represents one of two different structural arrangements. The most common framework is the oxazole-triene-amide18 also known as inthomycin derivative.19 Inthomycin A (16; Fig. 2) with (4Z,6Z,8E)-configurated triene is present in the structures of oxazolomycin A, oxazolomycin A2, neooxazolomycin, 16-methyloxazolomycin, curromycin A, curromycin B, KSM-2690 B and in the both segments of bisoxazolomycin. Inthomycin B (17) which possesses (4Z,6E,8E)-configurated triene is present in the structures of oxazolomycin C and KSM-2690 C. Finally, inthomycin C (18) with (4E,6E,8E)-configurated triene moiety represents left-hand fragment of oxazolomycin B. The second structural framework for left-hand fragment is tetraene-nitro-hydroxy-amide which is typical for all lajollamycins including lajollamycin, lajollamycin B, lajollamycin C and lajollamcin D.
The middle fragment represents pentadienylamine,18 which contains (E,E)-configurated diene moiety. This fragment is present in the structures of all members of oxazolomycin family. Indeed, middle fragment is a subunit of larger chain bounded at C5 and C4′ positions and it is absolutely same including stereochemistry for all oxazolomycins.
The right-hand fragment is structurally different for some members, however in general it can be designated as a pyroglutamate core. For almost all members of the oxazolomycin family right-hand fragment represents spiro-β-lactone-γ-lactam core, which can be further substituted with methyl or methoxymethyl group at the C16 position. Oxazolomycin A2 is the only natural member of oxazolomycins, which right-hand fragment contains monocyclic γ-lactam core. The unusual pyroglutamate core is incorporated in the structure of neooxazolomycin, which is the only natural oxazolomycin with bicyclic γ-lactone-γ-lactam moiety.
2 Biosynthesis of oxazolomycins
2.1 Feeding experiments
Thiericke et al. reported in 1992 feeding experiments on Streptomyces albus (strain JA3453) to investigate skeletal origin of oxazolomycin A (1) by using following [13C]-labelled precursors: sodium [1,2-13C2]acetate, sodium [1-13C]propionate, L-[methyl-13C]methionine, L-[1-13C]alanine and [1-13C]glycine.20 Single dose of labelled precursor was added to shake-cultures of Streptomyces albus (strain JA3453) 24 h after inoculation. Culture broth was harvested 72 h, isolated and purified samples were determined by 13C NMR spectroscopy. Thiericke et al. observed intact incorporation of acetate into the positions C1′/C2′, C3′/C4′, C5′/C6′, C7′/C8′, C9′/C10′ of left-hand fragment, C5/C6, C7/C8, C9/C10 positions near by middle fragment and C1/C2 positions of γ-lactam core.20 Methylation mediated by L-[methyl-13C]methionine proved the origin of all C-linked methyl groups, attached at positions C4′, 2×(C2′), C6 and at C2, as well as O-linked and N-linked methyl groups. Feeding experiments with [1-13C]glycine revealed incorporation into the positions C11 and C11′ of 1. Application of sodium [1-13C]propionate and L-[1-13C]alanine in feeding experiments had not confirmed incorporation of marked atoms into the structure of 1.20In the same work Thiericke et al. examined hypothetical incorporation of inthomycins into the structure of 1.20 Thus, [14C]-labelled inthomycins were prepared by feeding of [1,2-14C2]acetate to the inthomycin producing organism Streptomyces strain Gö2. However, feeding experiments of purified [14C]-labelled inthomycins to the oxazolomycin producing strain proved that only 0.4% of labelled intermediate was incorporated into the structure of 1. Moreover, amount of inthomycins during fermentation drastically decreased to 1%. Therefore, inthomycins themselves seem not to be intermediates involved in the biosynthesis of oxazolomycin A.20
2.2 Investigation of the oxazolomycin A gene cluster
Despite of significant results from labeling data,20 the origins of some carbons (C3, C4, C16 and C13′) remained unclear. However, chemical,21 biochemical22,23 and genetic investigations24 of some other natural products with similar moieties to the C3–C4 unit of oxazolomycins suggested that two-carbon moiety is probably derived from a metabolic intermediate of the glycolytic pathway, which serves as a precursor for the unusual methoxymalonyl-acyl carrier protein (methoxymalonyl-ACP) extender unit.25 These information enabled Zhao et al. to isolate the oxazolomycin A gene (ozm) cluster from Streptomyces albus JA3453 by cloning the methoxymalonyl-ACP biosynthetic locus and localizing the ozm cluster.7 The ozm cluster was confirmed by gene inactivation, affording mutant strains that had lost oxazolomycin production.26 The boundaries of the ozm cluster were evaluated by Zhao et al. and delimitated by gene inactivation of orf(−1), orf(−2) and orf(−3)–orf(−5) for the upstream boundary26 and orf(+1), orf(+2) and orf(+3)–orf(+5) for the downstream boundary.25 These experiments and bioinformatics analysis established that the ozm cluster spans at most 79.5 kb of DNA consisting of 20 open read frames (ORFs) designated ozmA to ozmU (Fig. 3).25–27 The functions of ORFs (Table 1) were predicted by comparing the deduced gene products with proteins of known function in the databases.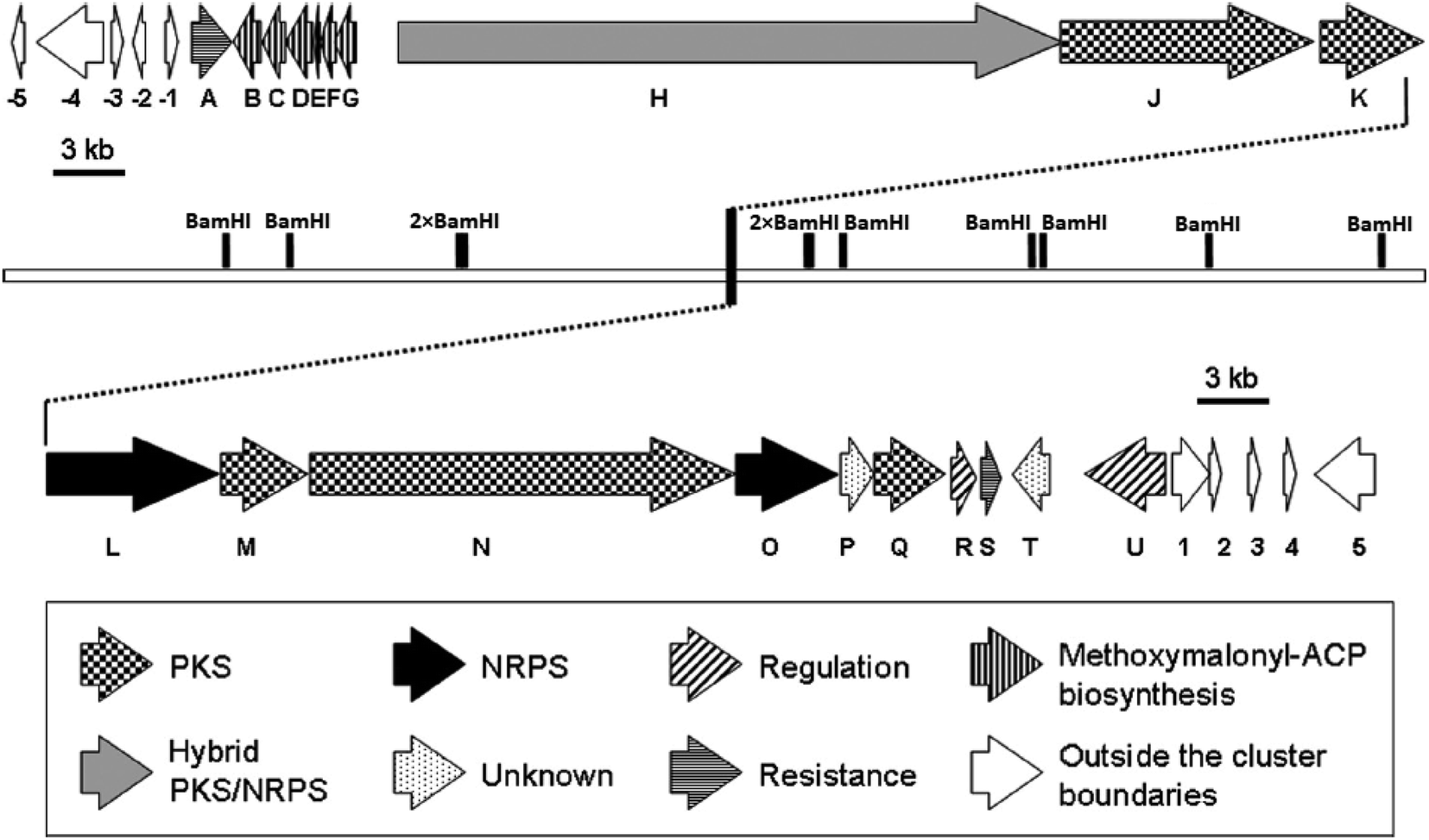 | ||
| Fig. 3 Genetic organization in the ozm cluster. Numbers refer to genes outside ozm cluster, letters refer to genes inside ozm cluster.25 | ||
| Gene | Sizea | Protein homologb | Proposed function | |
|---|---|---|---|---|
| a Size represents the number of amino acids.b Given in parentheses are accession numbers and percentage identity/percentage similarity. | ||||
| Upstream region of ozm cluster | orf(−5) | 335 | Franean1_1604(ABW11043, 52/39) | Transcriptional regulator |
| orf(−4) | 775 | SAV_1033(NP_822208 83/71) | Integral membrane protein | |
| orf(−3) | 158 | SAV_1074(NP_822249, 83/79) | Bacterioferritin comigratory protein | |
| orf(−2) | 339 | SACE_5658(CAM04844, 59/47) | Transcriptional regulator | |
| orf(−1) | 315 | CMS_2856(YP_001711489, 84/71) | Nucleoside hydrolase | |
| ozm cluster | ozmA | 482 | SgcB(AAF13999, 28/43) | Antibiotic efflux protein |
| ozmB | 366 | GdmH(ABI93783, 65/76) | Glyceryltransferase/phosphatase | |
| ozmC | 325 | DpsC(AAA65208, 25/39) | Acyltransferase | |
| ozmD | 366 | GdmI(ABI93784, 64/75) | Acyl-dehydrogenase | |
| ozmE | 93 | GdmJ(ABI93785, 63/78) | ACP | |
| ozmF | 221 | TtmC(AAZ08058, 60/77) | O-Methyltransferase | |
| ozmG | 287 | TtmB(AAZ08059, 63/72) | 3-Hydroxyacyl-CoA-dehydrogenase | |
| ozmH | 7737 | PksP(E69679, 35/51) | Hybrid NRPS/PKS | |
| ozmJ | 2926 | ObsC(AAS00421, 52/64) | PKS | |
| ozmK | 1202 | BryB(ABK51300, 34/50) | PKS | |
| ozmL | 1993 | McyA(AAF00960, 37/55) | NRPS | |
| ozmM | 1039 | MmpIII(AAM12912, 47/59) | Acyltransferase/oxidoreductase | |
| ozmN | 4971 | LnmJ(AF484556, 38/48) | PKS | |
| ozmO | 1196 | PedF(AAS47564, 42/56) | NRPS | |
| ozmP | 382 | — | Unknown | |
| ozmQ | 842 | NosB(AAF15892, 55/69) | PKS | |
| ozmR | 308 | Orf5(BAA32133, 53/64) | Transcriptional regulator | |
| ozmS | 214 | SC5F8.18(CAB93746, 71/78) | Transporter | |
| ozmT | 439 | SCH63.25(CAC10316, 77/83) | Thr-tRNA synthetase | |
| ozmU | 929 | AfsR(BAA14186, 33/45) | Transcriptional activator | |
| Downstream region of ozm cluster | orf(+1) | 445 | CypC(ABS73471, 42/59) | Cytochrome P450 |
| orf(+2) | 221 | — | Unknown | |
| orf(+3) | 282 | SC5F8.24(CAB93752, 70/81) | RNA polymerase sigma factor | |
| orf(+4) | 344 | — | Unknown | |
| orf(+5) | 700 | — | Unknown | |
The ozmA to ozmU genes encode corresponding proteins (OzmA to OzmU):25
• Nonribosomal peptide synthetases (NRPSs): OzmL, OzmO.
• Hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS): OzmH.
• trans-Acyltransferase (trans-AT) type I modular polyketide synthases (PKSs): OzmJ, OzmK, OzmN and OzmQ.
• Enzymes for methoxymalonyl-ACP biosynthesis: OzmB, OzmD, OzmE, OzmF and OzmG.
• Discrete AT enzymes: OzmM and OzmC.
• Hypothetical proteins as candidates for resistance: OzmA and OzmS.
• Proteins as candidates for regulation or postmodification: OzmR, OzmU and OzmT.
• Proteins with uncertain functions: OzmP.
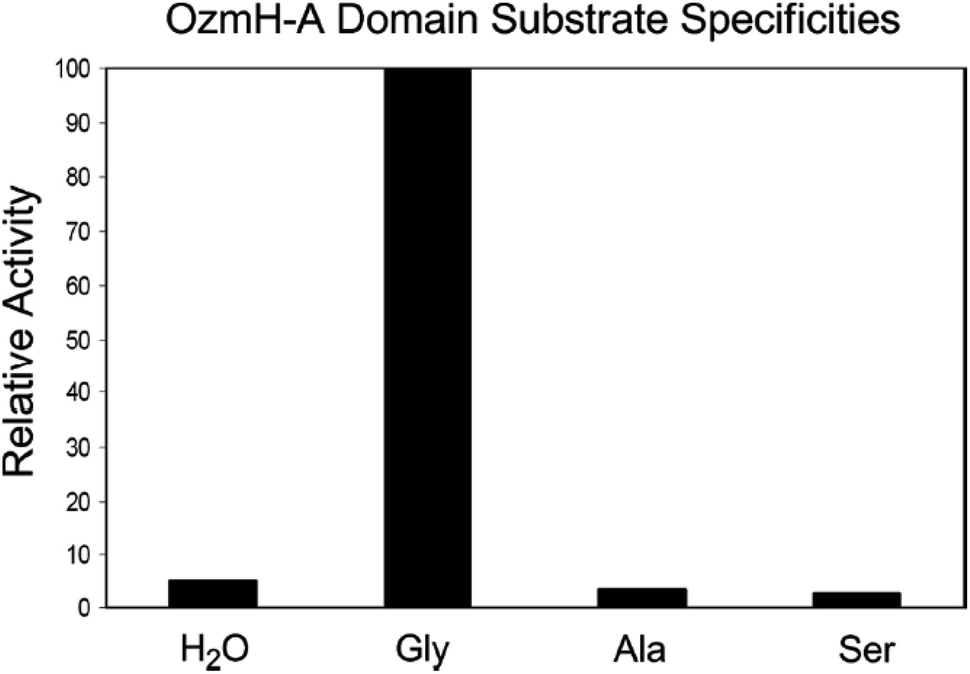 | ||
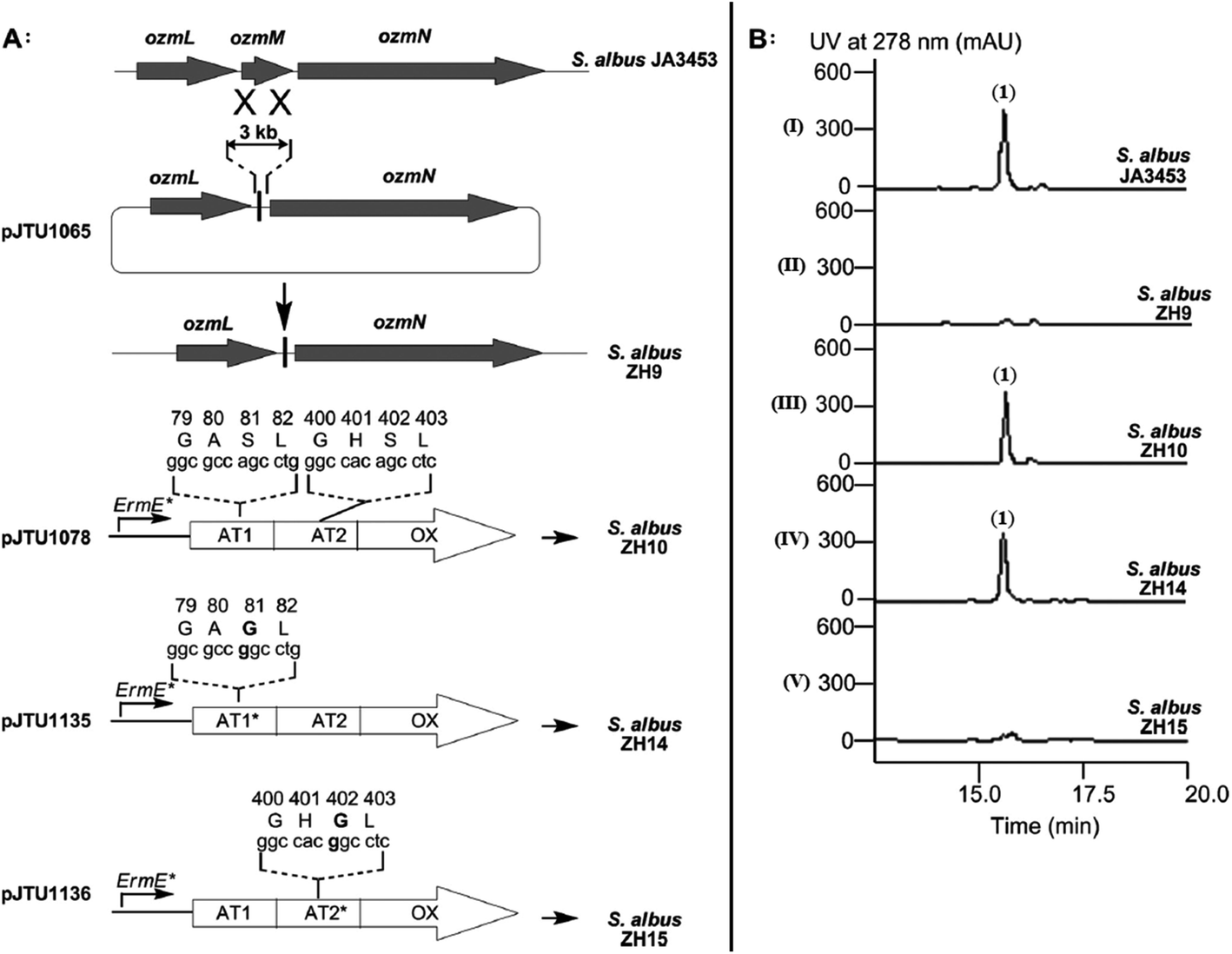
Fig. 4 Determination of OzmH A domain substrate specificities. The ATP-PPi exchange reactions were performed using amino acids Gly, Ala and Ser as substrates and H2O as a negative control (100% relative activity corresponds to 995![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 320 cpm).25 320 cpm).25 | ||
 | ||
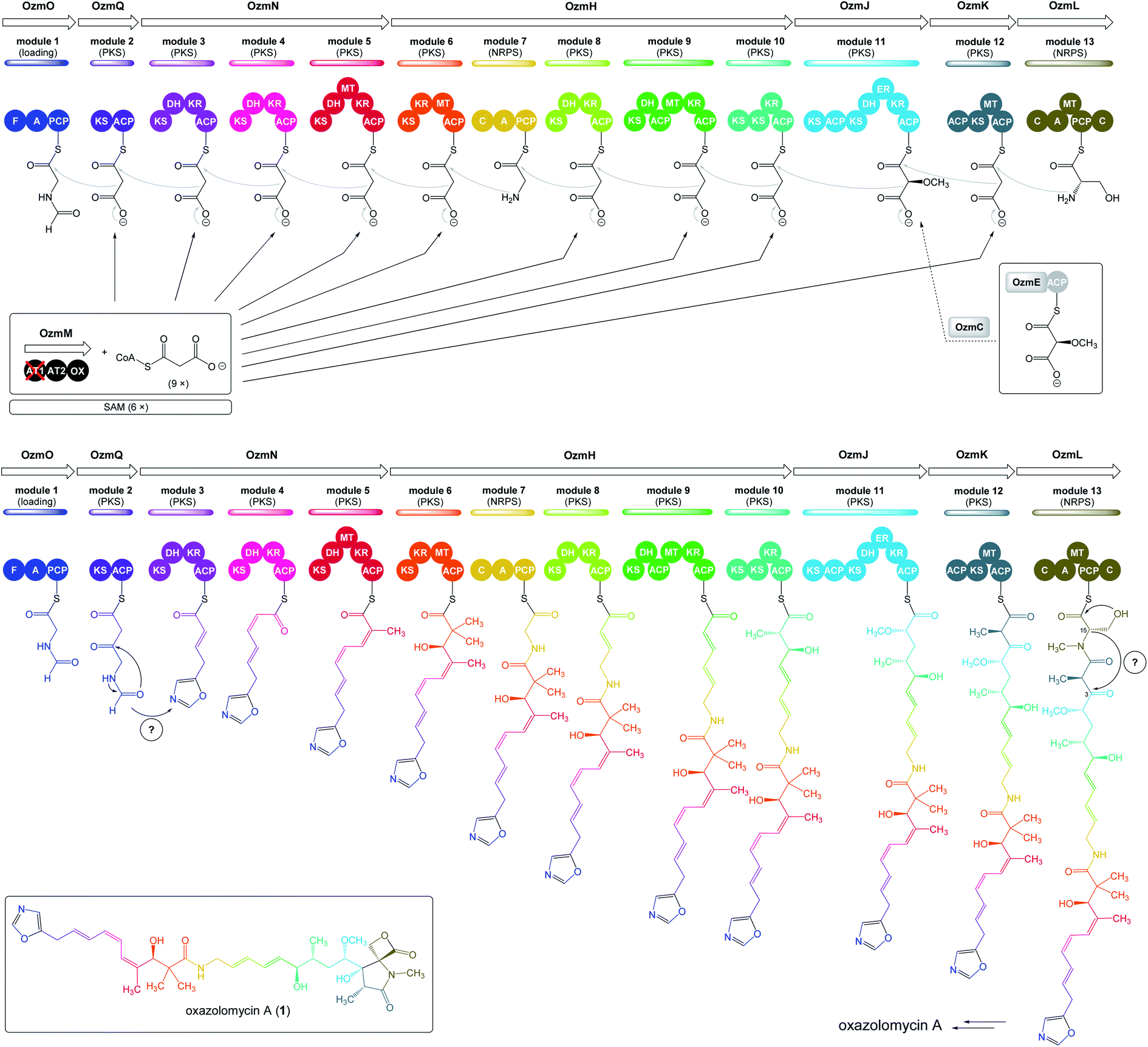
| Fig. 5 Deletion of ozmM and complementation of the ozmM mutant with either intact ozmM or ozmM-AT1 (Ser81 to Gly) or ozmM-AT2 (Ser402 to Gly) mutant and their effect on the biosynthesis of oxazolomycin A. (A) Schematic representation of constructs for the generation of the ZH9 deletion mutant strain and its genetic complementation strains ZH10, ZH14 and ZH15. Details of side-specific mutagenesis with OzmM-AT1 and OzmM-AT2 are shown. (B) HPLC analysis of oxazolomycin A (1) production in: (I) Streptomyces albus JA3453; (II) ZH9 mutant; (III) ZH10 mutant; (IV) ZH14 mutant and (V) ZH15 mutant.25 | ||
 | ||
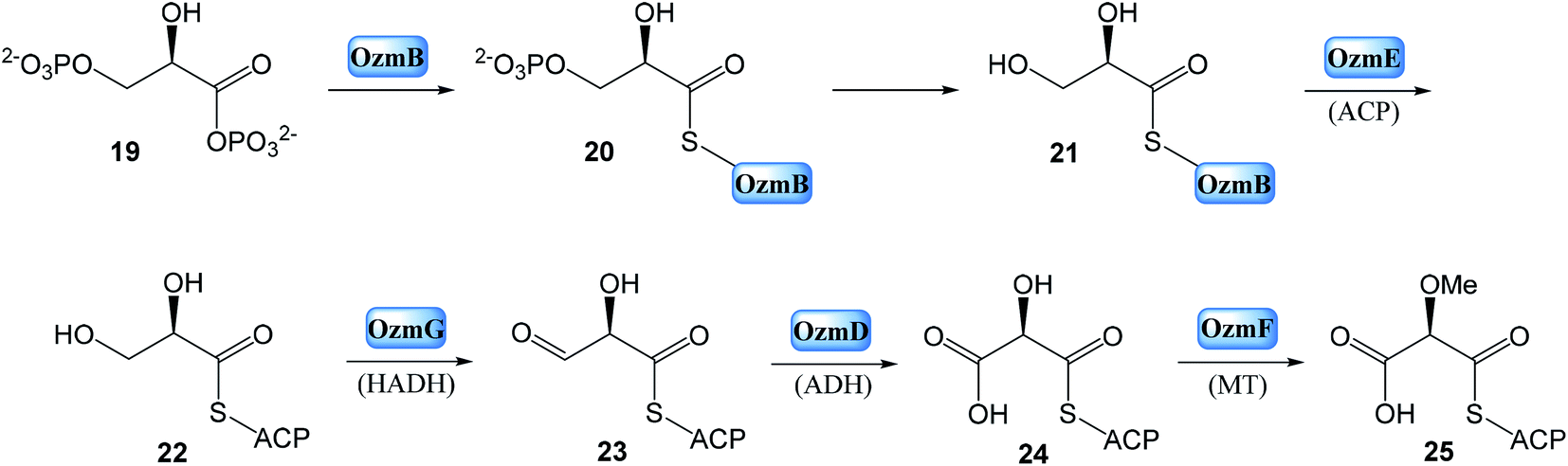
| Fig. 6 Proposed model of oxazolomycin A biosynthesis in Streptomyces albus JA3453. Used abbreviations: A, adenylation domain; ACP, acyl carrier protein domain; AT1, N-terminal acyl transferase domain of OzmM; AT2, central acyl transferase domain of OzmM; C, condensation domain; DH, dehydratase domain; ER, enoyl reductase domain; F, formylation domain; KR, ketoreductase domain; KS, ketosynthetase domain; MT, methyltransferase domain; OX, oxidoreductase domain; PCP, peptide carrier protein domain; ?, unknown domain; SAM, S-adenosyl methionine.25 | ||
 | ||
| Scheme 1 Proposed pathway for methoxymalonyl-ACP extender unit biosynthesis in ozm cluster. | ||
Bioinformatic analysis failed to predict such a cyclase gene or domain, within the ozm cluster. However, the OzmP, adjacent to and co-transcribed with OzmO, whose function could not be assigned to date, may be involved in oxazole ring biosynthesis. Further it is assumed, that intermediate could be condensed with malonyl-CoA on the OzmQ (module 2). Condensation with other three malonyl-CoAs takes place on OzmN (modules 3–5), along with C-methylation to C4′ position mediated by MT domain (module 5). Transportation to large protein OzmH involves condensation with malonyl-CoA and assumed double C-methylation to C2′ position by MT domain (module 6). After condensation with glycine (module 7) and with three malonyl-CoAs (modules 8–10), other C-methylation into C6 position occurs within OzmH. Although an MT domain should be present in module 10, as deduced from the structure of oxazolomycin A, it was unexpectedly identified in module 9. This gene organization may be explained as domain mispositioning, reflecting complex domain–domain interactions for polyketide-catalyzed methylation. Condensation with a methoxymalonyl-ACP is realized on OzmJ (module 11). After translocation to OzmK, a condensation with malonyl-CoA and C-methylation to C2 position takes place (module 12).
Finally, the biosynthesis of oxazolomycin A is terminated on OzmL. The amino acid serine is activated by the A domain and covalently bound to the PCP. Further, N-terminal C domain catalyzes formation of the peptide bond between seryl-S-PCP and the preceding acyl-S-ACP intermediate, followed by N-methylation by the MT domain. The C domain residing at the C terminus was proposed to release the full-length hybrid peptide polyketide product from PCP, giving the four-membered β-lactone ring, because no thioesterase domain was identified in the ozm cluster. Although the C-terminal C domain lacks a catalytic triad for condensation, it does have the Asp residue in both C domains and heterocyclase domains, which are subtypes of C domains. The heterocyclase domains catalyze not only peptide bond formation, but also subsequent cyclization of Cys, Ser or Thr, to afford a thiazoline or oxazoline ring. Although the C-terminal C domain in OzmL lacks the crucial His that is believed to be indispensable for condensation of aminoacyl-ACP and peptidyl-ACP, it may be still functional. Especially given, that this domain is not expected to catalyze peptide bond formation but only cyclization of the Ser side chain to form a β-lactone ring. Some of characterized C domains have been demonstrated39,40 to catalyze ester bond formation. A similar function would be envisioned for the OzmL C-terminal C domain to be involved in β-lactone ring formation. However, a cyclase may be required for C3–C15 bond formation to afford the heterocyclic γ-lactam ring, but no apparent candidate gene was identified by bioinformatic analysis in the ozm cluster (Fig. 6).25
This oxazolomycin A NRPS-PKS megasynthase was also characterized with many features that appear to violate the typical “co-linearity rule” for NRPS or PKS domain organization, including domain redundancy (presence of two ACPs flanking the MT and KR domain in module 9; tandem KSs in module 10; two ACPs and two KSs in module 11) and mispositioning (presence of MT domain in module 9 except assumed module 10).25,41,42 The example of domain redundancy has been confirmed experimentally where the catalytic Cys residue of the tandem KSs of OzmH module 10 were site-specifically mutated to Gly.25,35
According to results from labeling data20 and analysis of genes in the ozm cluster,25 presumably a whole structure of oxazolomycin A is constructed from 6 different building blocks (Fig. 7).
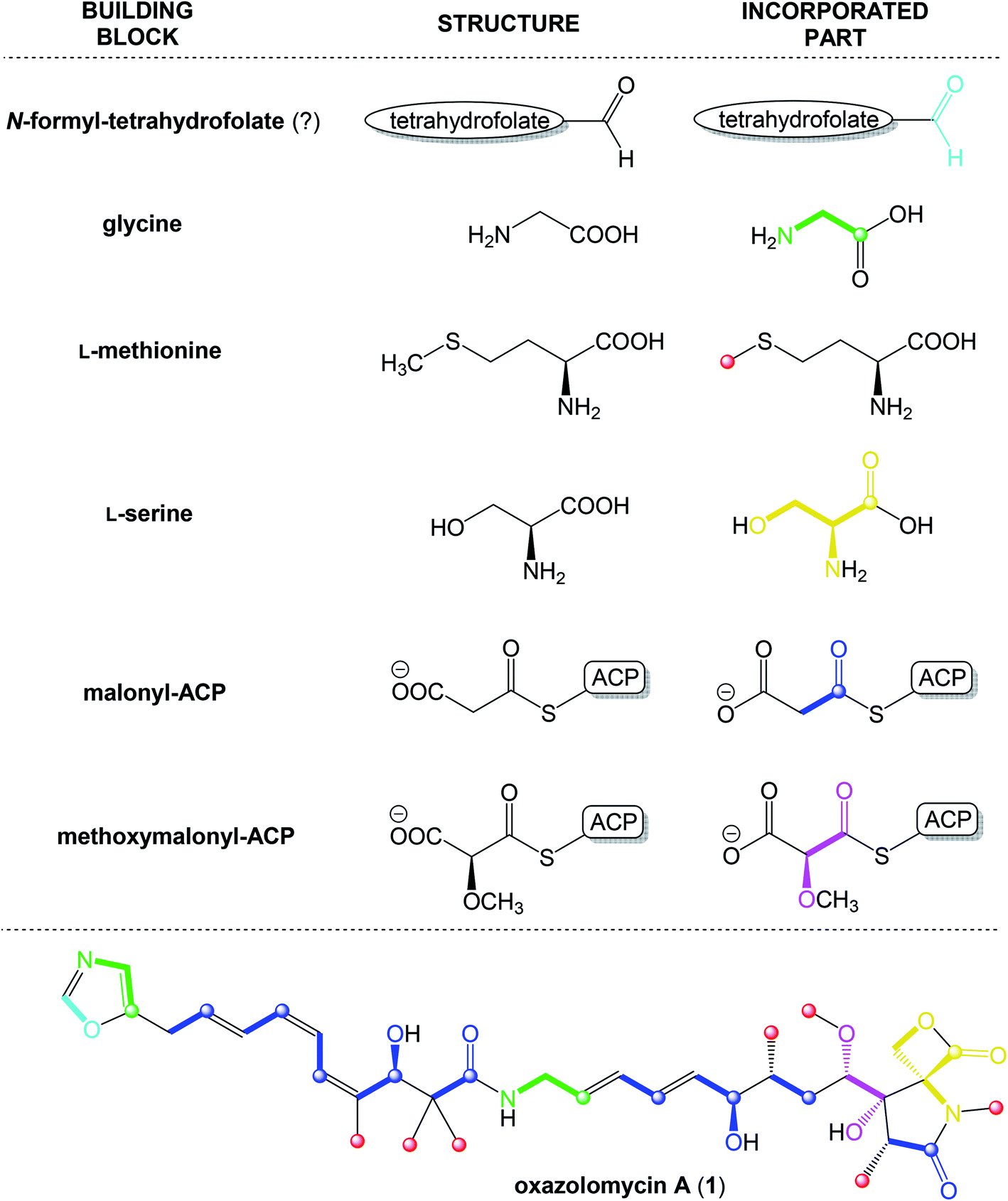 | ||
| Fig. 7 Proposed biosynthetic origin of oxazolomycin A (1) according to labeling data and analysis of genes in ozm cluster. | ||
3 Biological activity of oxazolomycins and their fragments
3.1 Biological activity of oxazolomycin fragments
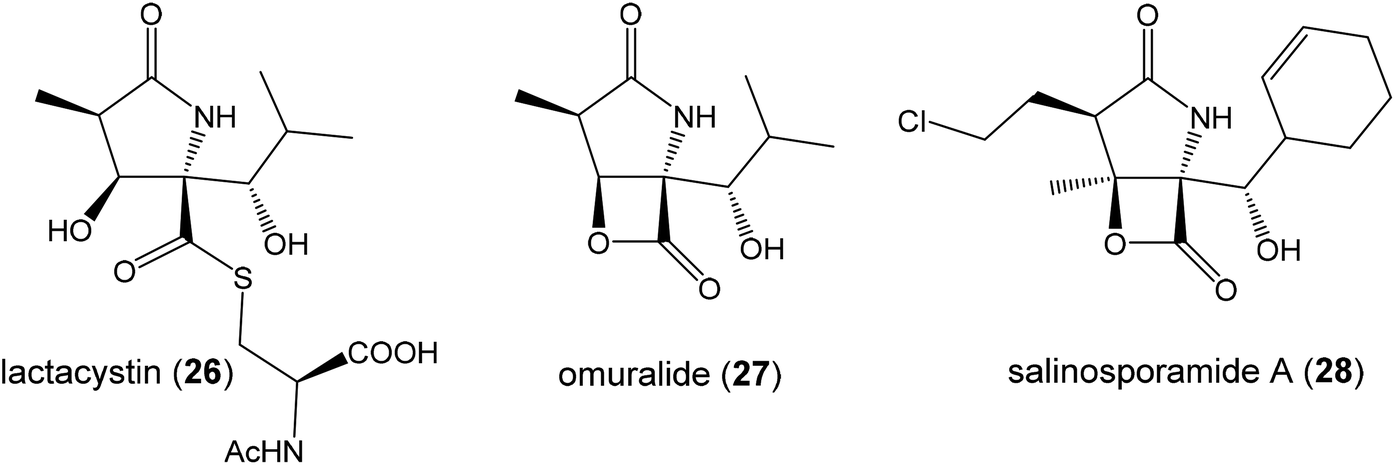 | ||
| Fig. 8 Known proteasome inhibitors with pyroglutamate core. | ||
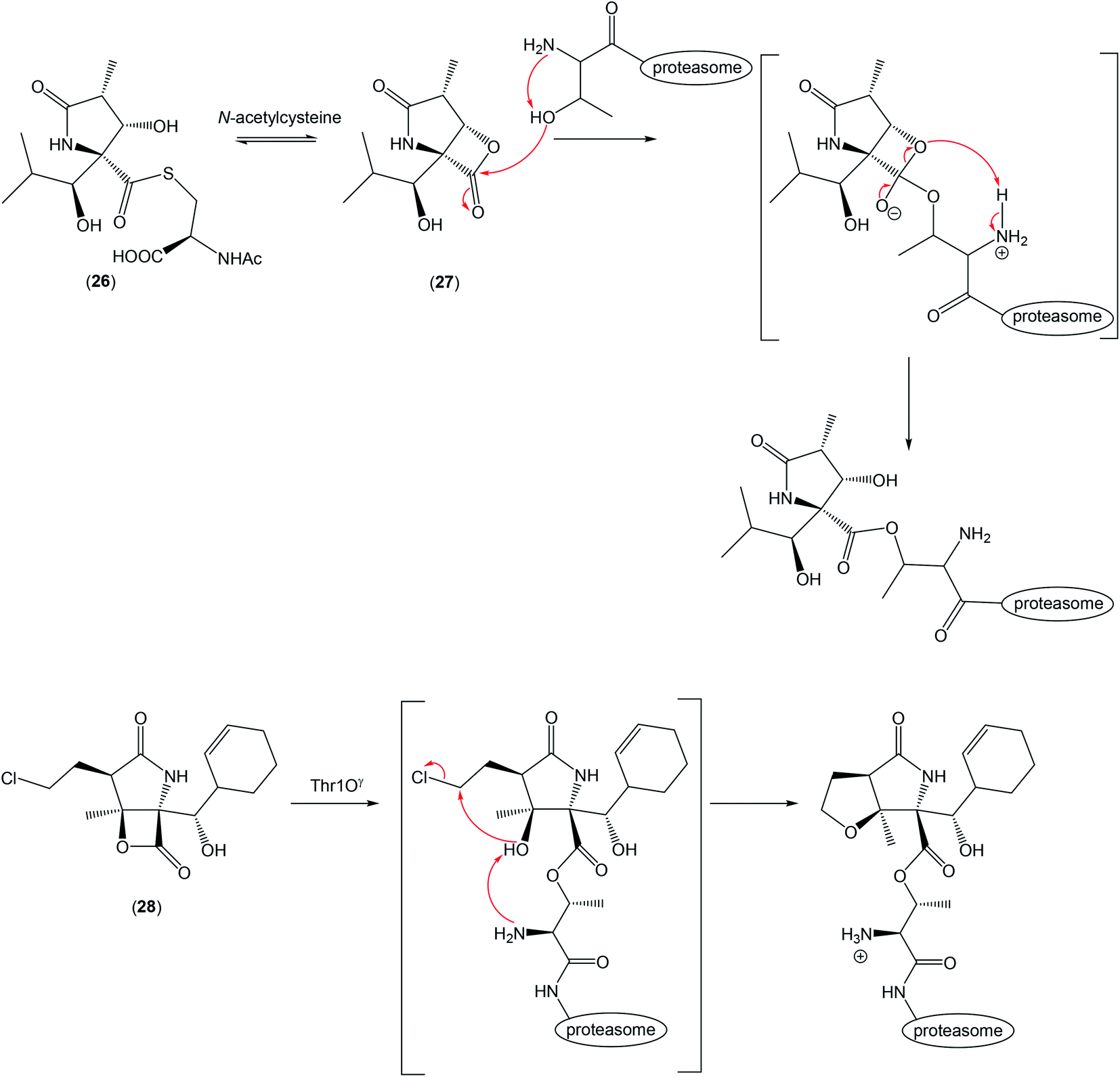 | ||
| Scheme 2 Pyroglutamate derivatives and their binding in 20S proteasome. | ||
Several studies evaluated SAR properties of right-hand fragment derivatives of oxazolomycins. It was reported, that the intrinsic antibacterial activity of simple pyroglutamates48,49 and tatramates50 is low. However further reports suggested,51,52 that homologation to longer chain side-units restores bioactivity. Anwar et al. observed that a change as small as the introduction of a methyl substituent improves bioactivity in tetramates.53 The right-hand fragment derivatives were evaluated against Staphylococcus aureus and Escherichia coli using the hole-plate method. Although quantitative MIC values are not available, these in vivo assays give outcomes enabling rapid assessment of the effect of structural variations of the fragment modification. Moreover, chemical informatics analysis of prepared derivatives was reported. Selected structures are shown in Fig. 9, their bioactivity and chemical informatics analysis are summarized in Table 2.
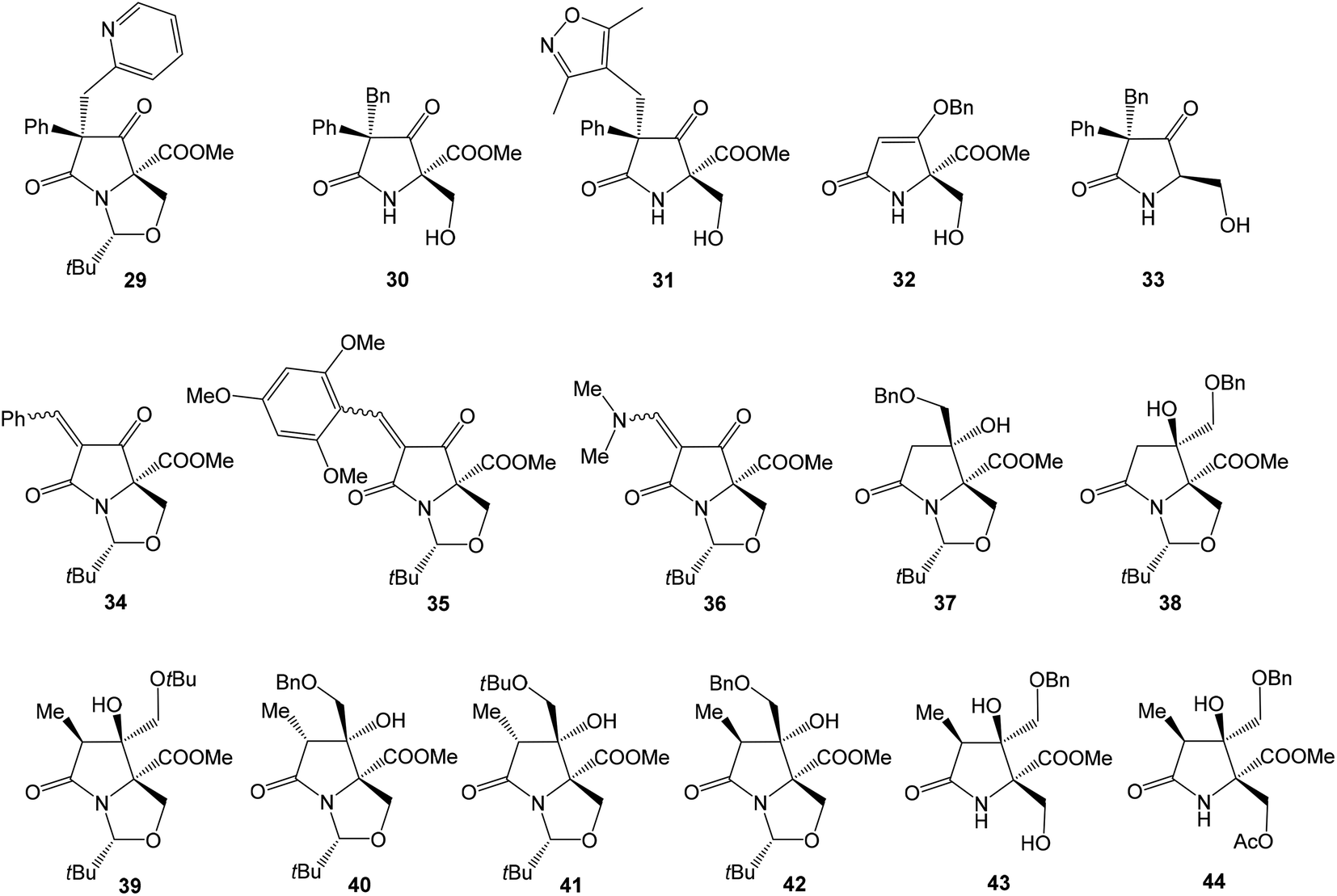 | ||
| Fig. 9 Selected bioactive right-hand fragment derivatives. | ||
| Compound | LogPa |
PSAa | % PSAb | Bioactivity | Reported by | |||
|---|---|---|---|---|---|---|---|---|
| S. aureus | E. coli | |||||||
| Zone size [mm] | Relative potencyc | Zone size [mm] | Relative potencyc | |||||
| a LogP, PSA and MSA calculated using Marvin.b % PSA = (PSA/MSA) × 100%.c Expressed as zone size per mg ml−1, relative to cephalosporin C standard.d Using 100 μl of 4 mg ml−1 solution (DMSO).e Using 100 μl of 4 mg ml−1 solution (6:4, DMSO:H2O).f Halo only. |
||||||||
| Oxazolomycin A (1) | 1.61 | 171.7 | 17.9 | — | — | — | — | Angelov et al.54 |
| Oxazolomycin B (3) | 1.61 | 171.7 | 17.9 | — | — | — | — | Angelov et al.54 |
| Oxazolomycin C (4) | 1.61 | 171.7 | 17.9 | — | — | — | — | Angelov et al.54 |
| 16-Methyloxazolomycin (5) | 2.03 | 171.7 | 17.1 | — | — | — | — | Angelov et al.54 |
| Curromycin A (6) | 1.72 | 180.1 | 16.6 | — | — | — | — | Angelov et al.54 |
| Curromycin B (7) | 2.10 | 171.7 | 16.5 | — | — | — | — | Angelov et al.54 |
| KSM-2690 B (8) | 2.03 | 171.7 | 17.1 | — | — | — | — | Angelov et al.54 |
| KSM-2690 C (9) | 2.03 | 171.7 | 17.1 | — | — | — | — | Angelov et al.54 |
| Lajollamycin (10) | 2.66 | 191.5 | 18.4 | — | — | — | — | Angelov et al.54 |
| 29d | 3.75 | 85.8 | 13.8 | 25 | — | Inactive | — | Holloway et al.52 |
| 30d | 2.45 | 92.7 | 18.8 | 15 | — | 18 | — | Holloway et al.52 |
| 31d | 1.15 | 118.7 | 22.8 | 14 | — | 19 | — | Holloway et al.52 |
| 32d | −0.04 | 84.9 | 22.4 | 15 | — | 19 | — | Holloway et al.52 |
| 33d | 2.45 | 66.4 | 15.8 | 12 | — | 14 | — | Holloway et al.52 |
| 34d | 3.34 | 72.9 | 14.8 | 15 | — | 18 | — | Holloway et al.52 |
| 35d | 2.87 | 100.6 | 15.7 | 15 | — | 19 | — | Holloway et al.52 |
| 36d | 1.33 | 76.2 | 16.1 | Inactive | — | 18 | — | Holloway et al.52 |
| 37e | 1.97 | 85.3 | 14.4 | 15 | 3.8 | 18 | 0.087 | Angelov et al.54 |
| 38e | 2.14 | 85.3 | 14.4 | 13 | 0.91 | 19 | 0.08 | Angelov et al.54 |
| 39e | 1.84 | 85.3 | 14.0 | 11f | 1.7 | 12f | 0.035 | Angelov et al.54 |
| 40e | 2.51 | 85.3 | 13.8 | 13 | 2.6 | 18 | 0.087 | Angelov et al.54 |
| 41e | 1.84 | 85.3 | 14.0 | 11f | 1.7 | 12f | 0.035 | Angelov et al.54 |
| 42e | 2.51 | 85.3 | 13.8 | 15 | 3.8 | 15 | 0.055 | Angelov et al.54 |
| 43e | −0.01 | 105.1 | 21.8 | 11 | — | 18 | 0.06 | Angelov et al.54 |
| 44e | 0.45 | 111.2 | 20.4 | 13 | 0.88 | 18 | 0.07 | Angelov et al.54 |
Holloway et al. prepared series of substituted tetramates 29–36 with antibacterial properties.52 Significant activity against S. aureus was observed for substituted tetramate 29, however against E. coli no bioactivity was observed. The tatramates 30, 31, 32 and bicyclic derivatives 34, 35 exhibited similar bioactivities, whereas activity of primary alcohol 33 was lightly reduced. Bicyclic enamine 36 was inactive against S. aureus, however the activity against E. coli was found. Chemical informatics analysis is shown in Table 2.52 Additionally, SAR analysis of pyroglutamate core derivatives was evaluated by Angelov et al. on different structural modifications of oxazolomycin's right-hand fragment.54 The compounds 37–44 (Fig. 9) were the only products with activity against both S. aureus and E. coli, although out of the compounds with the correct relative configuration for oxazolomycin A, only 39, 43 and 44 were active.
Most of the active compounds prepared by Angelov et al. have clogP values in the range 1.8–2.5 and % PSA values (polar surface area parameter (PSA)/molecular surface area parameter (MSA) × 100%) close to 14, with a potency of some 1–4% relative to the cephalosporin reference standard (Table 2). However, the compounds 43 and 44 were clearly different, being very much polar, with clogP values in the range −0.01 to 0.5 and % PSA values close to 21. It is noteworthy, that substitution of Bn group in benzyloxymethyls 37, 38, 40 and 42 to Me group afforded corresponding methoxymethyls, which might be considered as mimics structurally closest to the oxazolomycin's right-hand fragment. However, all of these methoxymethyls were inactive or only weakly active. It was assumed, that better activity of benzyloxymethyl structures 37, 38, 40 and 42 than corresponding methoxymethyls was a result of improved cell membrane permeability. It is interesting, that most active compounds prepared by Angelov et al. had chemical informatics values which correlated with the corresponding values for oxazolomycins described in Table 2. However, it is important to note, that only compounds 39, 43 and 44 have correct relative configuration to oxazolomycins.54
Moreover, Koomsiri et al. reported, that antibacterial activity (Fig. 16) of oxazolomycin A (1) is more potent than oxazolomycin A2 (14) and bisoxazolomycin (15).10 It is proposed, that β-lactone ring in right-hand fragment of oxazolomycin A plays an important role in the antibacterial activity.10 This assumption is in accordance with proposed β-lactone-γ-lactam core importance for biological activity.46,47
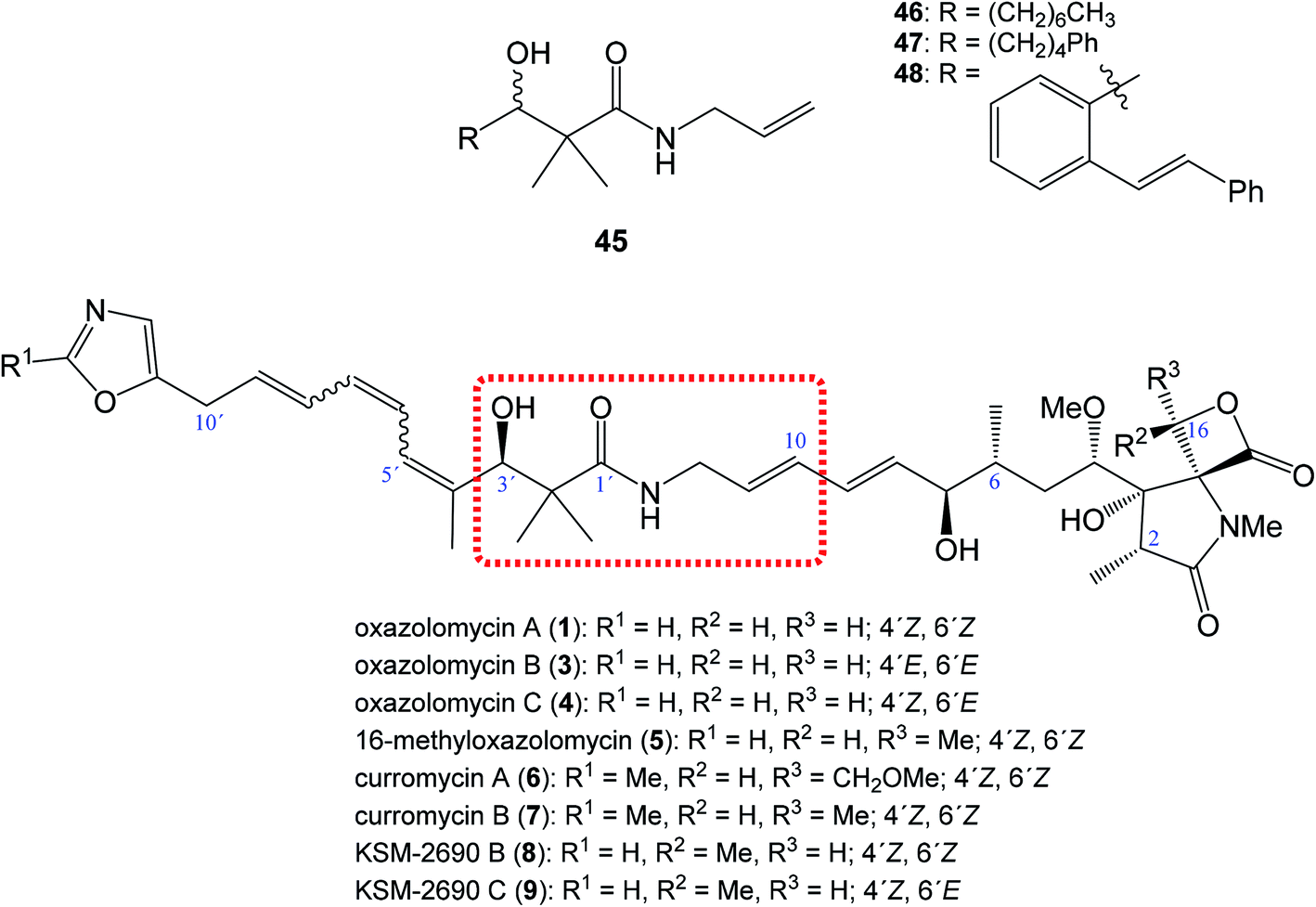 | ||
| Fig. 10 Position of structural subunit 45 in the structure of oxazolomycins responsible for U-shaped conformation and its derivatives 46–48 prepared by Bagwell et al. | ||
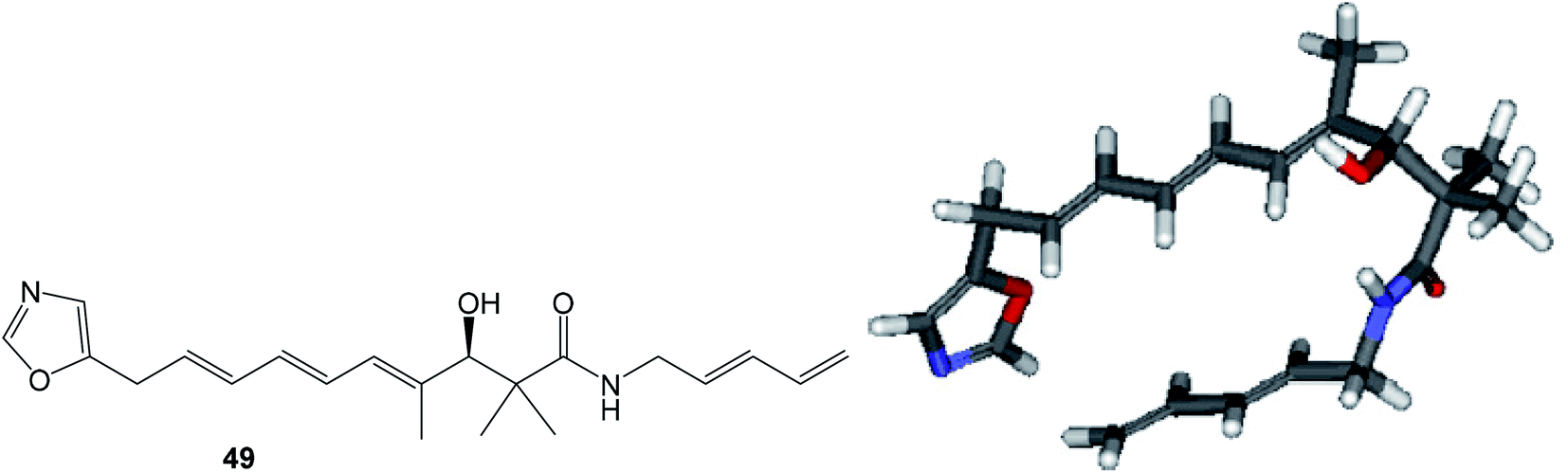 | ||
| Fig. 11 Structure and calculated minimum energy conformation of amide 49. | ||
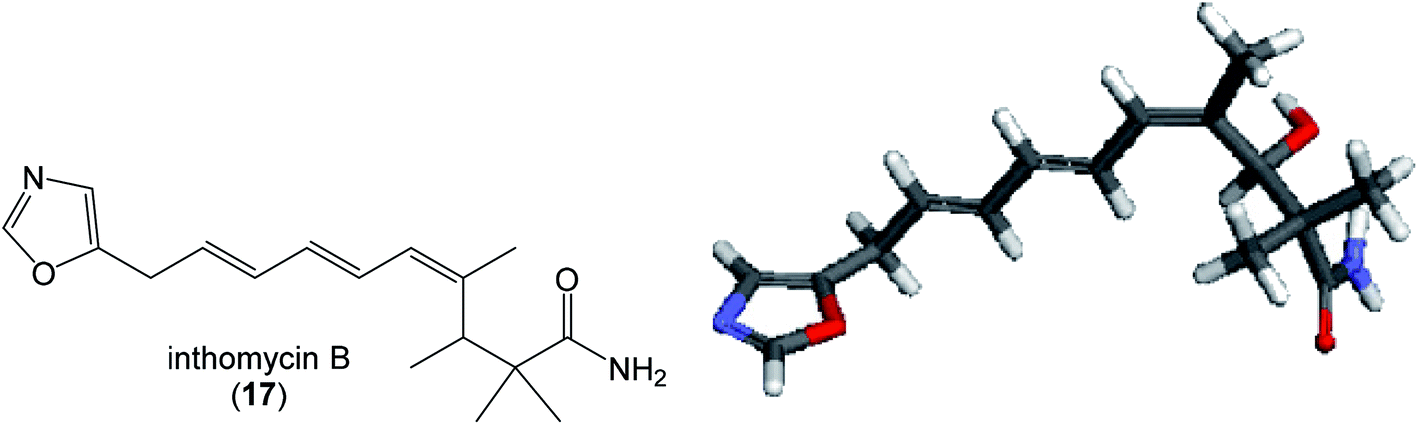 | ||
| Fig. 12 Structure and calculated minimum energy conformation of inthomycin B (17). | ||
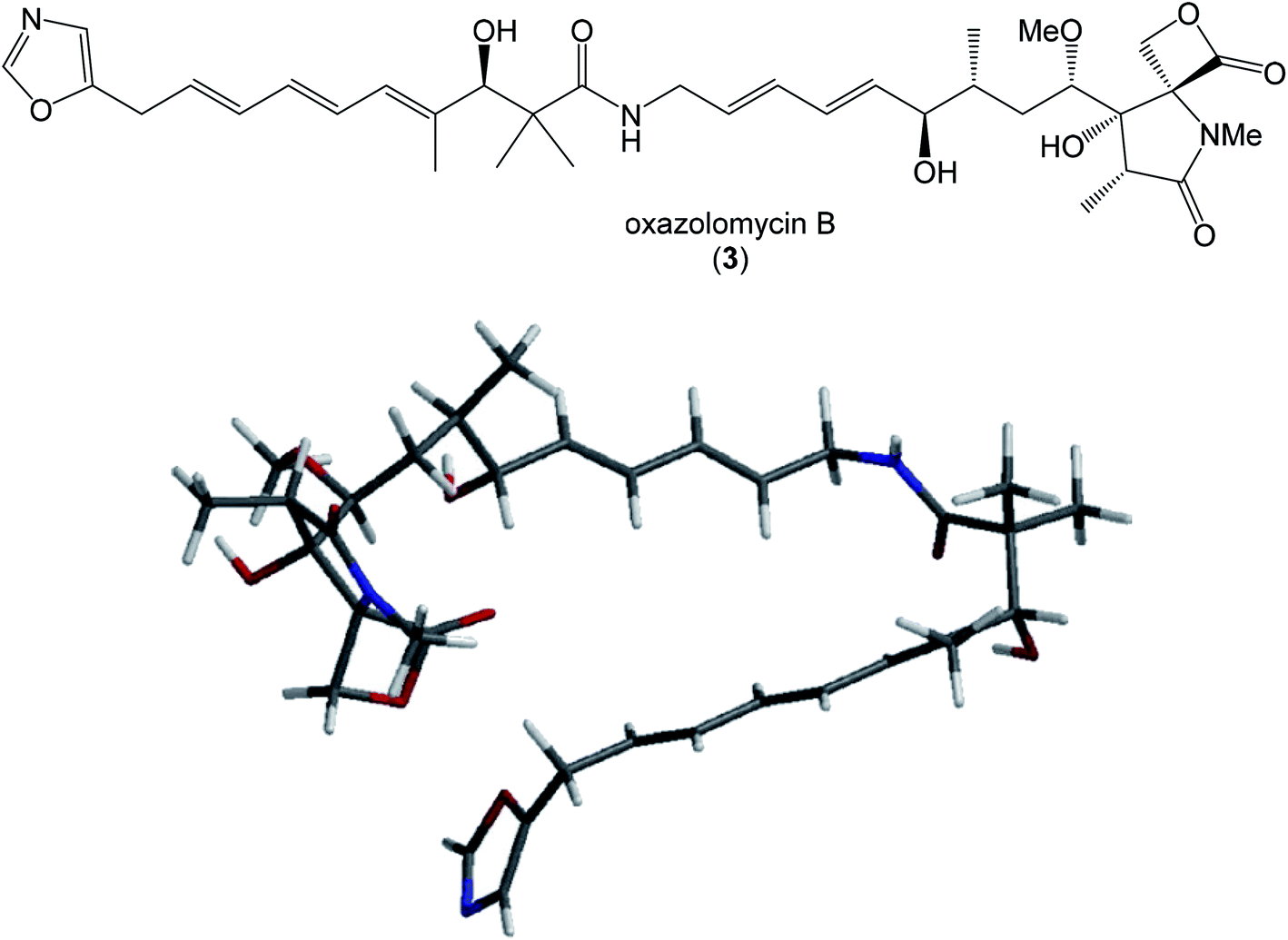 | ||
| Fig. 13 Structure and calculated minimum energy conformation of oxazolomycin B (3). | ||
Bagwell et al. further reported, that appropriately substituted 3-hydroxy-2,2-dimethylpropanamide motif present in oxazolomycins can exhibit antibacterial activity itself. Selected compounds 46, 47 and 48 (Fig. 10) were evaluated against Staphylococcus aureus and Escherichia coli to provide their bioactivity (Table 3).55 The bioassay against S. aureus showed, that compound 46 is inactive, however derivatives 47 and 48 were active. It was found, that the most active structure 47 (MIC 4 mg ml−1)55 is approximately 80-fold less active than KSM-2690 B (8) (MIC 50 μg ml−1).7 However, higher level of the activity were found against E. coli. The MIC of selected compounds 46–48 was found to be in range 0.5–1 mg ml−1.55 Since the reported MIC values for E. coli using oxazolomycins A–C (1, 3 and 4) were found 100 μg ml−6, the most active compounds 47 and 48 resulted approximately only 5–10 times weaker.55 These results suggest, that structural subunit 45 present in structure of oxazolomycins could plays an additional role for final bioactivity.
| Compound | LogPa |
Bioactivityb | |||||
|---|---|---|---|---|---|---|---|
| S. aureus | E. coli | ||||||
| Zone size [mm] | Relative potencyc | MICd | Zone size [mm] | Relative potencyc | MICd | ||
| a LogP calculated using ChemBioDraw Ultra 11.0.b Hole plate bioassay at 4 mg ml−1 (7:3, DMSO:H2O).c Expressed as zone size per mg ml−1, relative to cephalosporin C standard.d Estimated by seriál dilution until no inhibition zone is observed.e nd = not determined. |
|||||||
| 46 | 3.73 | Inactive | — | — | 22 | 0.01 | 1.0 |
| 47 | 4.08 | 17 | 0.08 | 4 | 23 | 0.01 | 0.5 |
| 48 | 4.74 | 14 | 0.06 | nde | 20 | 0.01 | 0.5 |
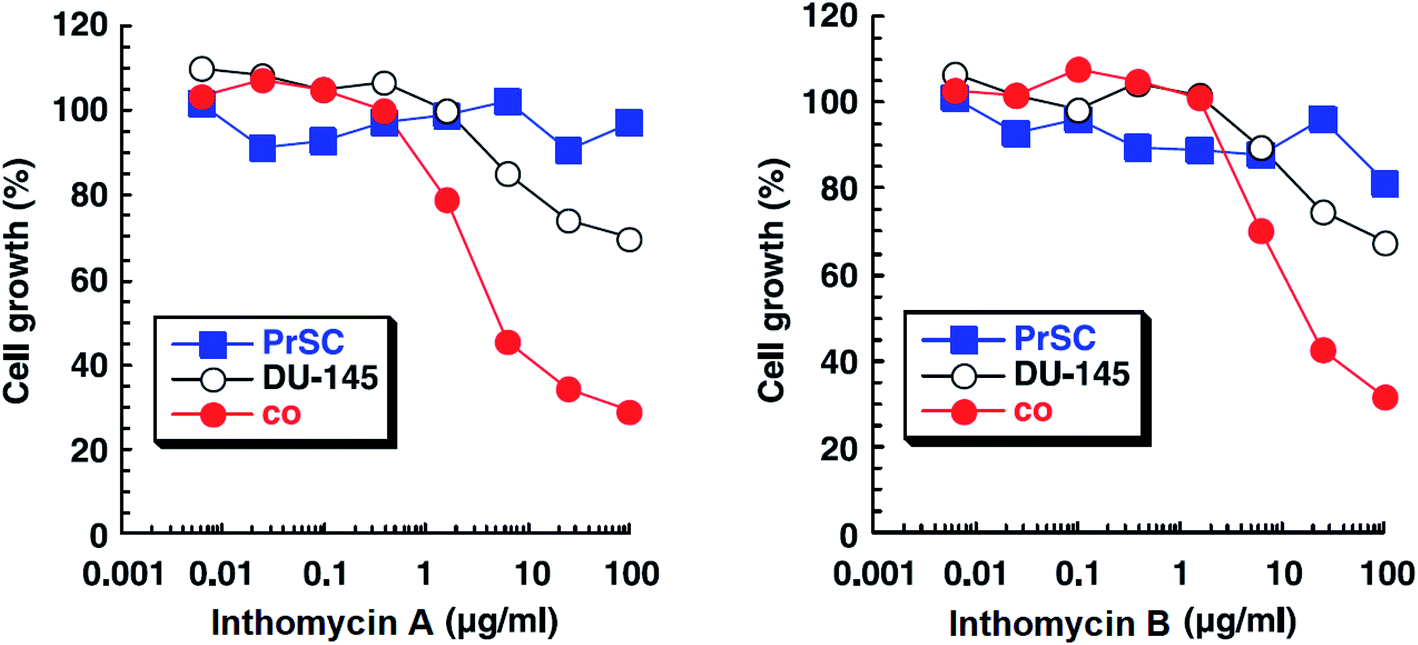 | ||
| Fig. 14 Effects of inthomycin A (16) and inthomycin B (17) on the growth of DU-145 cells alone (black open circles), DU-145 cells cocultured with PrSC (red filled circles) and PrSC alone (blue filled squares) were determined using rhodanile blue staining.59 | ||
Inthomycins as the structural subunits of oxazolomycins are interesting compounds with wide range of biological activities. It is assumed, that some of the beneficial biological properties of inthomycins could resemble to bioactivity of oxazolomycins. Unfortunately, direct and comprehensive evaluation of the biological properties of inthomycins and oxazolomycins is still missing to date.
3.2 Biological activity of oxazolomycins
The biological activities of oxazolomycin A (1), oxazolomycin B (3) and oxazolomycin C (4) were mutually compared by Kanzaki et al. on following bacteria: Agrobacterium tumefaciens IFO 13263, Agrobacterium tumefaciens EHA 101, Agrobacterium rhizogenes IFO 13257, Rhizobium loti IFO 13336, Escherichia coli and Bacillus subtilis (results are summarized in Table 4).6 As seen in Table 4, oxazolomycins 3 and 4 were inactive against tested bacteria, whereas 1 showed some activity against Agrobacterium rhizogenes IFO 13257 and significant activities against Agrobacterium tumefaciens IFO 13263 and Agrobacterium tumefaciens EHA 101. Noteworthy, oxazolomycins 1, 3 and 4 are geometric isomers with structural diversity in their left-hand fragment (inthomycin subunit). These results suggest biological importance of left-hand fragment considering identical central and right-hand fragment of tested oxazolomycins.
| Minimum inhibitory concentration (MIC) [μg ml−1] | |||||||
|---|---|---|---|---|---|---|---|
| Agrobacterium rhizogenes IFO 13257 | Agrobacterium tumefaciens EHA 101 | Agrobacterium tumefaciens IFO 13263 | Bacillus subtilis | Escherichia coli | Pseudomonas putida IFO 3738 | Rhizobium loti IFO 13336 | |
| Oxazolomycin A (1) | 25.0 | 6.3 | 3.1 | >100 | >100 | 50.0 | >100 |
| Oxazolomycin B (3) | >100 | 100 | >100 | >100 | >100 | — | >100 |
| Oxazolomycin C (4) | >100 | >100 | >100 | >100 | >100 | — | >100 |
Antibacterial activities of curromycin A (6) and B (7) were reported as very similar. Early in 1985 Ogura et al. reported,61 that biological activities of 7 were almost identical with 6.3 For example significant bioactivity of 7 against Bacillus subtilis IAM 1026 (MIC 3.9 μg ml−1) but lower against Pseudomonas cepacia M-0527 (MIC 50.0 μg ml−1) were reported.61 Similarly, identical weak antimicrobial activity for both 6 and 7 was reported by Ikeda et al. against Micrococcus luteus FDA16 (MIC 25 μg ml−1) and Pseudomonas aeruginosa A3 (MIC 50 μg ml−1).4 Solutions of 2.5 μg ml−1 of 6 and 7 in 1/15 M phosphate buffer (pH = 6.8) showed hazy inhibition zones 20.0 mm for 6 and 21.5 mm for 7 against Staphylococcus aureus FDA 209P using cylinder plate method.4 Kanzaki et al. also reported identical biological activities for both curromycins.62 No substantial bioactivity of 6 and 7 against Escherichia coli OP50 (MIC 100 μg ml−1) nor against Bacillus subtilis IFO 3007 (MIC >100 μg ml−1) was reported. The strong inhibitory activity was observed against Agrobacterium tumefaciens IFO 13263 (MIC 6.3 μg ml−1 for both curromycins), approximately half MIC when compared to oxazolomycin A. In addition, once both hydroxy groups in 6 and 7 (at C7 and C3′ positions) were acetylated, corresponding curromycin diacetates had no antibacterial activity against the three species of bacteria (MIC > 100 μg ml−1). And Kanzaki et al. compared effect of 7 and its diacetate against Agrobacterium tumefaciens growth on an inoculated potato tuber disk. No inhibitory effect was observed for curromycin B diacetate in contrary to active curromycin B (Fig. 15). The antibacterial activity of curromycins is summarized in Table 5.
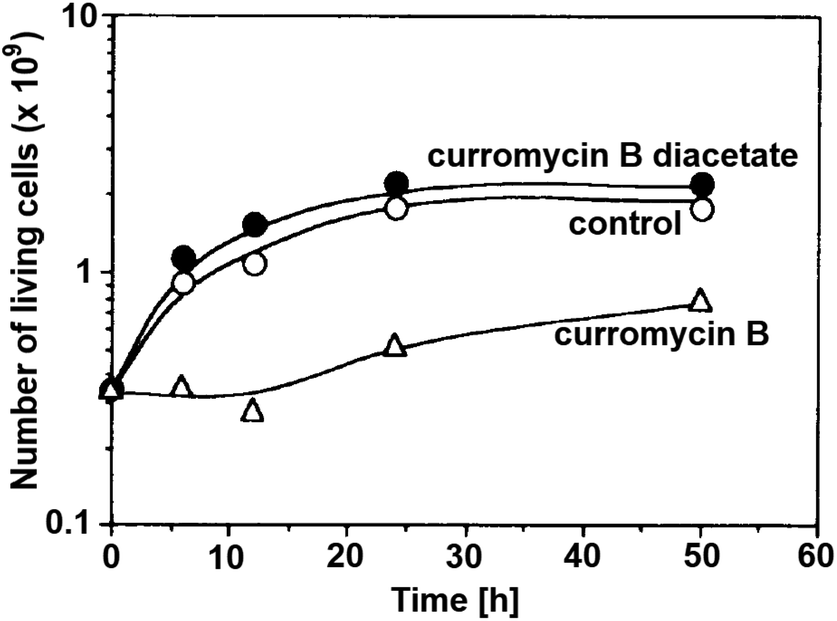 | ||
| Fig. 15 Effect of curromycin B (7) and curromycin B diacetate against Agrobacterium tumefaciens growth on an inoculated potato tuber disk. Bacterial cells in the absence (○) and the presence of curromycin B diacetate (●) or curromycin B (Δ) were counted 0, 6, 12, 24 and 48 h after inoculation.62 | ||
| Minimum inhibitory concentration (MIC) [μg ml−1] | Inhibition zoneb [mm] | |||||||
|---|---|---|---|---|---|---|---|---|
| Agrobacterium tumefaciens IFO 13263 | Bacillus subtilis IAM 1026 | Bacillus subtilis IFO 3007 | Escherichia coli OP50 | Micrococcus luteus FDA16 | Pseudomonas aeruginosa A3 | Pseudomonas cepacian M-0527 | Staphylococcus aureus FDA 209P | |
| a Values reported for 6 were described as “almost identical” with 7.b Using cylinder plate method with concentration 2.5 μl ml−1, reported as a diameter of inhibition zone. | ||||||||
| Curromycin A (6) | 6.3 | ∼3.9a | >100 | 100 | 25 | 50 | ∼50a | 20.0 |
| Curromycin B (7) | 6.3 | 3.9 | >100 | 100 | 25 | 50 | 50 | 21.5 |
Otani et al. examined KSM-2690B (8) and KSM-2690C (9) by using a paper disk diffusion method (concentration 50 μg ml−1, 8 mm in diameter).7 Interestingly, 8 showed antibacterial activity against Micrococcus luteus ATCC 9341 (15.0 mm, hazy zone), Bacillus subtilis PCI 219 (23.0 mm) and the highest activity against Staphylococcus aureus Smith (25.0 mm, hazy zone), whereas no growth inhibition of 9 was observed.7 Since 8 and 9 are geometric isomers, which differ only in their left-hand fragment, and thus there is apparent importance of left-hand fragment suitable orientation for increased antimicrobial activity.
Potts et al. revealed light-sensitivity of lajollamycin (10).8 For example, a DMSO solution of 10 (50 μM) degraded rapidly at room temperature when exposed to light, with only 25% of the parent compound remaining after 1 h. In contrast, 10 was stable in DMSO for at least 32 h, when protected from light. Thus, appropriate precautions were taken to protect the compound from light during bioassays. The results proved antimicrobial activity of 10 against both drug-sensitive and drug-resistant microorganisms. Remarkable bioactivity of 10 was observed against methicillin-sensitive (MIC 4 μg ml−1) and methicillin-resistant (MIC 5 μg ml−1) strains of Staphylococcus aureus. Higher activity was observed against penicillin-sensitive (MIC 2 μg ml−1) and penicillin-resistant (MIC 1.5 μg ml−1) Streptococcus pneumoniae strains. Lower bioactivity was observed against Enterococcus faecium for both vancomycin-sensitive (MIC 14 μg ml−1) and vancomycin-resistant (MIC 20 μg ml−1) strains. Lajollamycin (10) is also active against Escherichia coli IMP-type (MIC 12 μg ml−1). The antibacterial activity of 10 is summarized in Table 6.8
| Minimum inhibitory concentration (MIC) [μg ml−1] | |||||||
|---|---|---|---|---|---|---|---|
| Enterococcus faecalis vancomycin-resistant | Enterococcus faecalis vancomycin-sensitive | Staphylococcus aureus methicillin-resistant | Staphylococcus aureus methicillin-sensitive | Streptococcus pneumoniae penicillin-resistant | Streptococcus pneumoniae penicillin-sensitive | Escherichia coli IMP-type | |
| Lajollamycin (10) | 20 | 14 | 5 | 4 | 1.5 | 2 | 12 |
The antibacterial activity of lajollamycins 10, 11, 12 and 13 was evaluated by Oh et al. against various bacterial strains such as Bacillus subtilis ATCC 6633, Escherichia coli ATCC 35270, Kocuria rhizophila NBRC 12708, Proteus hauseri NBRC 3851, Salmonella enterica ATCC 14028 and Staphylococcus aureus ATCC 6538p, but no significant inhibitory activity (MIC > 128 μg ml−1) was observed.9
Comprehensive evaluation of oxazolomycin A (1), oxazolomycin A2 (14) and bisoxazolomycin (15) on bacterial strains was investigated by Koomsiri et al. using paper disk method.10 Oxazolomycins were applied on paper disc (diameter 6 mm) at the concentrations of 30, 10, 3 and 1 μg per disc (Table 7). Oxazolomycins were found inactive against Mycobacterium smegmatis ATCC 607 and Pseudomonas aeruginosa IFO 3080. The bioassays against Bacillus subtilis ATCC 6633 revealed that activity of 1 increases with higher concentration, but oxazolomycins 14 and 15 were completely inactive. Evaluation of Escherichia coli NIHJ showed bioactivity of 1 at all tested concentrations, whereas compound 14 displayed growth inhibition only at higher concentrations. Dimeric oxazolomycin 15 was inactive at all tested concentrations. Lower activity of 1, compared to activity against other tested strains was observed against Kocuria rhizophila ATCC 9341. No growth inhibition against this strain was observed for 14, however application of dimeric 15 resulted in some activity. The inhibition of Staphylococcus aureus ATCC 6538p with 1 was observed for all tested concentrations, whereas oxazolomycins 14 and 15 displayed activity only at higher concentrations (10 and 30 μg per disc). The highest antibacterial activity of 1 was obtained in bioassays against Xanthomonas campestris pv. oryzae KB 88. Interestingly, compounds 14 and 15 also displayed such activity, although apparently reduced.
| Concentration [μg per disc] | Diameter of inhibition zone [mm] | |||||||
|---|---|---|---|---|---|---|---|---|
| Bacillus subtilis ATCC 6633 | Escherichia coli NIHJ | Kocuria rhizophila ATCC 9341 | Mycobacterium smegmatis ATCC 607 | Pseudomonas aeruginosa IFO 3080 | Staphylococcus aureus ATCC 6538p | Xanthomonas campestris pv. oryzae KB 88 | ||
| Oxazolomycin A (1) | 30 | 31 | 28 | 16 | Inactive | Inactive | 26 | 45 |
| 10 | 30 | 27 | 15 | Inactive | Inactive | 24 | 42 | |
| 3 | 25 | 25 | 10 | Inactive | Inactive | 24 | 13 | |
| 1 | 18 | 20 | Inactive | Inactive | Inactive | 20 | 27 | |
| Oxazolomycin A2 (14) | 30 | Inactive | 15 | Inactive | Inactive | Inactive | 24 | 16 |
| 10 | Inactive | Inactive | Inactive | Inactive | Inactive | 17 | 12 | |
| 3 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | 10 | |
| 1 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | |
| Bisoxazolomycin (15) | 30 | Inactive | Inactive | 11 | Inactive | Inactive | 17 | 14 |
| 10 | Inactive | Inactive | 8 | Inactive | Inactive | 13 | 9 | |
| 3 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | 9 | |
| 1 | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | |
In general, the widest antibacterial activity was observed for oxazolomycin A (1). Oxazolomycins 14 and 15 displayed activity only at higher concentrations. The comparison of antibacterial activities at higher tested concentration (30 μg per disc) is demonstrated in Fig. 16. Oxazolomycin 15 is the only dimeric member of oxazolomycin family and as such its different antibacterial activity (lower in this case) from 1 or 14 can be presumed. However, compounds 1 and 14 are structurally related oxazolomycins. The right-hand fragment of 1 (spirocyclic β-lactone-γ-lactam) and 14 (monocyclic γ-lactam) represents the only structural difference. From this point of view, β-lactone ring in structure of oxazolomycin A (1) seems to play an important role for the increase of antibacterial activity.10
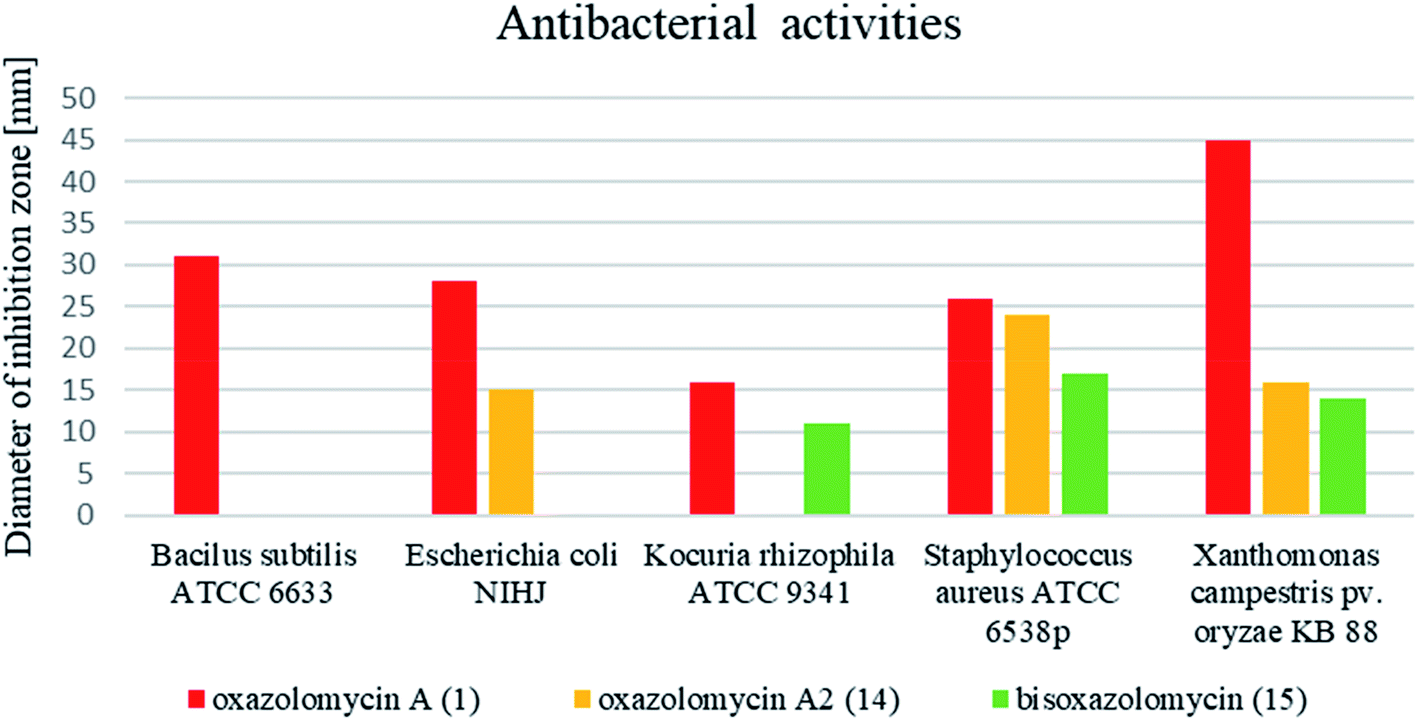 | ||
| Fig. 16 Comparison of antibacterial activity of oxazolomycins 1, 14 and 15 using disc diffusion method at the concentration 30 μg per disc. | ||
Curromycins were investigated by Ohno et al. against replication of human immunodeficiency virus (HIV) in both acute and chronic infection.64 Reverse transcriptase assay65 was performed to estimate the concentration of viral particles in the culture supernatant. MTT assay,66 based on the mitochondrial reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, was utilized to determine the number of living cells. The 50% effective concentrations (EC50) of 6 and 7 for HIV-1 IIIB replication (reverse transcriptase assay on primary infected human lymphoid cells) were determined as 2.5 μg ml−1 and 5 μg ml−1. Azidothymidine (AZT) treatment also showed concentration dependent inhibition in this assay system (Fig. 17). To exclude the possibility that curromycins directly inhibit reverse transcriptase in the reverse transcriptase assay system, p24 antigen was monitored by capture ELISA, to study the production of HIV antigens with treatment of curromycins. The OD values, which represent the antigen expression exhibited the same tendency with reverse transcriptase values (data not reported).64 Therefore, these agents did not have effect on reverse transcriptase directly. The 50% inhibitory concentration (IC50) values of MTT on primary infected human lymphoid cells were at 6.5 μg ml−1 for 6 and 20 μg ml−1 for 7. Furthermore, the antiviral activity of curromycins was examined in chronically infected U937 cells67 and it was proved that curromycins have anti-HIV activity in chronically infected cells. In this case the ratio of IC50 and EC50 was greater than 10. The results suggest that curromycins are more effective in chronically infected monocytoid cells than in primarily infected lymphoid cells. Lower effect was observed with AZT in chronic assay system (Fig. 18). The chronic assay results further indicated that curromycins affect the later steps in the virus replication cycle.64
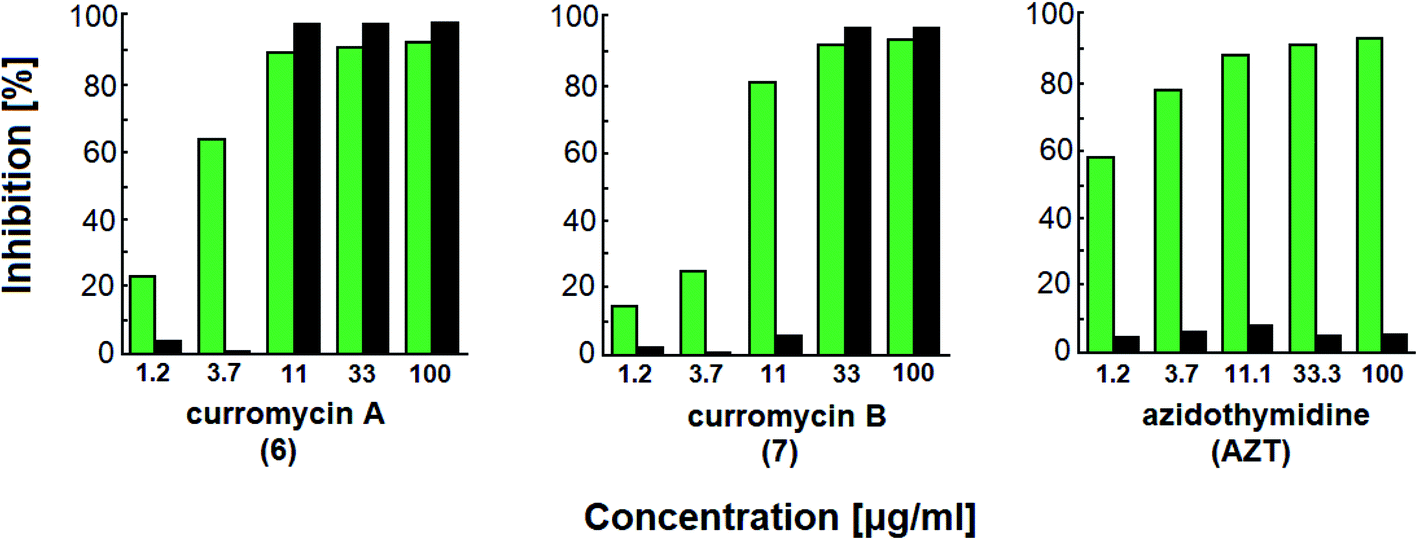 | ||
| Fig. 17 Effect of curromycin A (6), B (7) and azidothymidine (AZT) on primary infected human lymphoid cells. Green columns: % inhibition of reverse transcriptase; black columns: % inhibition of MTT.67 | ||
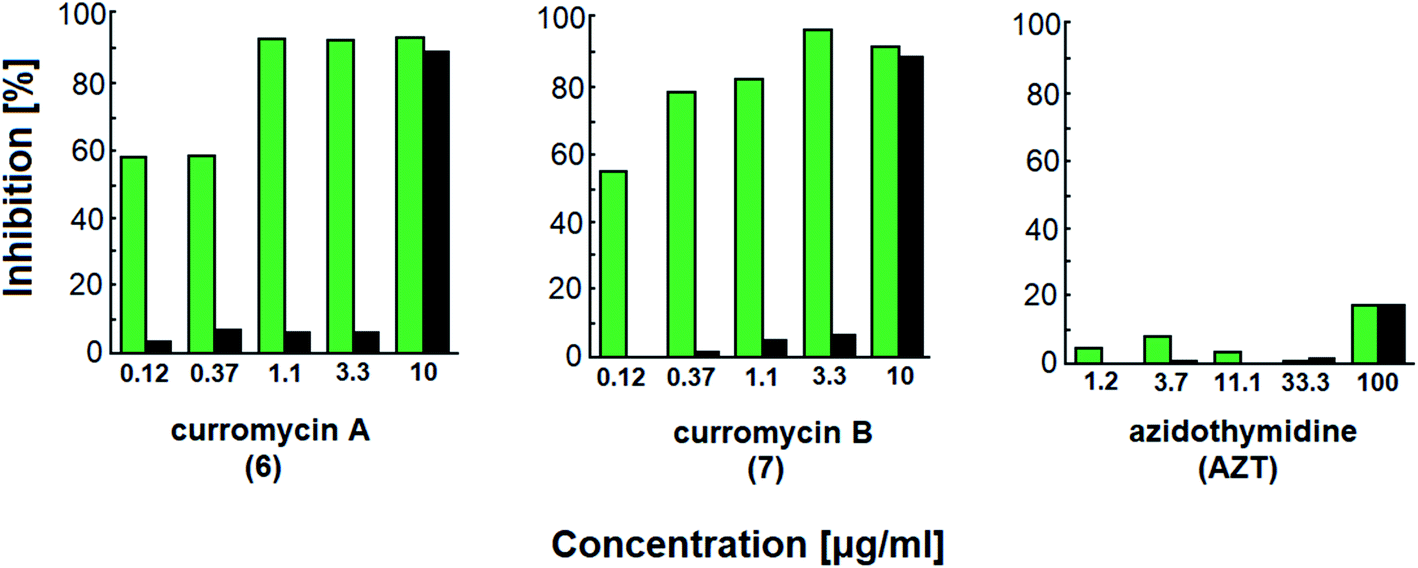 | ||
| Fig. 18 Effect of curromycin A (6), B (7) and azidothymidine (AZT) in chronic replication assay. Green columns: % inhibition of reverse transcriptase; black columns: % inhibition of MTT.67 | ||
Structural determination of curromycin A (6) and B (7) reported by Ogura et al. involved some biological assays.3,61 Curromycin 6 showed cytotoxic activity against Fried leukemia cells and against mouse melanoma B16. Significant bioactivity against mouse leukemia P388 (IC50 0.06 μg ml−1) was observed.3 Moreover, Hayakawa et al. evaluated the cytotoxic activity of 6 against human gastric carcinoma MKN45 in nutrient-deprived and normal medium using the MTT method.68 The bioassay in nutrient-deprived medium (Earle's salt solution supplemented with 10% fetal bovine serum) showed potency of 6 against MKN45 (IC50 15 ng ml−1). The growth of MKN45 cells in a normal medium (Dulbecco's modified Eagle's medium with 10% fetal bovine serum) was inhibited by 6 at the range of IC50 20-20000 ng ml−1, although no cell death was observed.68 Curromycin 7 showed cytotoxic activity against mouse melanoma B16 (IC50 2.5 μg ml−1). Further, cytotoxicity against mouse leukemia P388 for 7 (IC50 0.12 μg ml−1) was lower than reported for 6, but still significantly different from a control.3
Kim et al. reported cytotoxic activity of 16-methyloxazolomycin (5) against human lung adenocarcinoma A549 cells (IC50 4.6 μg ml−1).5 Mouse leukemia P388 was inhibited by 5 (IC50 0.23 μg ml−1). Despite lower bioactivity of 5 against P388 compared to curromycins, it seems that oxazolomycins express significant activity against selected leukemia cell line (Fig. 19).
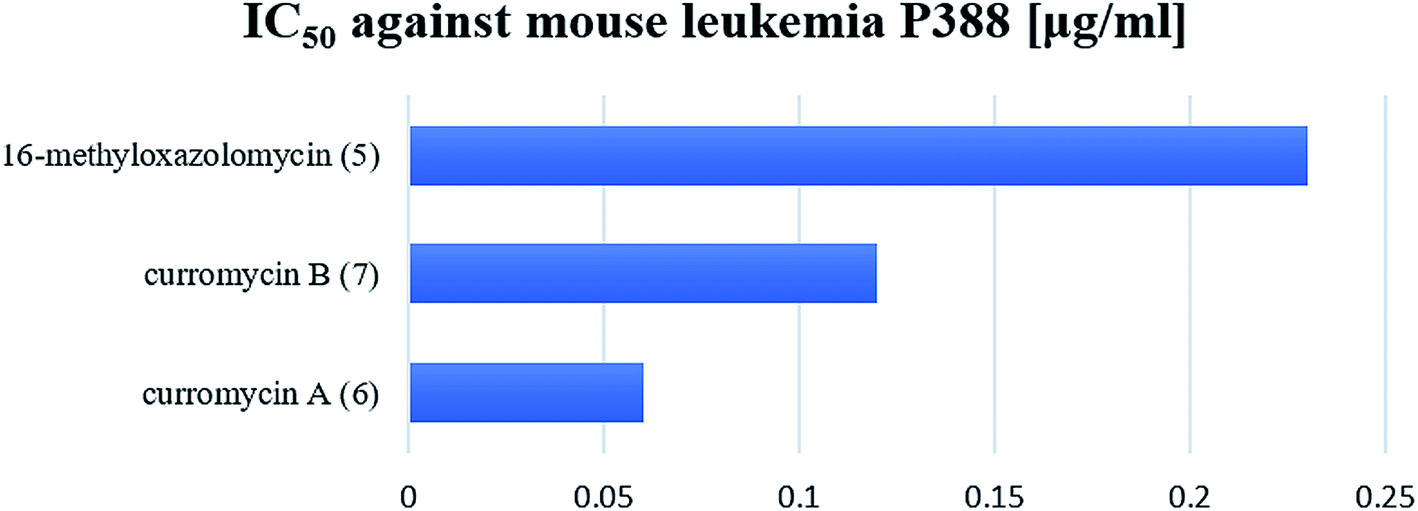 | ||
| Fig. 19 Cytotoxic activity of curromycin A (6), B (7) and 16-methyloxazolomycin (5) against mouse leukemia P388. | ||
Cytotoxic activity of KSM-2690 B (8) and C (9) was evaluated by Otani et al. against human bladder carcinoma T24 cells.7 Both oxazolomycins 8 and 9 were active and exhibited the same cytotoxic effects (IC50 10 μg ml−1). The cytotoxicity of lajollamycin (10) was demonstrated on mouse melanoma cell line B16–F10 growth (EC50 9.6 μM) reported by Potts et al.,8 whereas further study reported by Oh et al.9 showed that lajollamycins (10–13) did not exhibit cytotoxic activity against human breast carcinoma MDA-MB-231, colorectal carcinoma HCT 116, gastric carcinoma SNU-638, hepatic adenocarcinoma SK-HEP-1, leukemia K562 nor lung adenocarcinoma A549 cell lines (IC50 > 100 μM).
Koomsiri et al. compared the cytotoxicity of oxazolomycin A (1), A2 (14) and bisoxazolomycin (15).10 The highest cell growth inhibition activity against human leukemia HL60 cell line was observed for 1 (IC50 0.6 μM), followed by dimeric structure 15 (IC50 7 μM) and monocyclic oxazolomycin 14 (IC50 20 μM; Fig. 20). The results suggest spirocyclic β-lactone-γ-lactam core significance for cytotoxic activity, similarly to antibacterial activity discussed above, but further comprehensive experiments are needed to confirm this hypothesis.
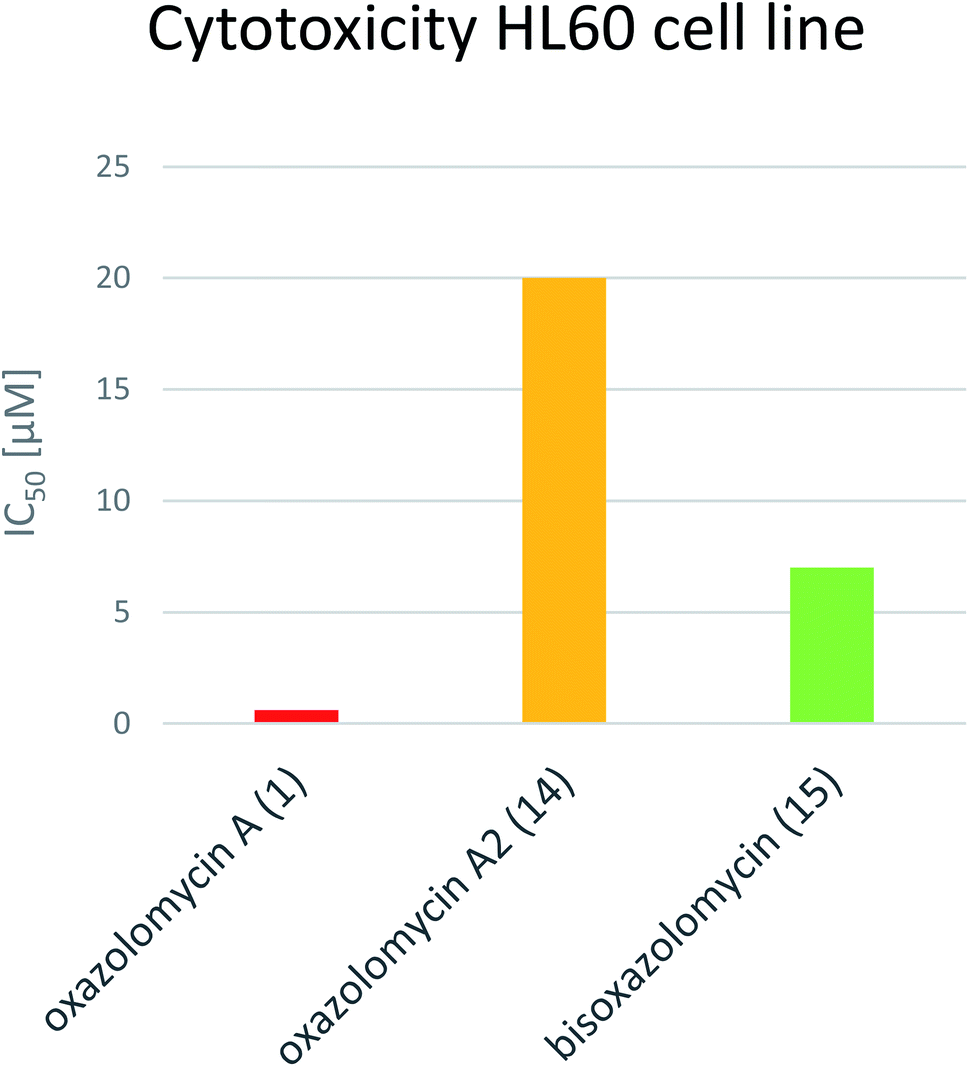 | ||
| Fig. 20 Comparison of IC50 values for oxazolomycin A (1), A2 (14) and bisoxazolomycin (15) against human leukemia HL60. | ||
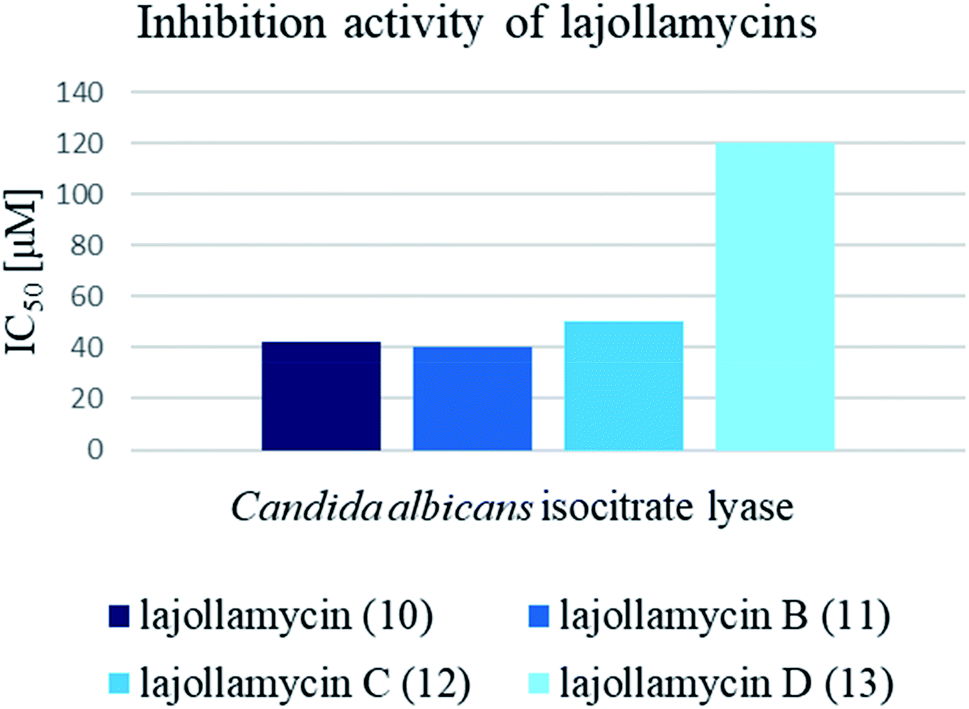 | ||
| Fig. 21 Inhibition activity Candida albicans isocitrate lyase by lajollamycins 10–13. | ||
Hayakawa et al. investigated68 the effect of curromycin A (6) on GRP78 gene expression by the luciferase reporter assay using human fibrosarcoma HT1080 cells transformed with the luciferase gene under the control of the GRP78 promoter (HT1080 G-L).70 It was reported that 6 dose-dependently inhibited the luciferase expression (IC50 4.3 ng ml−1) in the presence of 10 mM of 2-deoxyglucose (Fig. 22).68
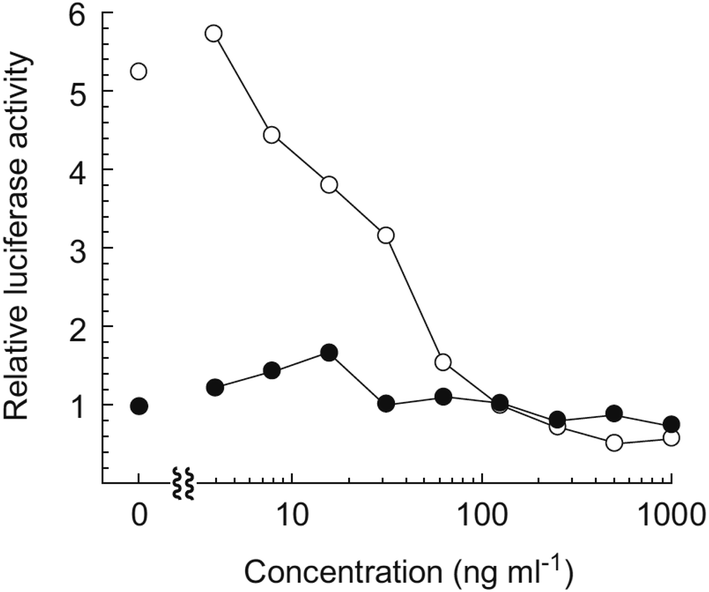 | ||
| Fig. 22 Effect of curromycin A (6) on the luciferase expression in HT1080 G-L cells. HT1080 G-L cells were treated with 6 in the presence (○) or absence (●) of 2-deoxyglucose (10 mM) for 18 h at 37 °C. The relative luciferase activity compared with non-treated control was measured with a luminometer. | ||
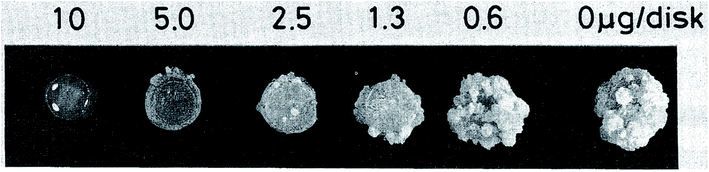 | ||
| Fig. 23 Effect on crown gall formation of oxazolomycin A (1) administered simultaneously with the inoculation of Agrobacterium tumefaciens.60 | ||
Kawai et al. further examined effect on crown gall formation of oxazolomycin A (1) administrated a certain period after the inoculation of Agrobacterium tumefaciens (Table 8).60 It was observed that administration of oxazolomycin A (1) 3 h after the inoculation inhibited crown gall formation at a dose of 5.0 μg per disc and the surface of the potato disc was necrosed at a dose of 10.0 μg per disc. Increase of the interval between the inoculation and the administration of the agent resulted in observation of small crown gall formation at a dose of 10.0 μg per disc, the potato disc being no longer necrosed and forming as many crown galls as the control. The necrosis was also observed for cycloheximide at 1.3 μg per disc, where no formation of crown gall was found. The administration of cycloheximide at any time after the inoculation caused no necrosis of the potato disc at a dose of 2.5 μg per disc. As a result of these data, Kawai et al. concluded, that cycloheximide was strongly phytotoxic to both the nontransformed and transformed plant cells. In contrast to cycloheximide, oxazolomycin 1 had toxicity against nontransformed plant cells as well as against A. tumefaciens, while it had no toxicity toward transformed plant cells whose genome integrated the T-DNA of A. tumefaciens. This indicates, that 1 might be used for distinguishing between the transformed and nontransformed plant cells.
| Dose [μg per disc] | Time interval between the inoculation and administration | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 h | 3 h | 6 h | 12 h | 24 h | 72 h | 120 h | 192 h | ||
| Oxazolomycin A (1) | 10.0 | 100% | 75% | 50% | 50% | 0% | 0% | 0% | 0% |
| 5.0 | 100% | 75% | 0% | 0% | 0% | 0% | 0% | 0% | |
| 2.5 | 75% | 75% | 0% | 0% | 0% | 0% | 0% | 0% | |
| Tetracycline | 10.0 | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 50% |
| 5.0 | 100% | 100% | 100% | 100% | 100% | 75% | 75% | 25% | |
| 2.5 | 75% | 50% | 50% | 100% | 50% | 0% | 0% | 0% | |
| Chloramphenicol | 10.0 | 100% | 100% | 100% | 100% | 100% | 100% | 75% | 100% |
| 5.0 | 100% | 100% | 75% | 100% | 75% | 50% | 25% | 0% | |
| 2.5 | 75% | 25% | 25% | 25% | 25% | 25% | 25% | 0% | |
The similar experiments were accomplished with the strong antibacterial agents against A. tumefaciens, tetracycline and chloramphenicol. These two antibiotics administrated 72 h after the inoculation still inhibited crown gall formation and they inhibited the multiplication of crown gall even when administered after 8 days (192 h; Table 8). These results suggested that both antibiotics were toxic toward not only A. tumefaciens, but also toward transformed cells. The results reported by Kawai et al. suggested that oxazolomycin A (1) inhibited the earliest step in crown gall formation, therefore 1 may be used as a chemical probe in studying the early stages of crown gall formation. Kawai et al. further examined, that neooxazolomycin (2) is inactive against crown gall formation, probably due to the lack of a β-lactone ring.60
Kawai et al. further provided application of oxazolomycin A (1) for a sterilization of crown gall in experiments on the transformation of plant cells by A. tumefaciens.60 In these experiments, transformed cells must be firstly purged from A. tumefaciens by culturing on Murashige–Skoog medium containing antibiotics (i.e. carbenicilin). Despite the ability of carbenicillin to completely remove the bacterium, a high concentration of the antibiotic is needed, because of its weak activity against A. tumefaciens. Kawai et al. proposed, that 1 as the selective antibiotic against A. tumefaciens and with no toxicity toward transformed plant cells could be useful for removing the bacterium. This possibility was examined by transferring crown gall to a solid Murashige–Skoog medium containing 1. It was observed, that 1 inhibited growth of any bacteria at a concentration of 100 μg ml−1, while commonly used carbenicillin at 1000 μg ml−1. A nearly equal rate of crown gall growth was obtained on the medium containing the respective concentrations of these compounds (Fig. 24). However, 1 at concentration 50 μg ml−1 had a bactericidal effect only on the growth of A. tumefaciens and the resulting infestation of other bacteria spoiled the crown galls. Therefore Kawai et al. concluded, that this antibiotic could be useful for sterilizing the transformed plant cells, but would be better used together with other antibiotics.60
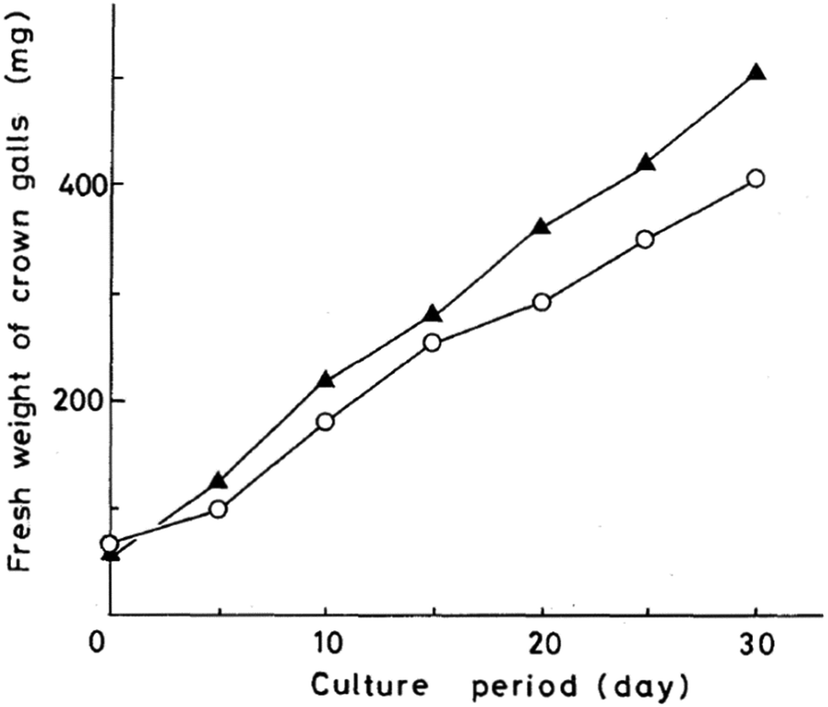 | ||
| Fig. 24 Growth curves of crown gall tissue on solid Murashige–Skoog medium. Oxazolomycin A (○) at 100 μg ml−1; carbenicillin (▲) at 1000 μg ml−1. | ||
Kanzaki et al. evaluated the inhibitory activity of oxazolomycin B (3) and C (4) against crown gall formation.6 It was observed that both geometric isomers 3 and 4 of 1 showed comparable bioactivity (MID 0.8 μg per disc). These results indicate that the oxazolomycin's geometry of left-hand fragment is not significant for the inhibition of crown gall formation.
The inhibitory effect of curromycin A (6) and B (7) against crown gall formation was investigated by Kanzaki et al.62 The both curromycins 6 and 7 showed strong, but little reduced activity (MID 3.2 μg per disc) against crown gall formation than 1. Kanzaki et al. suggested, that strong and selective activity of 6 and 7 against A. tumefaciens possibly resulted in an inhibition of crown gall formation (Fig. 25).
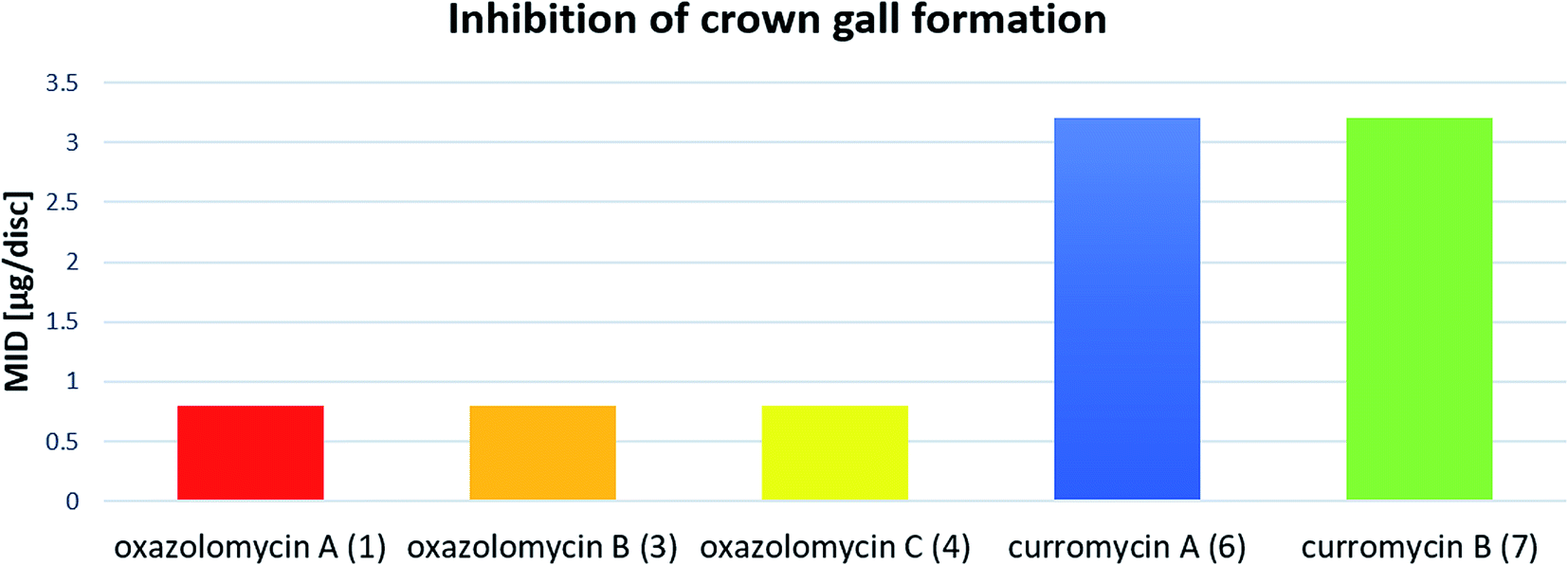 | ||
| Fig. 25 Effect of oxazolomycins and curomycins against crown gall formation.62 | ||
Kanzaki et al. further examined inhibition of crown gall formation by diesters derived from 6 and 7, acylated on hydroxyl groups at C7 and C3′.62 Curromycin diesters had similar (curromycin A diacetate; MID 3.2 μg per disc) or lower (curromycin B diacetate and curromycin B dipropionate; MID 6.3 μg per disc for both) bioactivity compared to natural curromycins. In contrast to curromycins, the curromycin esters had no inhibitory antibacterial activity against A. tumefaciens (Fig. 15). These results indicated that the inhibitory activity of the curromycin esters against crown gall formation was not related to growth inhibition of A. tumefaciens.62
Moreover, Kawazu et al. similarly examined the inhibition of crown gall formation by diesters derived from 1, acylated on hydroxyl groups at C7 and C3′.71 All evaluated esters: oxazolomycin A diacetate, oxazolomycin A dipropionate, oxazolomycin A dibutyrate including monoester oxazolomycin A monobutyrate (acylated at hydroxyl group at C7 position) showed half inhibitory activity (MID 1.6 μg per disc) if compared to natural oxazolomycin A (1). Similarly, oxazolomycin A esters had no antibacterial activity against A. tumefaciens, in contrast to 1.71 These findings suggest that the curromycin esters62 and oxazolomycin A esters71 specifically inhibited some steps of plant transformation (but not A. tumefaciens) and therefore could be promising chemical probes for studying the mechanism of plant transformation mediated by A. tumefaciens.62
Kawazu et al. want to investigated, if the oxazolomycin A ester administrated to potato tuber disc was converted to oxazolomycin A (1) or other compounds.71 The decay of the oxazolomycin A dipropionate administered to potato tuber disc inoculated with A. tumefaciens was examined and compared with 1. Both compounds were rapidly decomposed, decreasing to about a half content after 6 h and to one tenth content after 24 h in the potato disc, irrespective of the presence or the absence of A. tumefaciens. However, these compounds decayed more slowly to about one fourth content after 24 h in the culture of A. tumefaciens (Fig. 26). Under examined conditions, oxazolomycin A dipropionate was not converted to 1 or vice versa. The results suggested that the unique activity of the esters was not caused due to biological deesterification.71
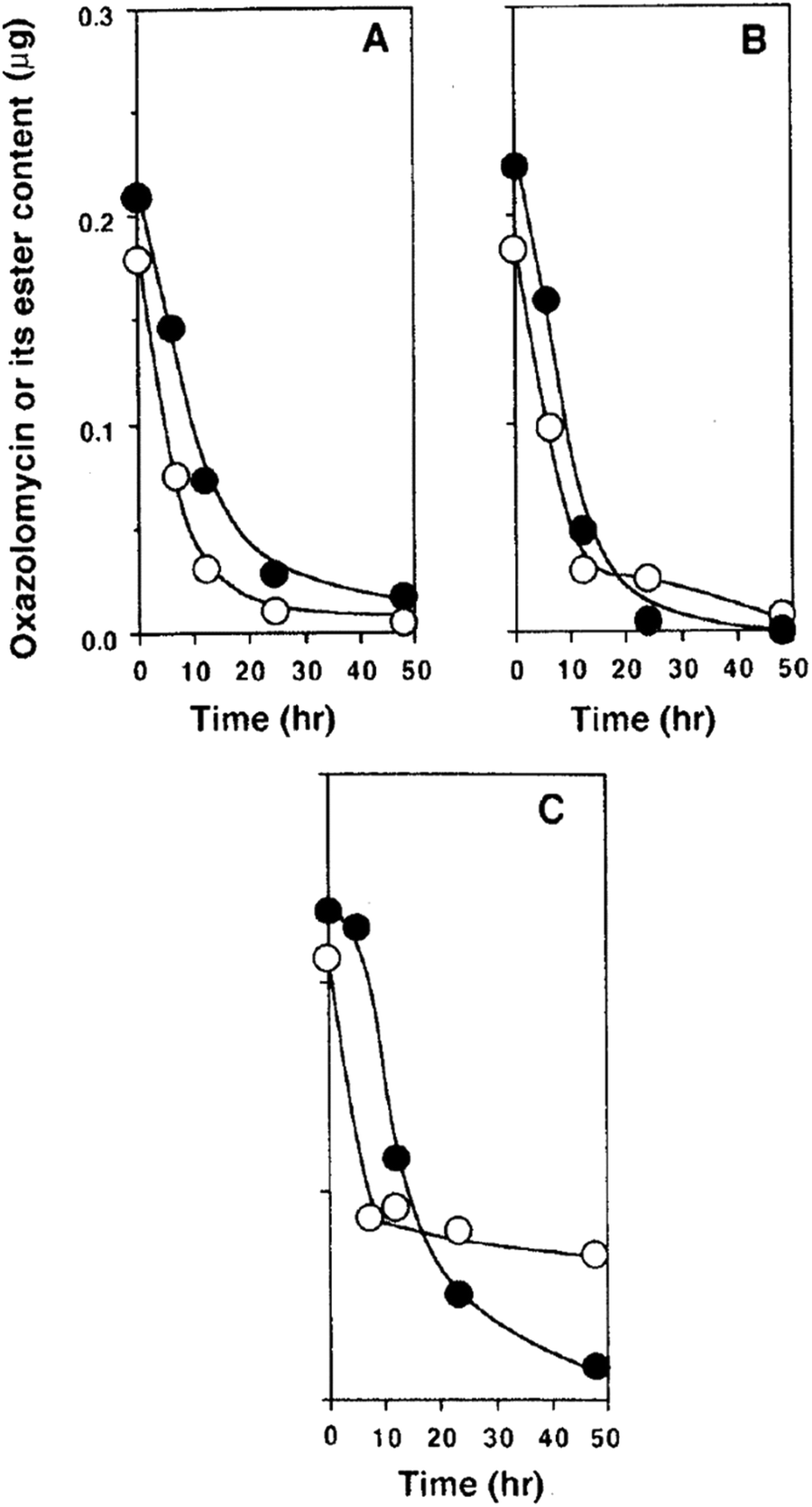 | ||
| Fig. 26 Decay of oxazolomycin A (●) and oxazolomycin A dipropionate (○) in the process of crown gall formation. The compounds were administered to potato tuber discs inoculated with A. tumefaciens (A), to potato tuber discs (B) or to a culture of A. tumefaciens (C).71 | ||
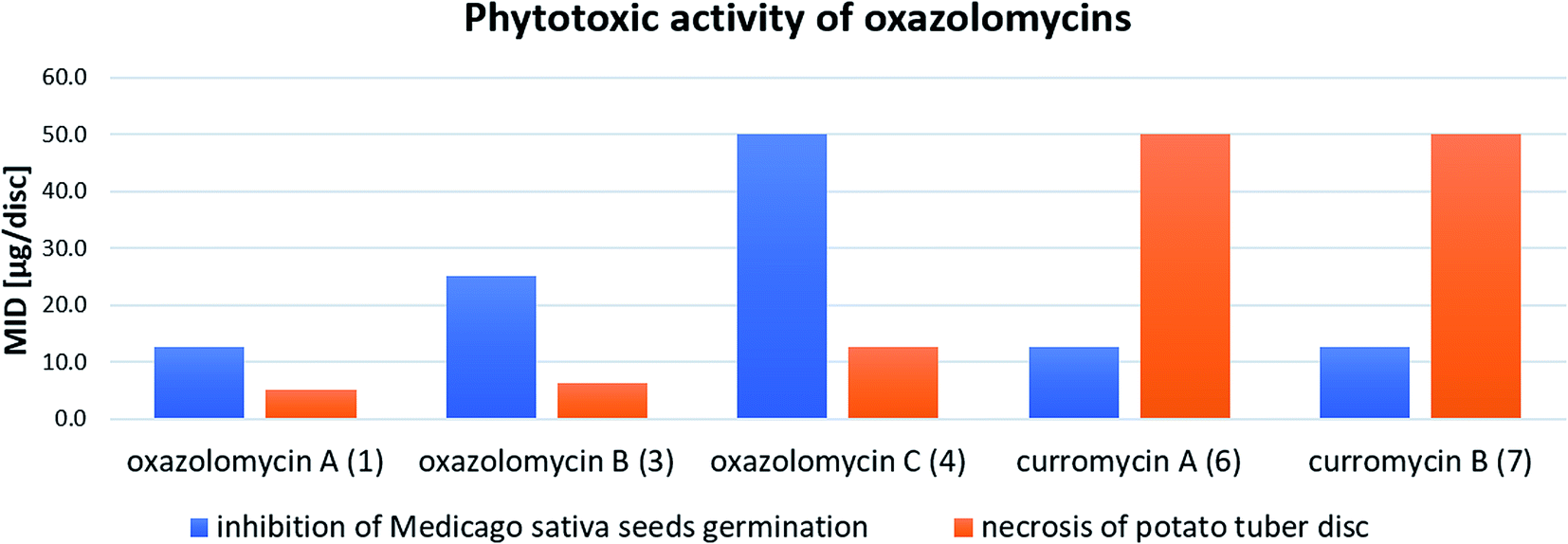 | ||
| Fig. 27 Summary of reported phytotoxic activity of oxazolomycins. | ||
Kawazu et al. evaluated phytotoxic activity of following oxazolomycin A diesters (acylated on hydroxyl groups at C7 and C3′): oxazolomycin A diacetate, oxazolomycin A dipropionate, oxazolomycin A dibutyrate and mono ester oxazolomycin A monobutyrate (acylated on hydroxyl group at C7) against seeds germination of Medicago sativa, but no activity (MID > 100 μg per disc) was observed.71 Any of oxazolomycin A esters caused necrosis on potato tuber disc even at dose of 50 μg per disc. Following curromycin diesters (acylated on hydroxyl groups at C7 and C3′): curromycin A diacetate, curromycin B diacetate and curromycin B dipropionate, were investigated by Kanzaki et al. toward phytotoxic activity, but no necrosis of potato tuber disc nor activity against seeds germination of Medicago sativa were observed.62
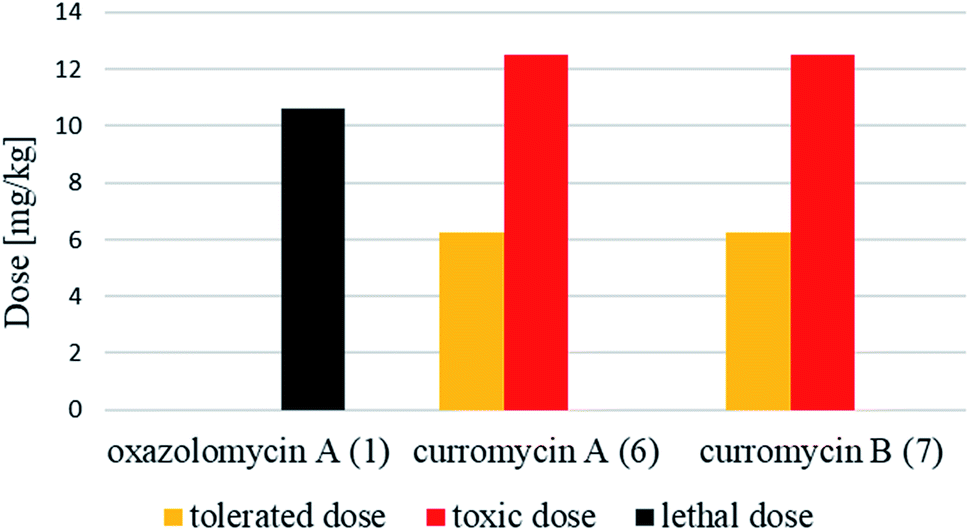 | ||
| Fig. 28 Tolerated, toxic and lethal doses of oxazolomycin A (1) and curromycins A (6) and B (7). | ||
4 Total syntheses of oxazolomycins
4.1 Total synthesis of neooxazolomycin by Kende
:1 ratio. The mixture of carboxylic acids 58 and 59 was hydrolyzed with LiOH, to provide acid 59 in 85% yield. Homochiral Evans oxazolidinone was recovered in 69% yield. Transformation of carboxylic acid 59 to enantiomerically pure (ee > 99%) methyl ester 60 was achieved with CH2N2 in Et2O. Enantiomeric purity of ester 60 was determined by use of chiral shift reagent Eu(hfc)3 employing racemic mixture 60 as standard. After protection of secondary hydroxy group in ester 60 with TBSOTf and 2,6-lutidine, corresponding silyl ether 61 underwent iodination reaction in presence of n-BuLi and I2 in THF to iodide 62. Diimide reduction77 of iodoacetylene 62 gave (Z,Z)-diene iodide 63 in 72% yield after 3 steps from 60 (Scheme 3).
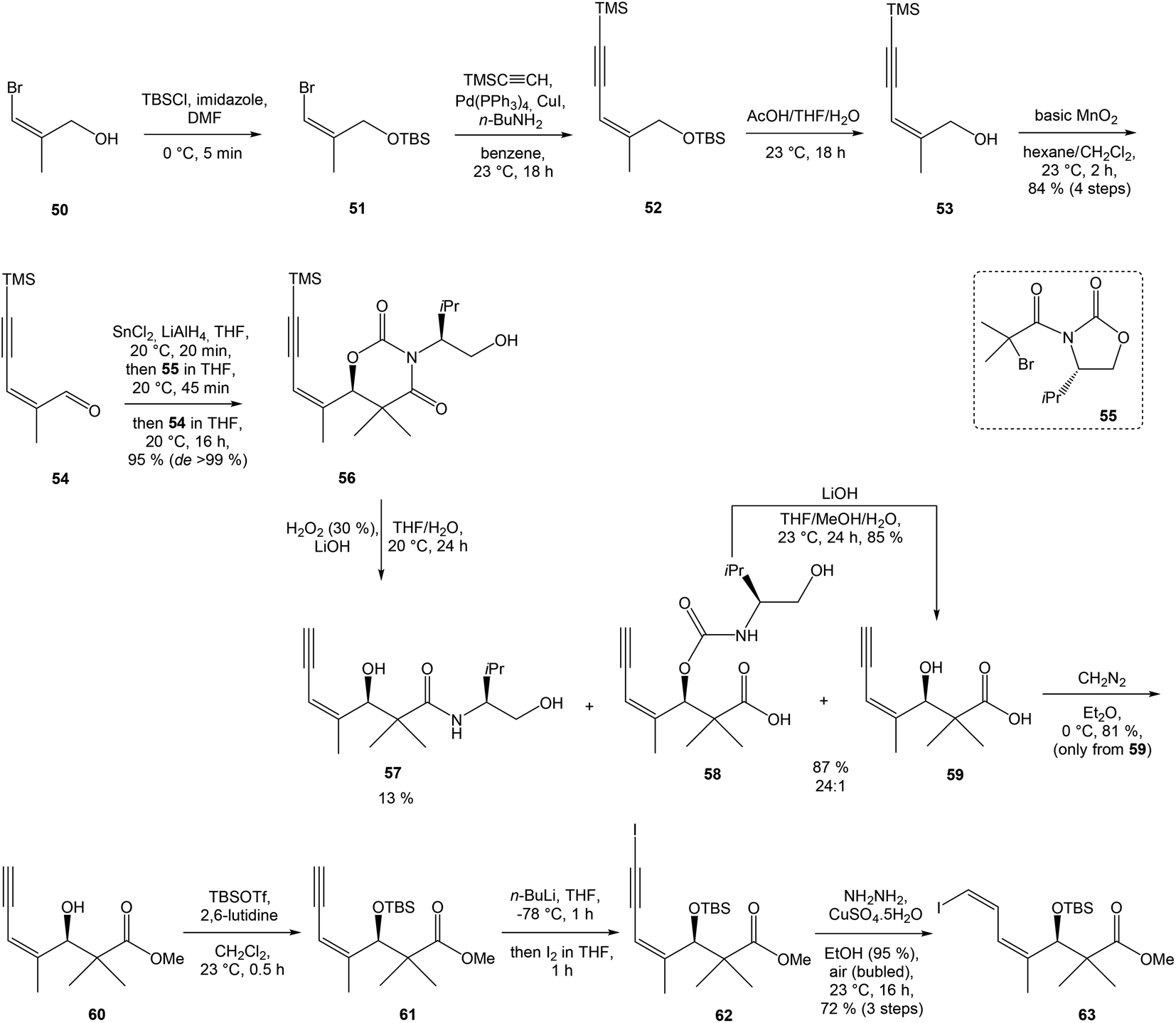 | ||
| Scheme 3 Synthesis of Stille cross coupling partner 63 for left-hand fragment of neooxazolomycin by Kende. | ||
To introduce the oxazole ring into the structure, ethyl diethoxyacetate and lithiated methyl isocyanide (LiCH2NC) under the conditions of Schöllkopf condensation78 provided 5-substituted oxazole acetal 64 in 86% yield. Acidic hydrolysis of 64 with HCl, followed by reduction with NaBH4 led to the oxazole methanol 65 in 68% yield. Alcohol was transformed in the Appel reaction by use of NBS and Ph3P to the unstable bromide 66, which was directly conjugated with the organocuprate 6779 to give the (E)-configurated vinyl stannane 68 in 49% yield. The Stille cross coupling conditions80 applied on the mixture of (E)-stannane 68 and (Z,Z)-diene iodide 63 provided desired (Z,Z,E)-configurated triene ester 69 with complete retention of all alkene geometries in 79% yield. Desilylation of silyl ether group with 50% HF/CH3CN gave secondary alcohol 70 in 92% yield, without any isomerization of the conjugated triene system. Hydrolysis of ester 70 under basic conditions with LiOH in the mixture THF/MeOH/H2O led to the β-hydroxy acid 71 in 94% yield. This hydrolysis required hydrogen bonding from free β-hydroxyl group, while TBS-protected ester 69 could not be hydrolyzed. Acetylation of hydroxyl group in β-hydroxy acid 71 was carried out using Ac2O in pyridine to give corresponding protected acid 72 in excellent 99% yield. Acid 72 represents the left-hand fragment of the target neooxazolomycin structure (Scheme 4).
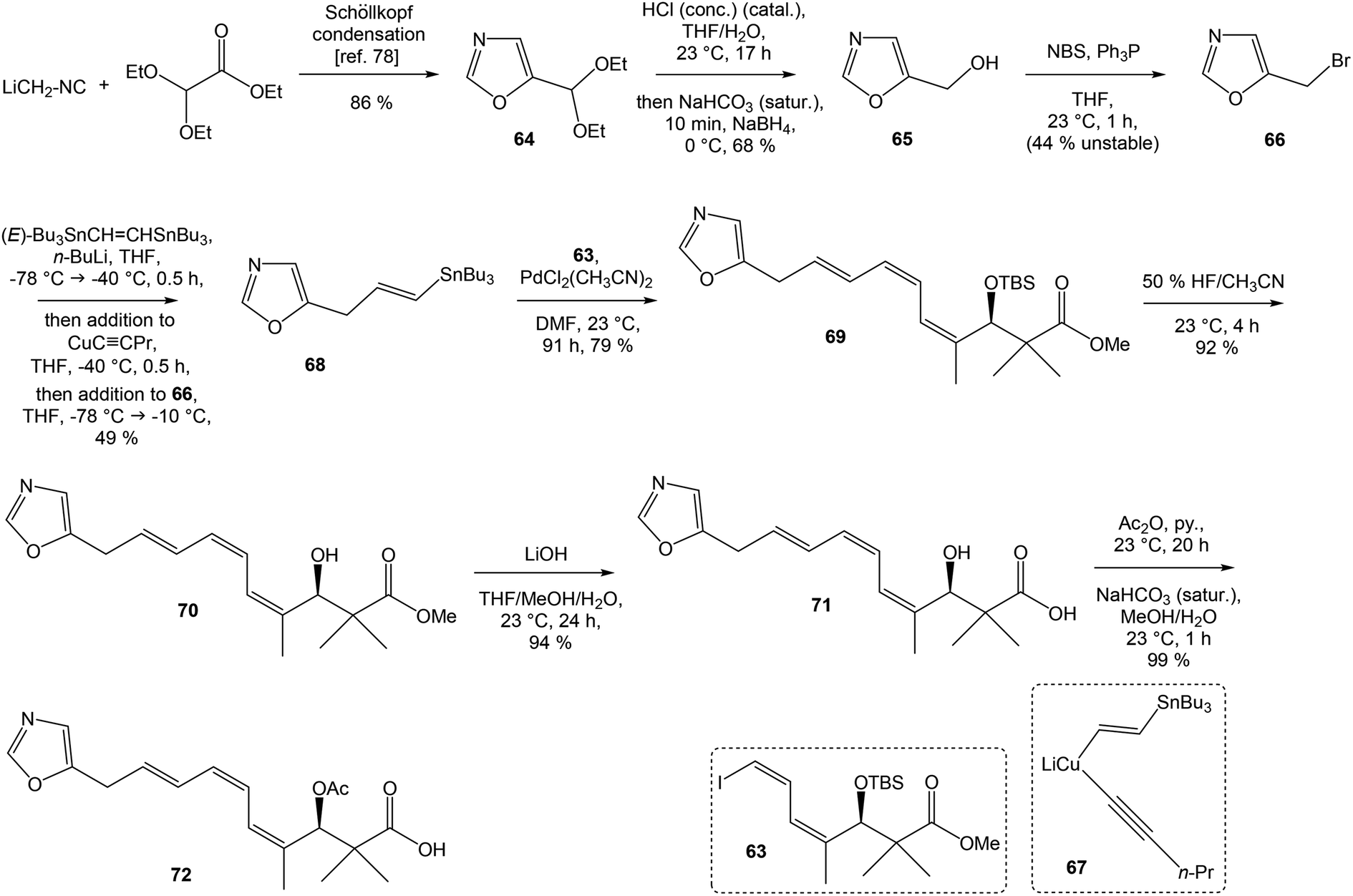 | ||
| Scheme 4 Synthesis of the left-hand fragment derivative 72 of neooxazolomycin by Kende. | ||
81 was used as the starting material, as well as the source of chirality on the route to synthesis of the right-hand fragment of neooxazolomycin. Methyl group introduction and contemporary trans-diaxial epoxide opening was carried out with MeLi and MeMgCl in THF/Et2O82 to produce diol 74 in 95% yield. No Payne rearrangement product was observed in this reaction. Glycoside 74 subsequently underwent four reaction steps including: (I) selective equatorial hydroxyl group silylation with i-Pr3SiOTf and 2,6-lutidine, (II) transformation of the axial hydroxyl group to the imidazole thiocarbamate with S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Im)2 followed by radical deoxygenation,83 (III) desilylation of silyl ether with TBAF, (IV) formation of acetonide with acetone/FeCl3,84 to provide deoxy acetonide 75 in 64% overall yield in 4 steps.
C(Im)2 followed by radical deoxygenation,83 (III) desilylation of silyl ether with TBAF, (IV) formation of acetonide with acetone/FeCl3,84 to provide deoxy acetonide 75 in 64% overall yield in 4 steps.Further four reaction steps: (I) benzyl protecting group removal under conditions of Pd(OH)2-catalyzed reductive debenzylation, (II) oxidation of primary hydroxyl group under Swern conditions to corresponding aldehyde, (III) following oxidation with KMnO4 buffered by 5% NaH2PO4 in t-BuOH to carboxylic acid, (IV) diazomethane esterification led to methyl ester 78 in 70% yield over four steps from 75. Cyclocondensation85 of ester 78 and dianion of amidomalonate 79 with t-BuLi and TMEDA in THF at −78 °C gave a mixture of γ-lactames α-80 and β-80 in ratio 1:1.4 and 82% yield (cumulative yield based on recovered ester 78). Desired γ-lactam α-80 was separated and transformed to bicyclic lactam–lactone 81 with EtSH and catalytic amount of HCl in almost quantitative yield. Following O-silylation of secondary hydroxyl with TBSOTf and 2,6-lutidine, basic hydrolysis of methyl ester with LiOH and Fujisawa reduction86 provided diol 84. In order to confirm the structure, intermediate 84 was converted to the known triacetate, prepared by Uemura et al.2 from 92. The silylation of primary alcohol group with TBSOTf and 2,6-lutidine led to thioacetal 85 in 59% overall yield from 81. Modification of thioacetal group 85 to the aldehyde 86 was performed with HgCl2 and CaCO3 in CH3CN/H2O in excellent 99% yield. The reaction performed on the aldehyde 86 with CH3I and CrCl2 gave a 70% yield of desired (E)-vinyl iodide 87.87 Completely desilylation with TBAF in THF provided triol 88 in quantitative yield. Following coupling of the (E)-vinyl triol 88 under Stille conditions80 and the Fmoc-amino protected propenylstannane 89 prepared from propargylamine was made in following sequence: (I) FmocCl, pyridine, CH2Cl2 and (II) Bu3SnH/AIBN,88 and afforded (E,E)-dienylamide 90 in 84% yield. Double O-acetylation with Ac2O in pyridine produced diacetate 91 in 96% yield, which represents combined right-hand and middle fragment of the neooxazolomycin structure (Scheme 5).
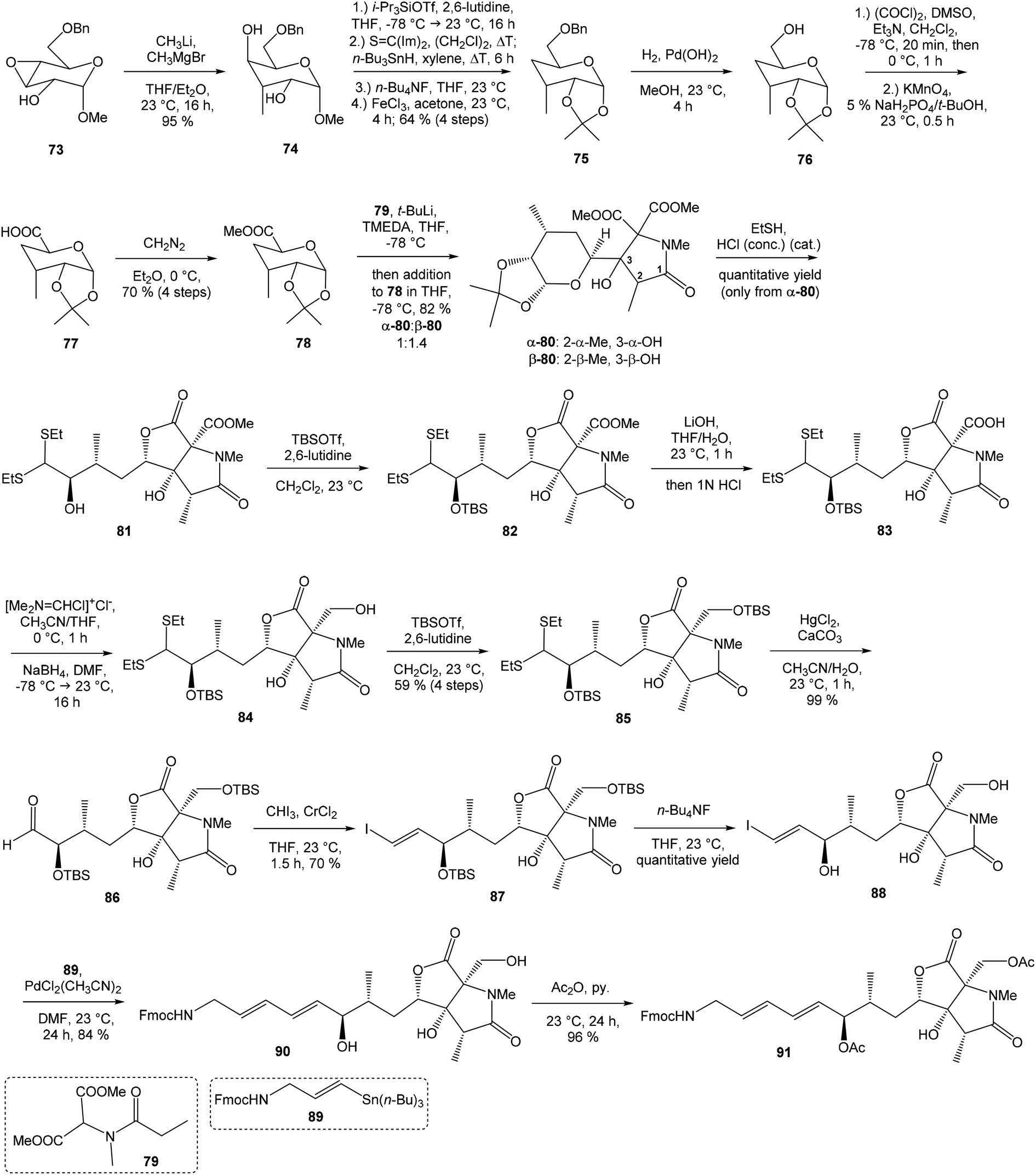 | ||
| Scheme 5 Synthesis of the right-hand and middle fragment derivative 91 of neooxazolomycin by Kende. | ||
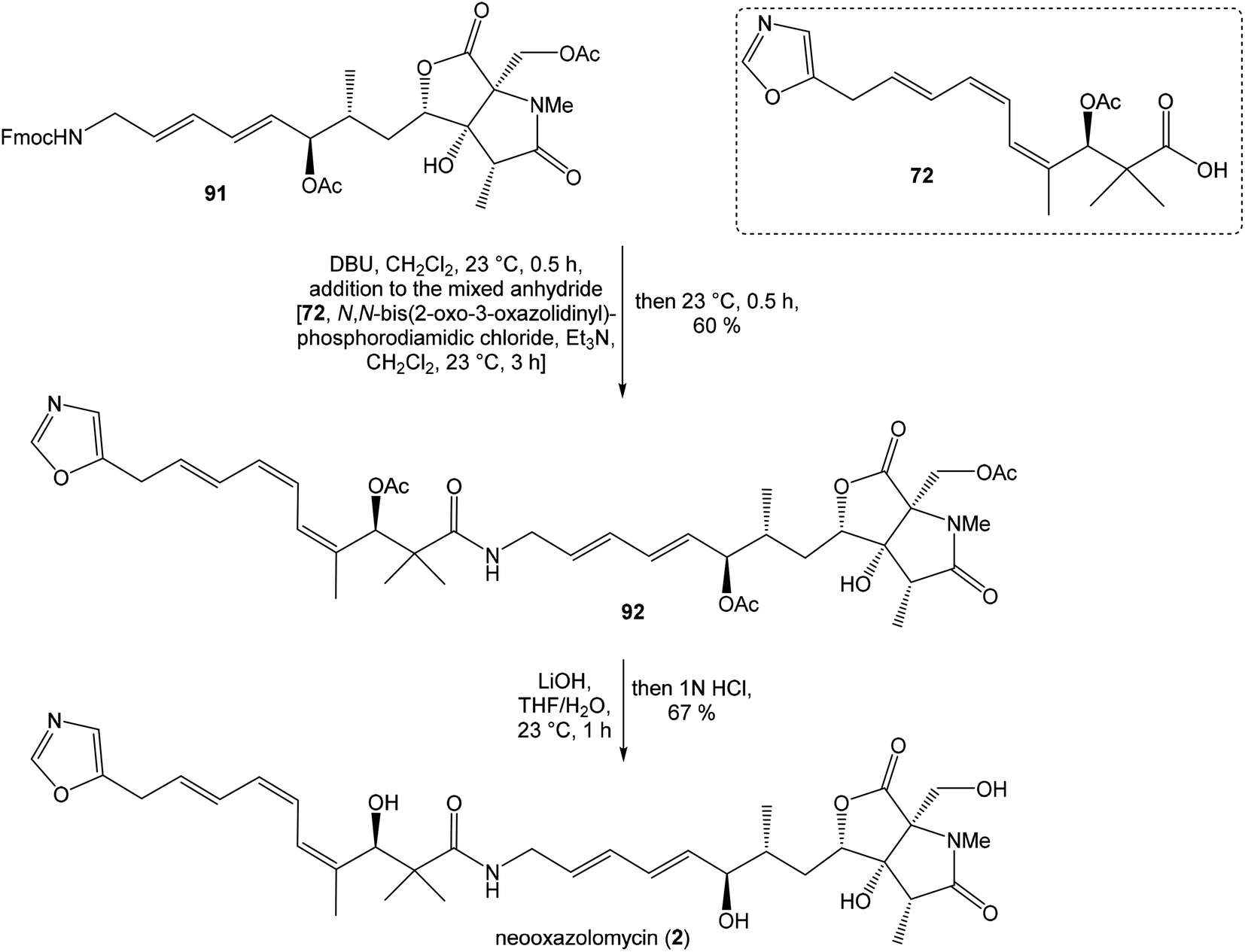 | ||
| Scheme 6 Finalization of the total synthesis of neooxazolomycin (2) by Kende. | ||
4.2 Total synthesis of neooxazolomycin by Hatakeyama
Kende's total synthesis of neooxazolomycin, reported in 1990,13 remained for many years the only total synthesis of oxazolomycins. However, Hatakeyama et al.16 reported in 2007 second total synthesis of the neooxazolomycin (2), in which the major challenge consisted in the stereoselective construction of the right-hand fragment.91,92 2,5-disubstituted oxazole 95 in 94% yield was isolated. Following desulfurization with RANEY® Ni provided 5-substituted oxazole 96 in 92% yield. Desilylation performed with AgOTf gave 73% yield of alkyne 97. The hydrostannylation under Bu3SnH/AIBN conditions led to stannane 98 in 88% yield, as a mixture of (E/Z)-isomers in ratio 6:1. The Stille cross coupling of stannane 98 and (Z,Z)-diene iodide 63 prepared by Kende procedure,13 afforded an inseparable mixture of (E/Z)-isomers of triene 99 in 79% yield. After desilylation with 47% HF/CH3CN, followed by recrystallization, a geometrically pure (Z,Z,E)-configurated triene ester 70 was obtained. Basic hydrolysis of ester 70 and acetylation, previously reported by Kende13 afforded protected acid 72 with 80% yield in 3 steps from 99 (Scheme 7).
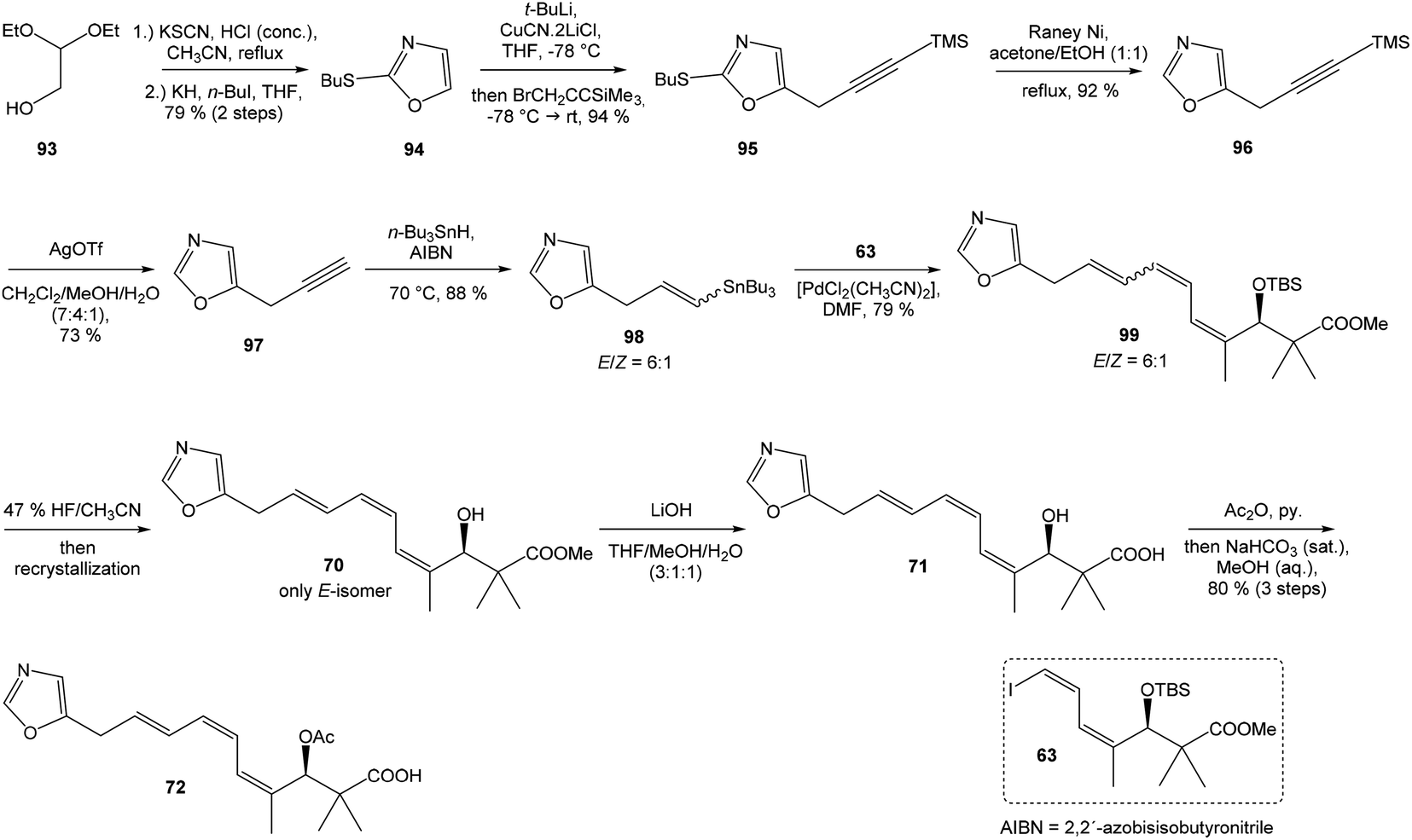 | ||
| Scheme 7 Synthesis of the left-hand fragment derivative 72 of neooxazolomycin by Hatakeyama. | ||
93 with triflate 102,94,95 both prepared from (S)-hydroxy-2-methylpropanoate 100. Alkynol 103 was prepared via desilylation with TBAF in THF in 81% yield after 2 steps. Protection of the primary hydroxyl group with tetramethyldisilazane to corresponding hydrodimethylsilyl ether 104 and following hydrosilylation catalyzed with [Pt(dvds)]96 in THF provided siloxane 105. Exposure of the siloxane 105 to the mixture of I2 and CsF in DMF/MeOH (5:1) gave (E)-iodoalkenol 106 with perfect stereoselectivity in 82% yield after 3 steps. Such combination of solvents and additives in the iodination step was found to be crucial for the selectivity within the preparation of desired (E)-configured iodoalkenol 106 (Scheme 8).
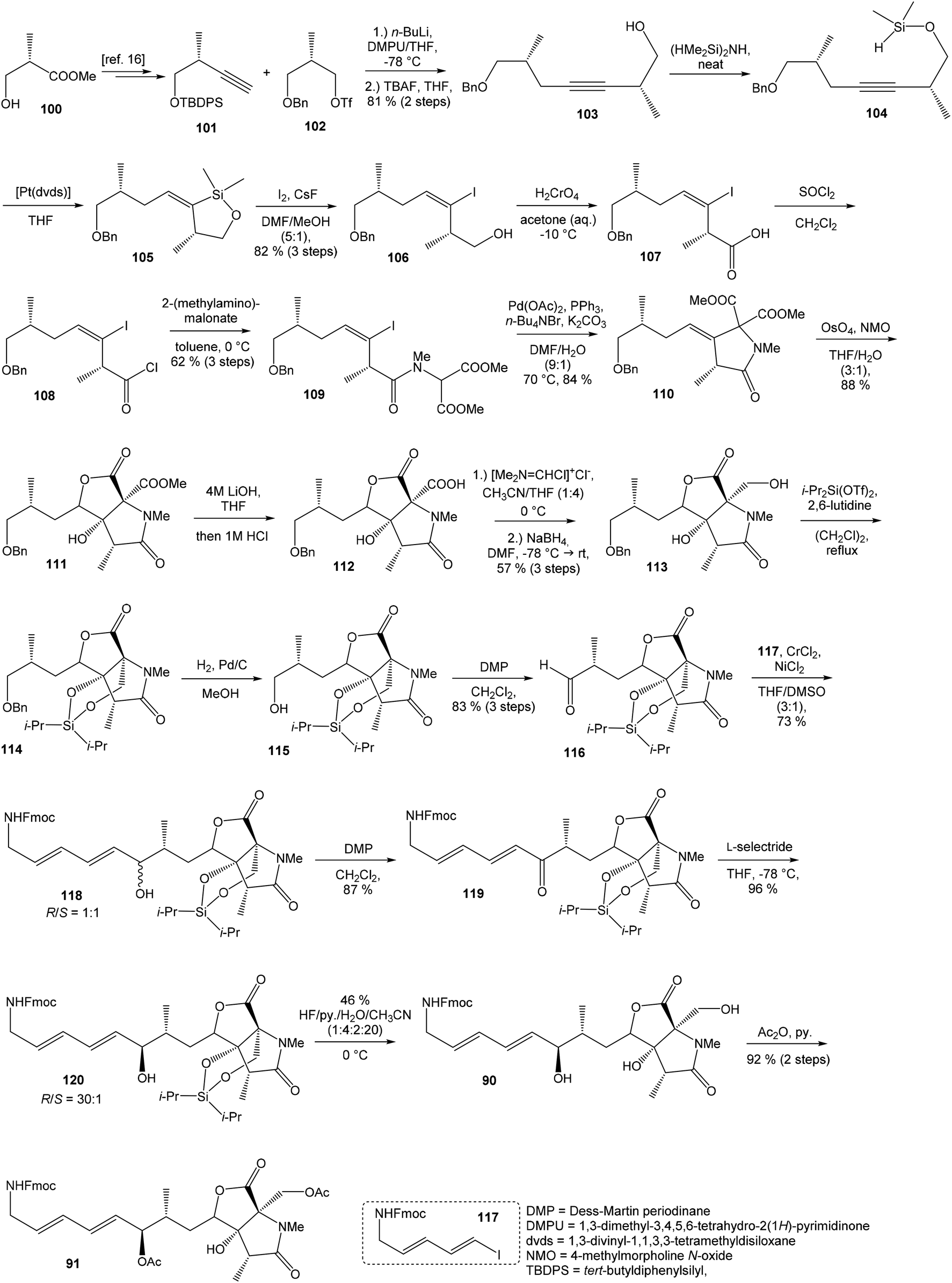 | ||
| Scheme 8 Synthesis of the right-hand and middle fragment derivative 91 of neooxazolomycin by Hatakeyama. | ||
Oxidation of primary hydroxyl group to the carboxylic acid 107 was carried out under Jones oxidation conditions. Carboxylic acid was transformed using SOCl2 in CH2Cl2 to the acyl chloride 108 and following reaction with dimethyl 2-(methylamino)malonate97 provided amide 109 in 62% yield in 3 steps. Amide 109 was converted to pyrrolidinone 110 by treatment with Pd(OAc)2/Ph3P in the presence of K2CO3 and TBABr in DMF/H2O at 70 °C in 84% yield.98 Double bond dihydroxylation under OsO4/NMO conditions and concomitant lactonization yield to lactone 111 as the sole product in 88% (Scheme 8). Formation of other diastereomer was not observed. The high stereoselectivity can be explained by the preferred conformer of pyrrolidinone 110, where the method using OsO4 is restricted to the α-face. This hypothesis was supported by NOE experiments and molecular mechanics calculations.16
Hydrolysis of 111 was promoted with 4 M LiOH leading to carboxylic acid 112 and following Fujisawa reduction86 give the diol 113 in 57% yield after 3 steps. To confirm the configuration of diol 113, X-ray analysis of the corresponding mono-tert-butyldimethylsilyl ether was accomplished.16 Protection of diol 113 to the dioxasilinane 114, following reductive debenzylation with H2 catalyzed with Pd/C in MeOH and Dess–Martin oxidation led to aldehyde 116 in 83% yield after 3 steps. Reaction between aldehyde 116 and diene iodide 117 under Nozaki–Hiyama–Kishi conditions99 provided product 118 in 73% yield as a mixture of diastereomers (R/S = 1:1).87 Although no diastereoselectivity was observed in Nozaki–Hiyama–Kishi reaction, high stereoselective formation (R/S = 30:1) of desired (R)-configurated secondary alcohol 120 was observed in two following reaction steps (oxidation with Dess–Martin periodinane, followed by reduction with L-selectride) in very good yield. Exposure of 120 to HF/pyridine mixture provided triol 90, which after acetylation with Ac2O in pyridine gave compound 91 in 92% yield after 2 steps, as a combined right-hand and middle fragment of neooxazolomycin (Scheme 8).16
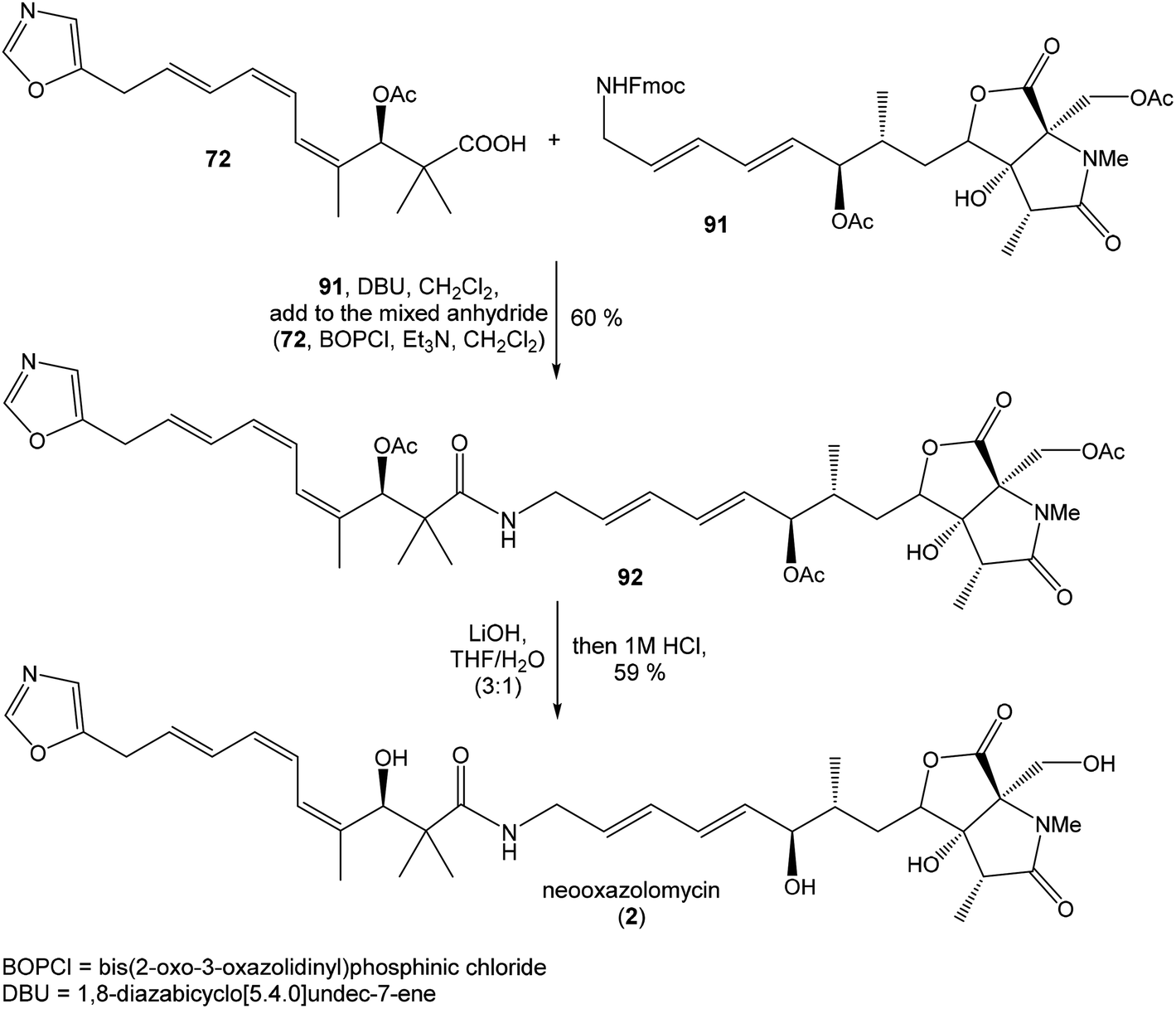 | ||
| Scheme 9 Finalization of the total synthesis of neooxazolomycin (2) by Hatakeyama. | ||
4.3 Total synthesis of neooxazolomycin by Kim
Kim et al. reported in 2019 a total synthesis of neooxazolomycin (2) using a chirality-transfer strategy.17 The six stereocenters of right-hand fragment were derived from D-serine by a series of chirality-transfer processes.104 afforded required oxazol triene 129 in 70% yield without significant isomerization of any double bond. The unidentified geometric isomer was detected at trace levels (<1:30). Then TBS protecting group of oxazol triene was removed to produce corresponding primary alcohol 130 in 90% yield. The Swern oxidation led to aldehyde 131 in 96% yield, which after following Pinnick oxidation provided carboxylic acid 132 in 84% yield (Scheme 10).
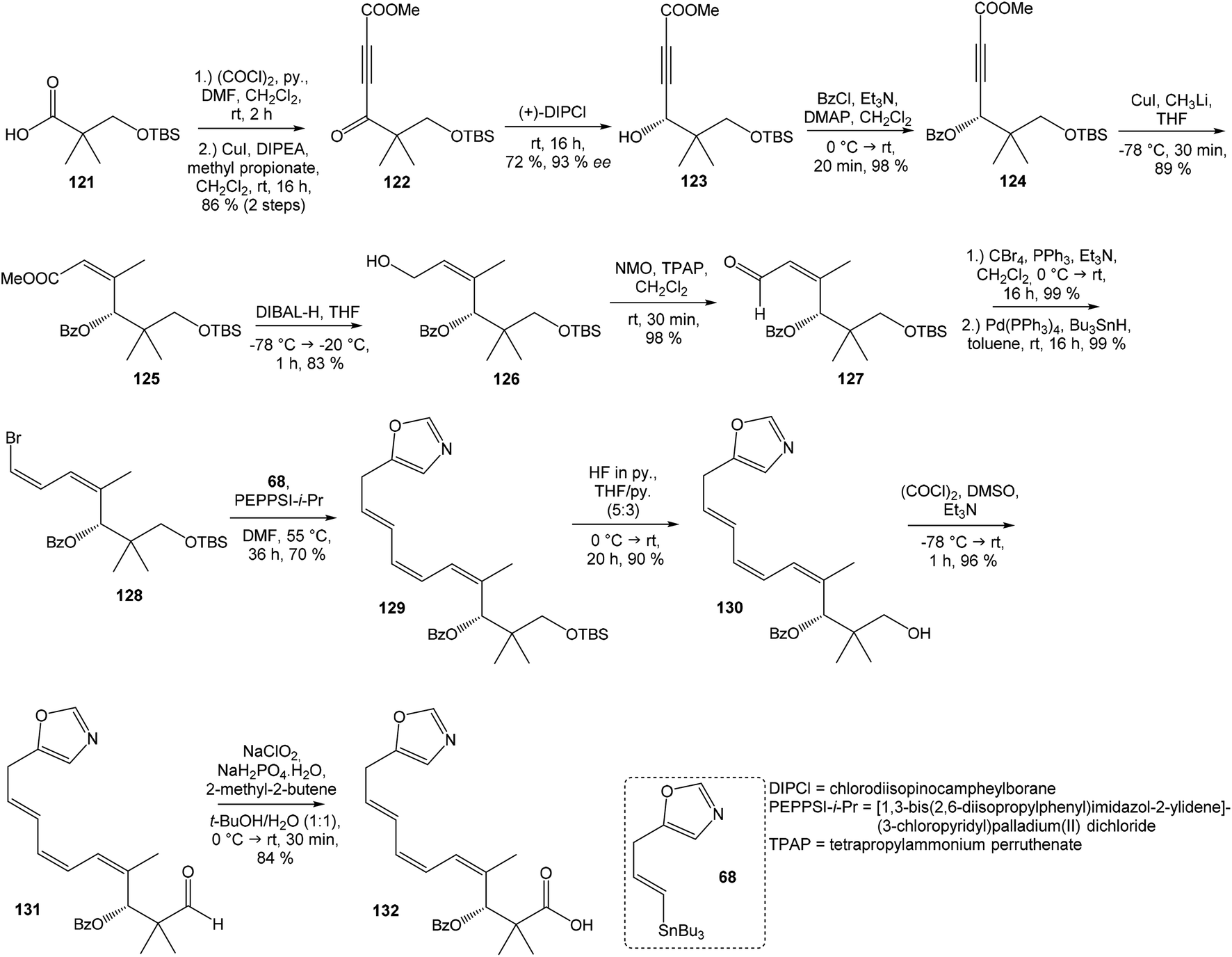 | ||
| Scheme 10 Synthesis of the left-hand fragment derivative 132 of neooxazolomycin by Kim. | ||
:1 (Scheme 11).
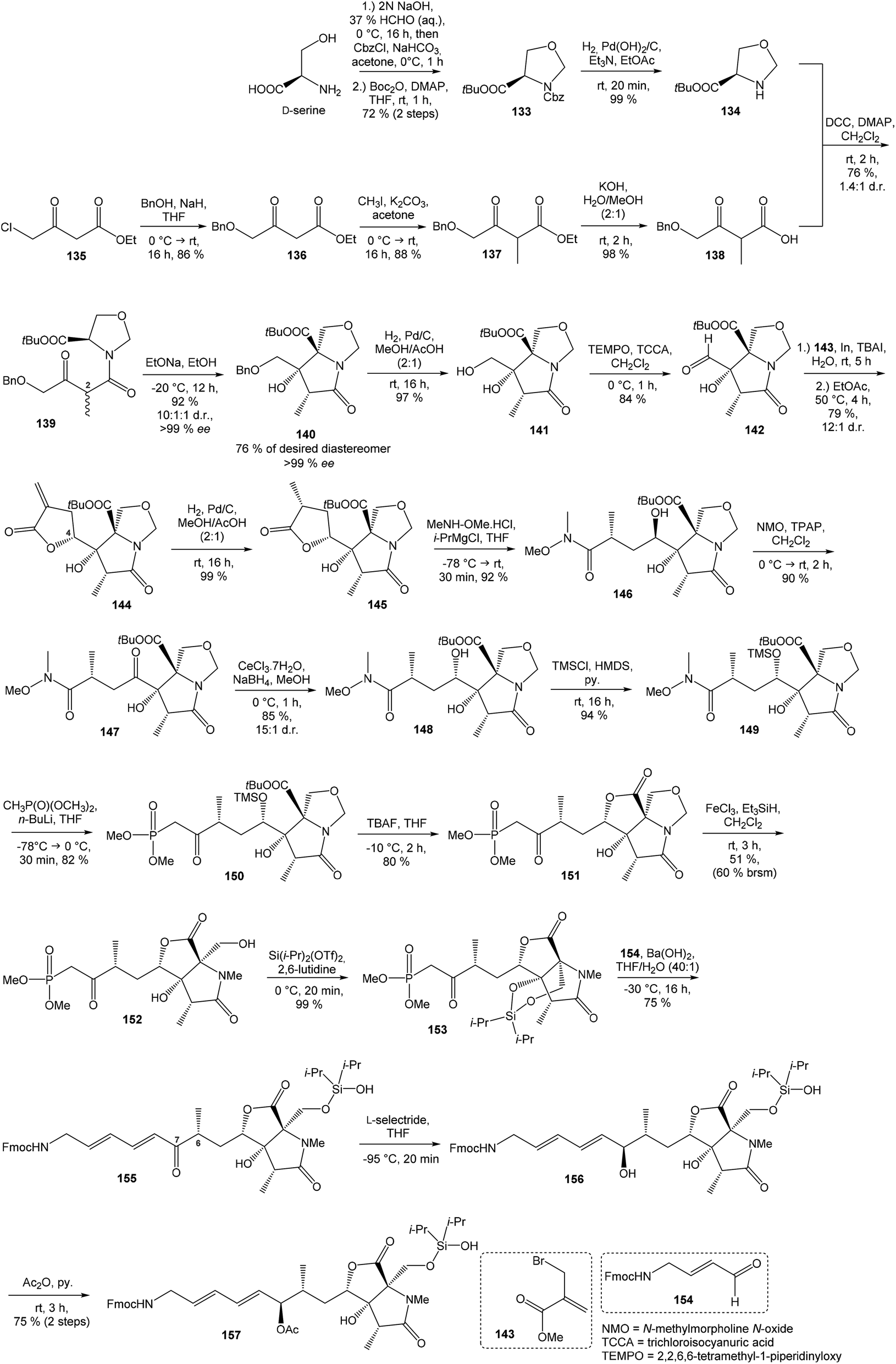 | ||
| Scheme 11 Synthesis of the right-hand and middle fragment derivative 157 of neooxazolomycin by Kim. | ||
Intramolecular aldol reaction was carried out with EtONa in EtOH and desired aldol product 140 was obtained with perfect enantioselectivity (>99% ee) in 61% yield. The configurations of the three contiguous stereocenters were unambiguously confirmed by XRD analysis.17 To introduce the exocyclic carbon chain, the benzyl protection group was removed under conditions of catalytic hydrogenation with H2, Pd/C in MeOH/AcOH (2:1) to produce primary alcohol 141 in 97% yield. Following oxidation with TEMPO, TCCA in CH2Cl2 gave aldehyde 142 in 84% yield. Various nucleophilic addition reactions were examined in following step. The best result was obtained when the Barbier reaction was conducted using In and TBAI in water to give 144 in 79% yield, in d.r. = 12:1 after spontaneous lactonization in EtOAc (Scheme 11). The stereocenter at C4 is epimeric to that of target natural product 2, however this stereochemistry was useful in the induction of the desired C6 stereocenter by hydrogenation.107,108
Reduction of 144 with H2, Pd/C in MeOH/AcOH (2:1) produced compound 145 in 99% yield. The stereochemistry of reduced product 145 was confirmed by XRD analysis.17 The lactone ring opening was performed with N,O-dimethylhydroxylamine hydrochloride and i-PrMgCl in THF to give compound 146 in 92% yield. Inversion of hydroxyl group on C4 was achieved by sequential oxidation with NMO, TPAP in CH2Cl2 to ketone 147 followed by stereoselective reduction with CeCl3·7H2O, NaBH4 in MeOH, to produce required epimer 148 in d.r. 15:1. Protection of secondary alcohol required careful selection of reaction conditions to avoid undesired lactonization. The protection was carried out with TMSCl and HMDS in pyridine and gave compound 149 in 94% yield without undesired lactonization. Next reaction was performed with dimethyl methylphosphonate, n-BuLi in THF and produced β-keto phosphonate 150 in 82% yield. Desilylation of 150 with TBAF in THF proceeded with concomitant lactonization to tricyclic 151 in 80% yield (Scheme 11).
The reductive ring opening of the oxaproline moiety was achieved with Et3SiH/FeCl3 to provide N-methylated bicyclic diol 152 in 51% yield.109 The structure of bicyclic diol was confirmed by XRD analysis.17 The hydroxyl groups were protected with Si(iPr)2(OTf)2 and 2,6-lutidine to corresponding dioxasilinane 153 in 99% yield.16 The Horner–Wadsworth–Emmons reaction of 153 with the aldehyde 154 in presence of Ba(OH)2 in THF/H2O (40:1) at −30 °C proceeded in 75% yield and produced ketone 155 with partially hydrolyzed dioxasilinane group. Under such reaction conditions, no isomerization at C6 was observed. The selective reduction of ketone 155 with L-selectride in THF was followed by immediate acetylation with Ac2O in pyridine to give 157 in 75% yield (after 2 steps) in d.r. 4:1 (Scheme 11). Notably, no diastereoselectivity was observed in the formation of the C7 stereocenter when the primary hydroxy group was not protected.
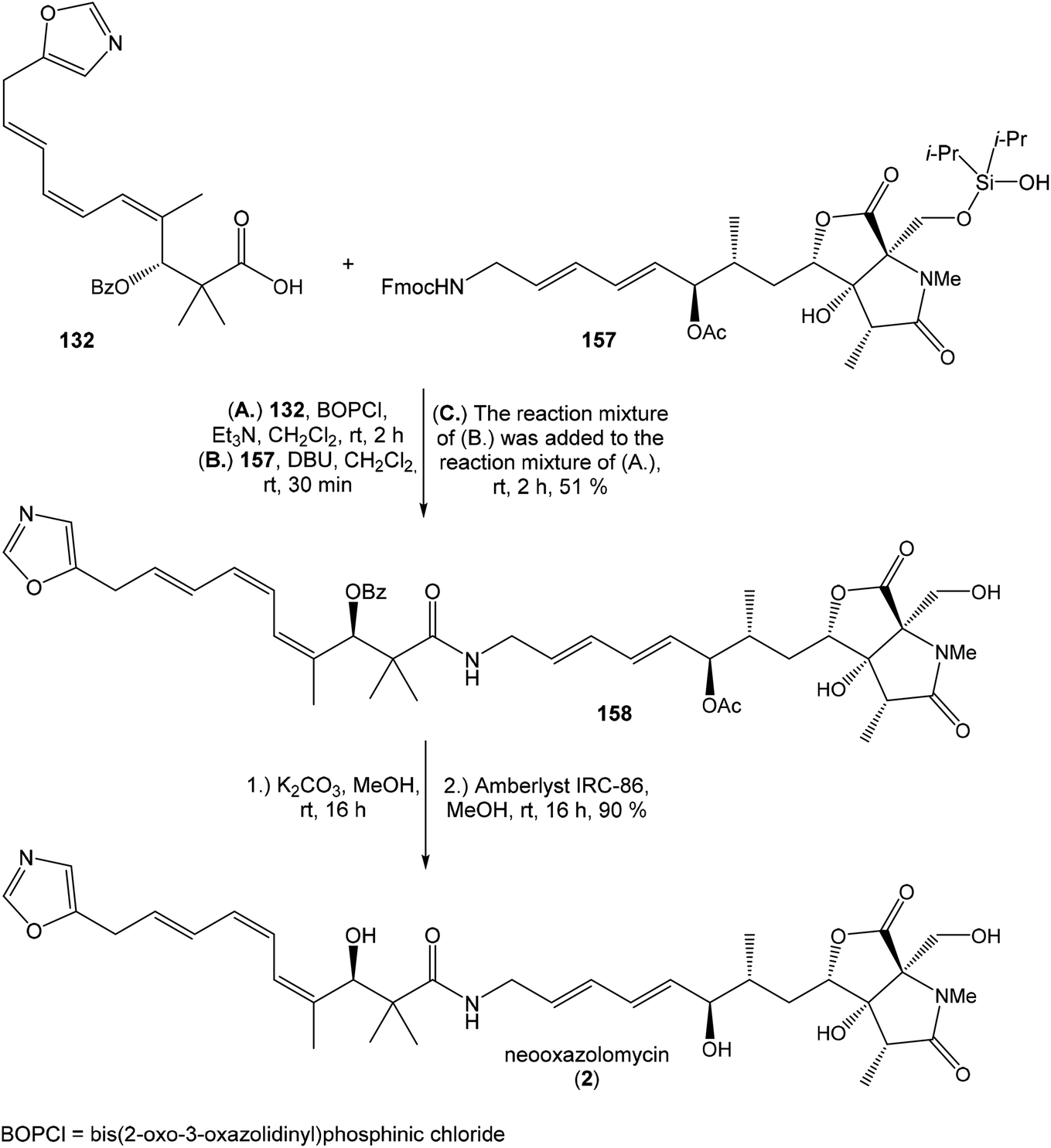 | ||
| Scheme 12 Finalization of the total synthesis of neooxazolomycin (2) by Kim. | ||
4.4 Total synthesis of oxazolomycin A by Hatakeyama
Hatakeyama et al. reported in 2011 a total synthesis of other member of the oxazolomycin family. The total synthesis of oxazolomycin A (1) was based on the methodology previously developed by Hatakeyama et al.16 in total synthesis of neooxazolomycin. Moreover, penultimate crude product in the Hatakeyama's total synthesis was in 2017 reported by Koomsiri et al.10 as a novel member of oxazolomycin family named oxazolomycin A2. Thus, Hatakeyama et al. elaborated suitable methodology for total synthesis of oxazolomycin A2 (14) as well.115 using Pd(PPh3)4, CuI and CsF116 in DMF provided (Z,Z,E)-triene 69 in 83% yield as a pure geometric isomer. Using a Pd(0) catalyst alone in Stille cross coupling always resulted in isomerization of triene system. Finally, desilylation, saponification and acetylation give to protected left-hand fragment 72 in 88% yield (after 3 steps) (Scheme 13).
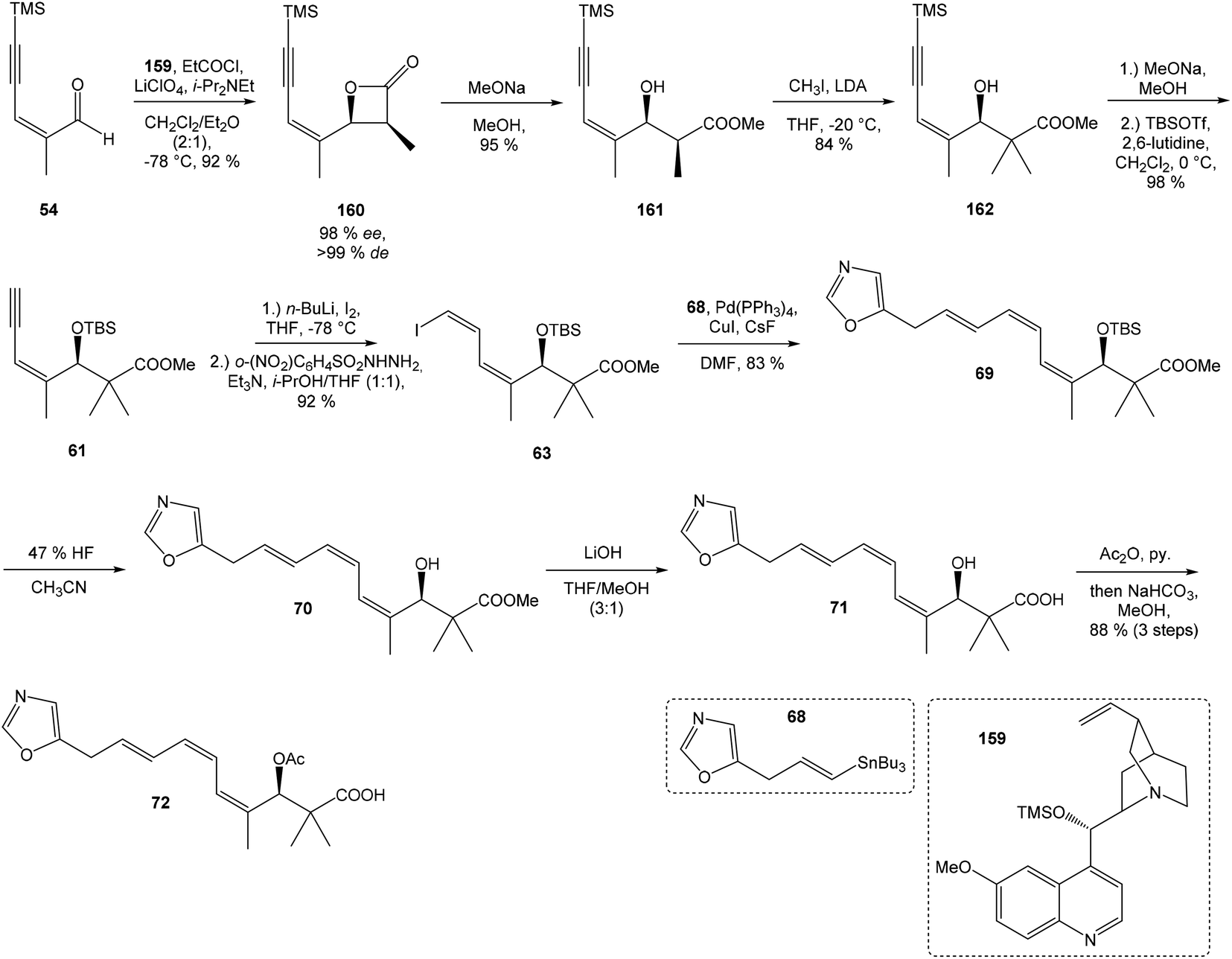 | ||
| Scheme 13 Synthesis of the left-hand fragment derivative 72 of oxazolomycin A by Hatakeyama. | ||
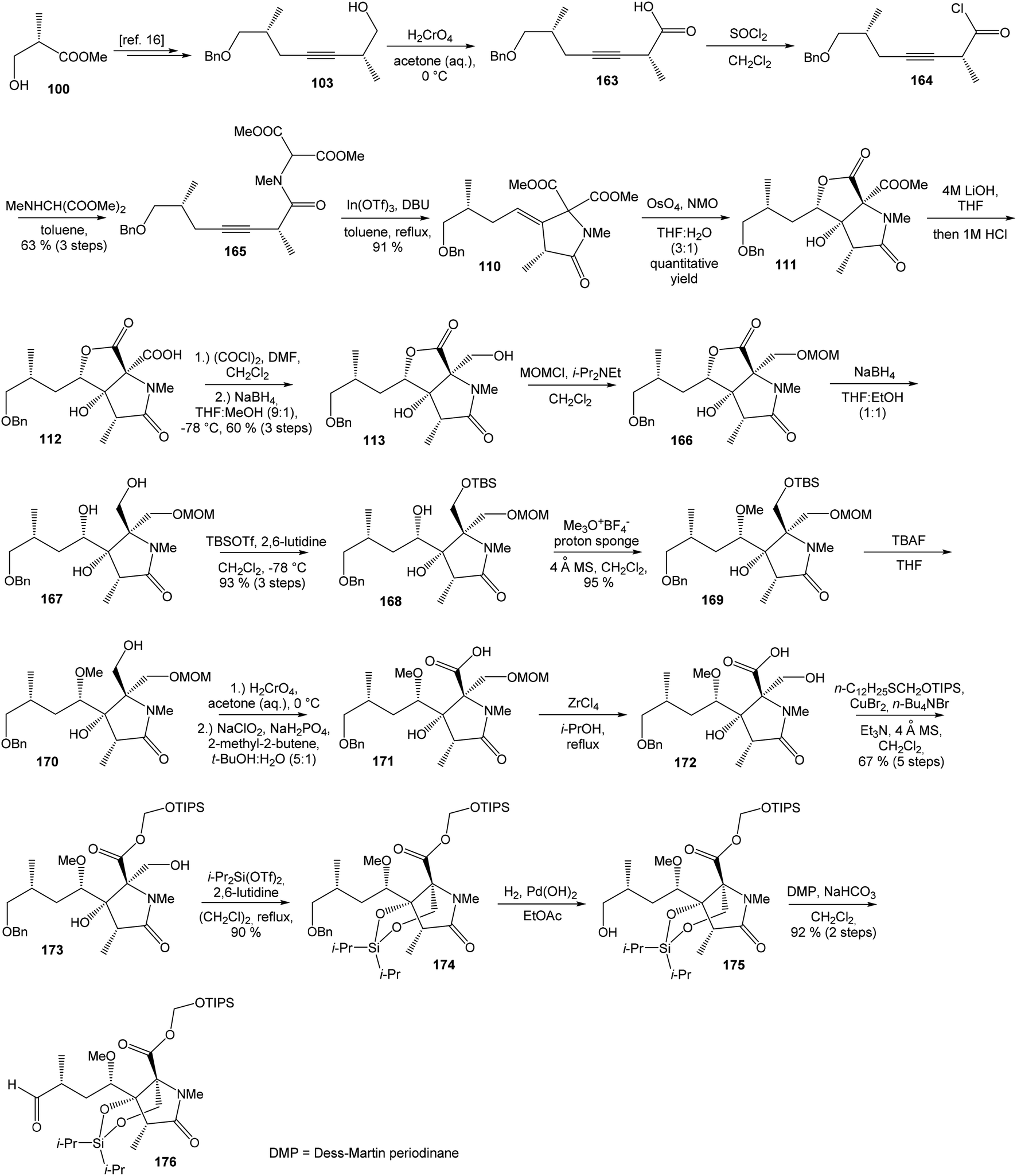 | ||
| Scheme 14 Synthesis of the right-hand fragment derivative 176 of oxazolomycin A by Hatakeyama. | ||
Further, primary hydroxyl group was protected with MOMCl, lactone ring reduced with NaBH4 and selective silylation with TBSOTf led to diol 168 that was prepared in 93%. Subsequent secondary hydroxyl group methylation of diol 168 using Meerwein reagent and proton sponge118 provided alcohol 169 in 95% yield. Compound 169 was desilylated with TBAF, primary hydroxyl group underwent Jones oxidation followed by Pinnick oxidation that led to acid 171. Cleavage of the MOM protecting group mediated with ZrCl4119 gave β-hydroxy acid 172, which was treated with triisopropylsilyloxymethyl(dodecyl)sulfane120,121 in the presence of CuBr2, TBABr and Et3N and allowed the selective esterification yielding to ester 173 in 67% (in 5 steps). Remarkably, esterification reaction did not proceed selectively in the absence of Et3N, although both the carboxylic acid and the primary alcohol groups were protected. The reaction of ester 173 with i-Pr2Si(OTf)2 and 2,6-lutidine in DCE led to dioxasilinane 174 in 90% yield. After reductive debenzylation of the compound 174 catalyzed with Pd(OH)2, followed by Dess–Martin oxidation, aldehyde 176 was prepared in 92% yield (in 2 steps) (Scheme 14).
:1 (Scheme 15). Geometrically pure (E)-isomer 117 was isolated by recrystallisation from EtOAc. This reaction sequence involved only 3 reaction steps in good overall yield (54%), compared to previously reported16 preparation of compound 117 from propargyl alcohol in 9 steps with only 18% overall yield.
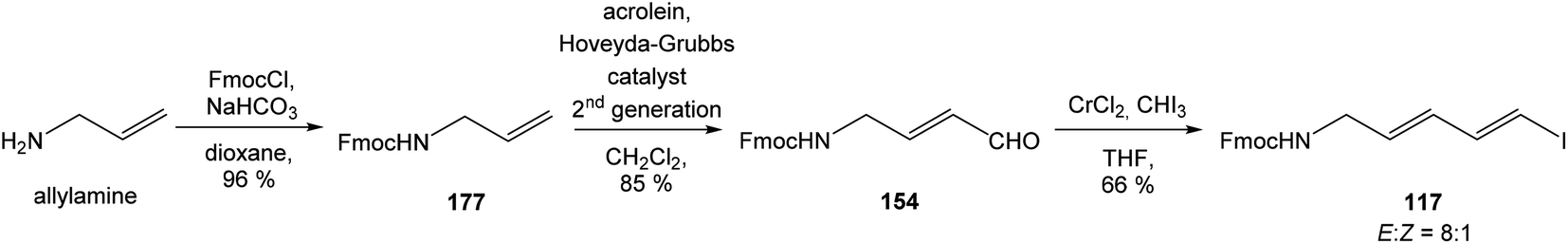 | ||
| Scheme 15 Synthesis of the middle fragment derivative 117 of oxazolomycin A by Hatakeyama. | ||
:2. Utilization of Dess–Martin oxidation followed by reduction with L-selectride provided desired (7R)-configurated alcohol 180 and its (7S)-epimer in ratio 4:1. Undesired (7S)-configurated alcohol was separated and again recycled by above mentioned oxidation–reduction procedure. Application of this sequence allowed to isolate desired alcohol 180 in 53% yield from aldehyde 176 (Scheme 16).
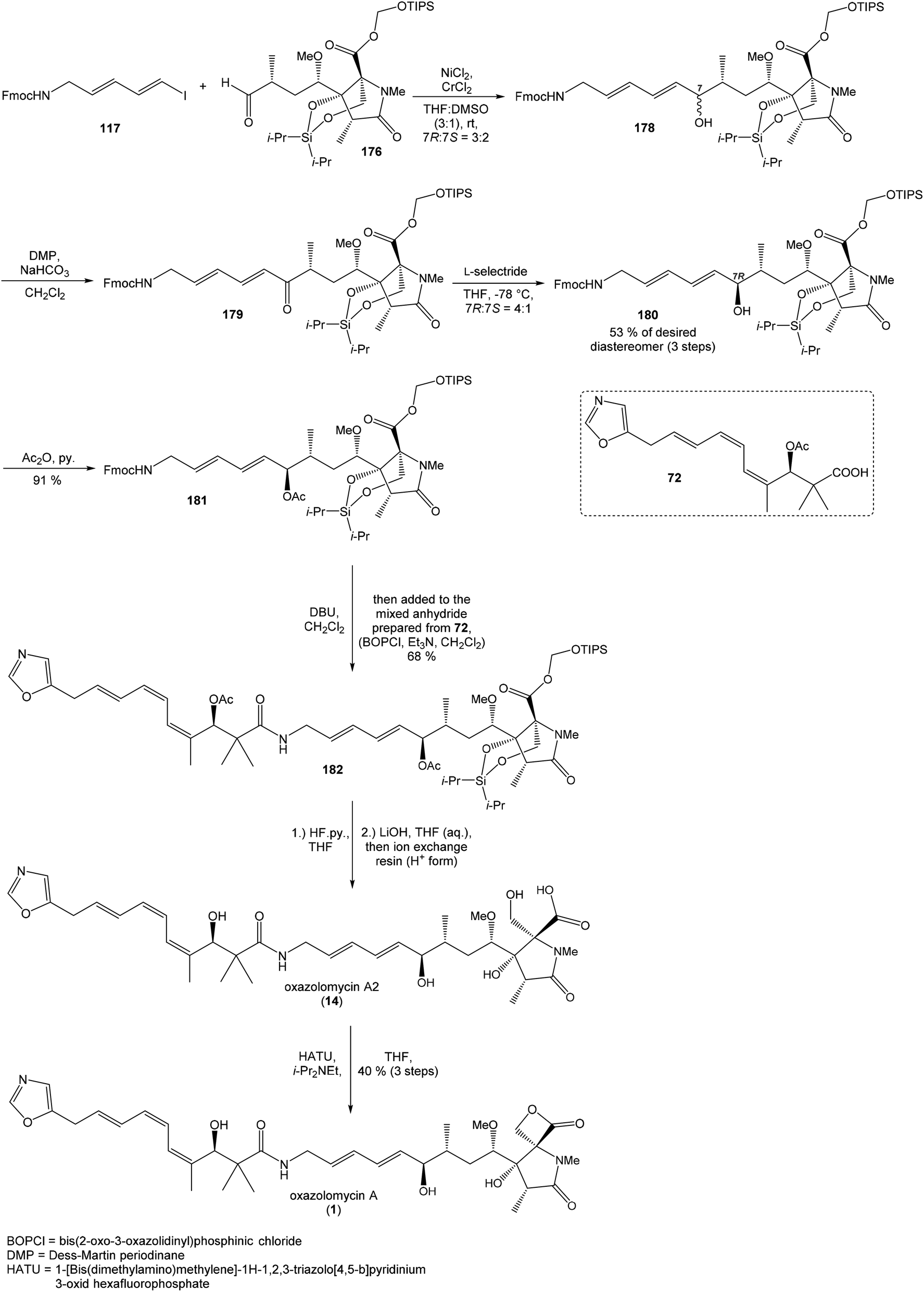 | ||
| Scheme 16 Finalization of the total synthesis of oxazolomycin A (1) by Hatakeyama. | ||
Protection of hydroxyl group with Ac2O in pyridine led to protected compound 181 in 91% yield. Exposure of 181 to DBU in CH2Cl2 provided free amine which was directly condensed with the left-hand fragment 72 in presence of BOPCl and produced amide 182 in 68% yield. Deprotection of amide 182 with HF-pyridine, followed by basic hydrolysis with LiOH and subsequent acidification with ion-exchange resin gave tetrahydroxy acid, which was later isolated by Koomsiri et al. as oxazolomycin A2 (14).10 Then final treatment of crude 14 with HATU125 in the presence of i-Pr2NEt in THF provided final oxazolomycin A (1) in 40% yield from amide 182 (Scheme 16). The spectroscopic data of the prepared oxazolomycin 1 were identical with the previously reported data for natural oxazolomycin A.1,6,60 The structure of final product 1 was confirmed by comparison of the spectral data of its diacetate with those reported previously.1,60
The first total synthesis of oxazolomycin A reported by Hatakeyama et al. involves 34 steps of the longest linear sequence in 1.4% overall yield from methyl (S)-3-hydroxy-2-methylpropionate. Moreover, synthesis of oxazolomycin A2 was achieved in 33 steps of the longest linear sequence in 1.4–3.5% overall yield from methyl (S)-3-hydroxy-2-methylpropionate. Unfortunately, oxazolomycin A2 in the Hatakeyama's total synthesis was not isolated but used as a crude product directly for next reaction. However, Hatakeyama et al. elaborated first consecutive total synthesis, which involved two different members of oxazolomycin family.10,12
4.5 Total synthesis of lajollamycin B by Hatakeyama
Hatakeyama et al. reported in 2019 the first total synthesis of lajollamycin B (11), nitrotetraene spiro-β-lactone-γ-lactame member of the oxazolomycin family.15 This convergent synthesis involves preparation and conjugation of three fragments. Left-hand fragment represents nitrodienylstannane 188, middle fragment 191 constitutes 7-iodoheptadienoic acid, right-hand fragment 181 has already been prepared for previous synthesis of oxazolomycin A reported by Hatakeyama et al.12:2. Corresponding nitroketone 186 was prepared in 87% using oxidation with Dess–Martin periodinane. Following Grignard reaction with MeMgBr in THF at 0 °C gave tertiary alcohol 187 in 88% yield as a 4:1 mixture of diastereomers (Scheme 17).
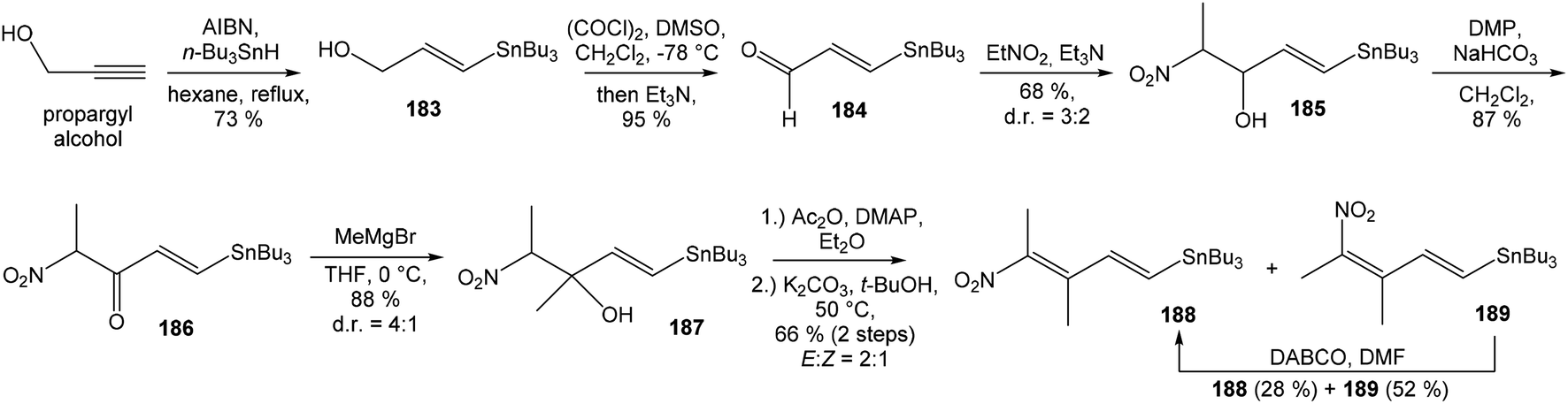 | ||
| Scheme 17 Synthesis of the left-hand fragment precursor 188 of lajollamycin B by Hatakeyama. | ||
Compound 187 was moved to next dehydration reaction to produce desired left-hand fragment 188. Unfortunately, all examined experiments using MsCl/Et3N and SOCl2/pyridine as well as various other acidic conditions totally failed. Thus, compound 186 was dehydrated128,129 via corresponding acetate, prepared with Ac2O and DMAP in Et2O, followed by using K2CO3 in t-BuOH at 50 °C. This procedure afforded desired (E)-isomer 188 and its (Z)-isomer 189 in 66% yield as a 2:1 mixture of (E/Z)-geometric isomers. After separation, undesired (Z)-isomer 189 was converted in reaction with DABCO to the mixture of (E)-isomer 188 in 28% yield and initial (Z)-isomer 189 in 52% yield (Scheme 17). It was observed that isomerization carried out at higher temperature led to largely decomposition of both products 188 and 189. Unambiguously determination of the geometries of 188 and 189 was achieved by NOESY spectra.15
Hatakeyama and co-workers examined other synthetic strategies to prepare desired nitrodienylstannane 188, e.g. synthesis starting with commercially available (E)-4-(dimethylamino)-3-buten-2-one or synthesis beginning from ethyl pyruvate. Unfortunately, such strategies did not provide satisfactory results.15
19 was under conditions of saponification using LiOH in THF/MeOH/H2O (3:1:1), followed by acetylation with Ac2O in pyridine, quantitatively transformed to carboxylic acid 191, which represents the middle fragment in total synthesis of lajollamycin B. Formerly prepared Fmoc-protected right-hand fragment 18112 was deprotected using DBU in CH2Cl2 to corresponding free amine, which was directly condensed with middle fragment 191, using BOPCl and Et3N and provided amide 192 in 62% yield. Following desilylation with HF/pyridine in THF and deacetylation using LiOH in THF (aq.) afforded tetrahydroxyacid 193, which was treated with HATU125 in presence of i-Pr2NEt in THF and provided the key precursor 194 in 44% yield after 3 steps (Scheme 18).
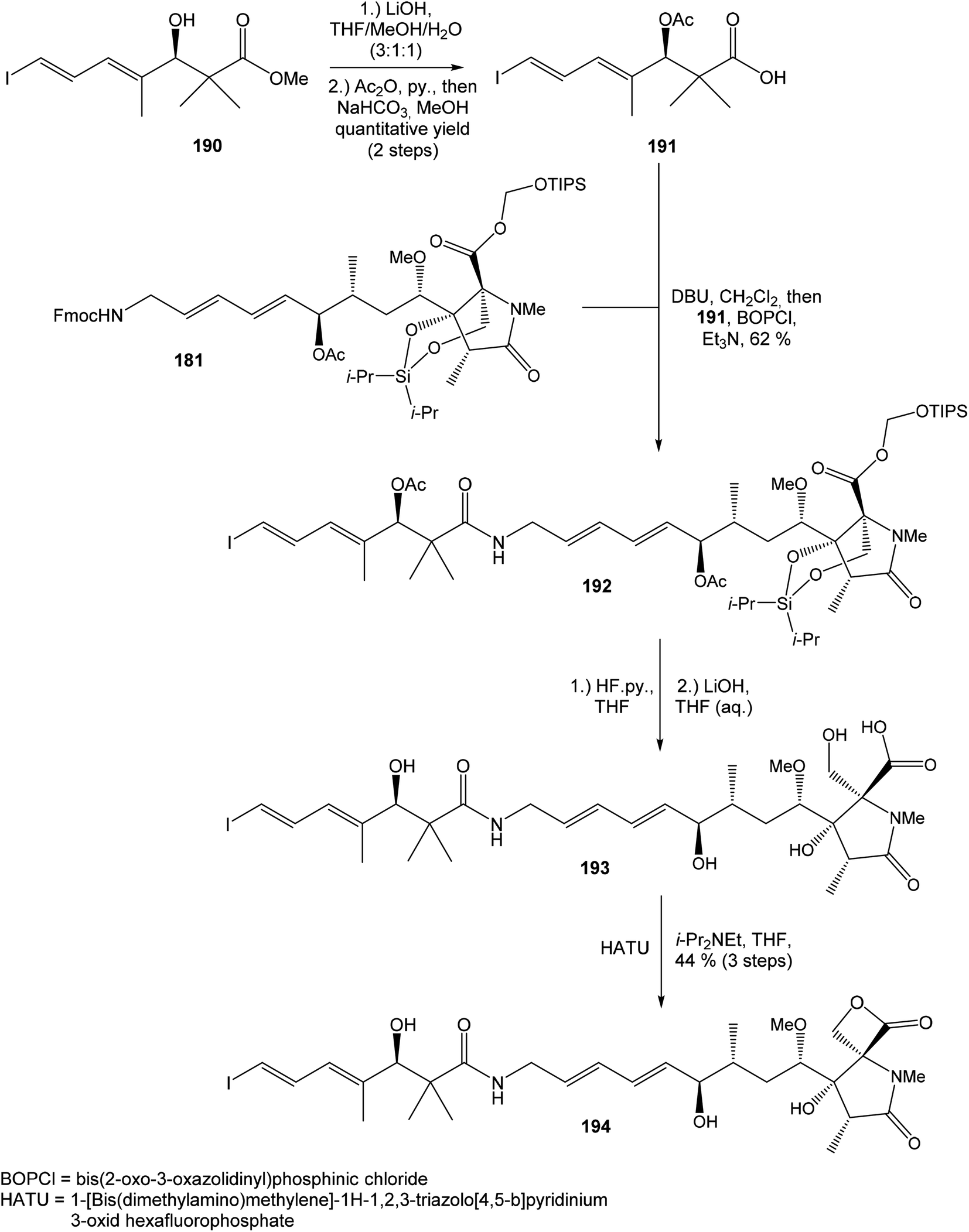 | ||
| Scheme 18 Synthesis of the right-hand and middle fragment derivative 194 of lajollamycin B by Hatakeyama. | ||
19 with (E)-nitrodienylstannane 188 and (Z)-nitrodienylstannane 189, served as a model study for the final step. These reactions were examined under Baldwin's conditions,116,130 which had been found to cause no isomerization of the similar conjugated triene systems during the Stille coupling.12,115 However, (E)-configurated tetra-substituted terminal double bond isomerization of 188 took place to produce mixture of (E)-isomer 196 and (Z)-isomer 195 in 38% yield, in ratio 5:4. When amounts of Pd(PPh3)4 and CsF were decreased in the reaction with (E)-configurated 188, it was found that yield increased to 73%, although isomerization of the terminal double bond was still presented for 196:195 in ratio 2:1. Similar reaction conditions applied on (Z)-configurated 189 provided mixture of 196 and 195 in 71% yield in ratio of 2.5:1. When the mixture of 2:3 of 188 and 189 was used as the starting material, mixture of products 196 and 195 was obtained in 76% yield in ratio 2:1 (Scheme 19). The results of these reactions are summarized in Table 9. Unfortunately, the mixture of 196 and 195 was not separable via chromatographic methods. Thus, geometries of the products 196 and 195 were determined directly from the prepared mixture, using COSY, HSQC, HMBC and NOESY spectral analysis.15
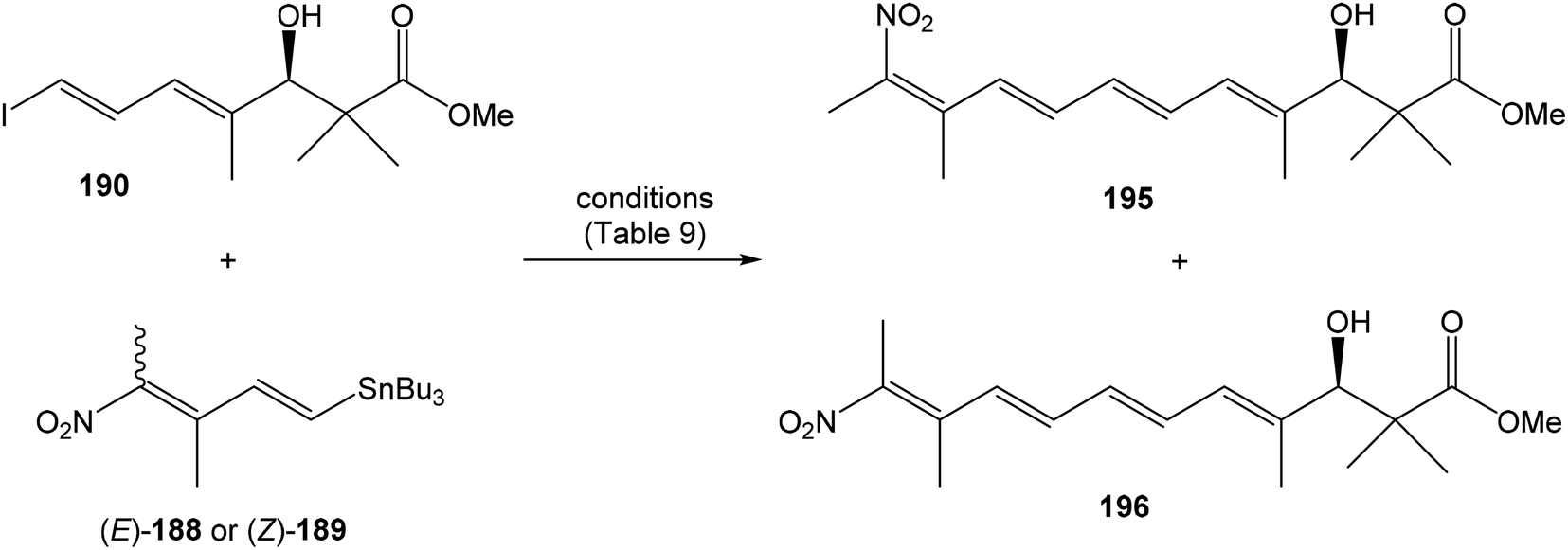 | ||
| Scheme 19 Hatakeyama's model study for the final Stille coupling in the synthesis of lajollamycin B. | ||
| Entry | Conditions | Yielda | 195:196b |
|---|---|---|---|
| a Yield of the (E/Z)-mixture.b Determined by 1H NMR. | |||
| 1 | 188 (1.1 equiv.), Pd(PPh3)4 (10 mol%), CuI (10 mol%), CsF (2 equiv.), DMF, rt, 1.5 h | 38% | 4:5 |
| 2 | 188 (1.1 equiv.), Pd(PPh3)4 (2 mol%), CuI (10 mol%), CsF (20 mol%), DMF, rt, 26 h | 73% | 1:2 |
| 3 | 189 (1.1 equiv.), Pd(PPh3)4 (1 mol%), CuI (10 mol%), CsF (20 mol%), DMF, rt, 5 h | 71% | 2:5 |
| 4 | 2:3 mixture of 188 and 189 (1.1 equiv.), Pd(PPh3)4 (2 mol%), CuI (10 mol%), CsF (20 mol%), DMF, rt, 71 h |
76% | 1:2 |
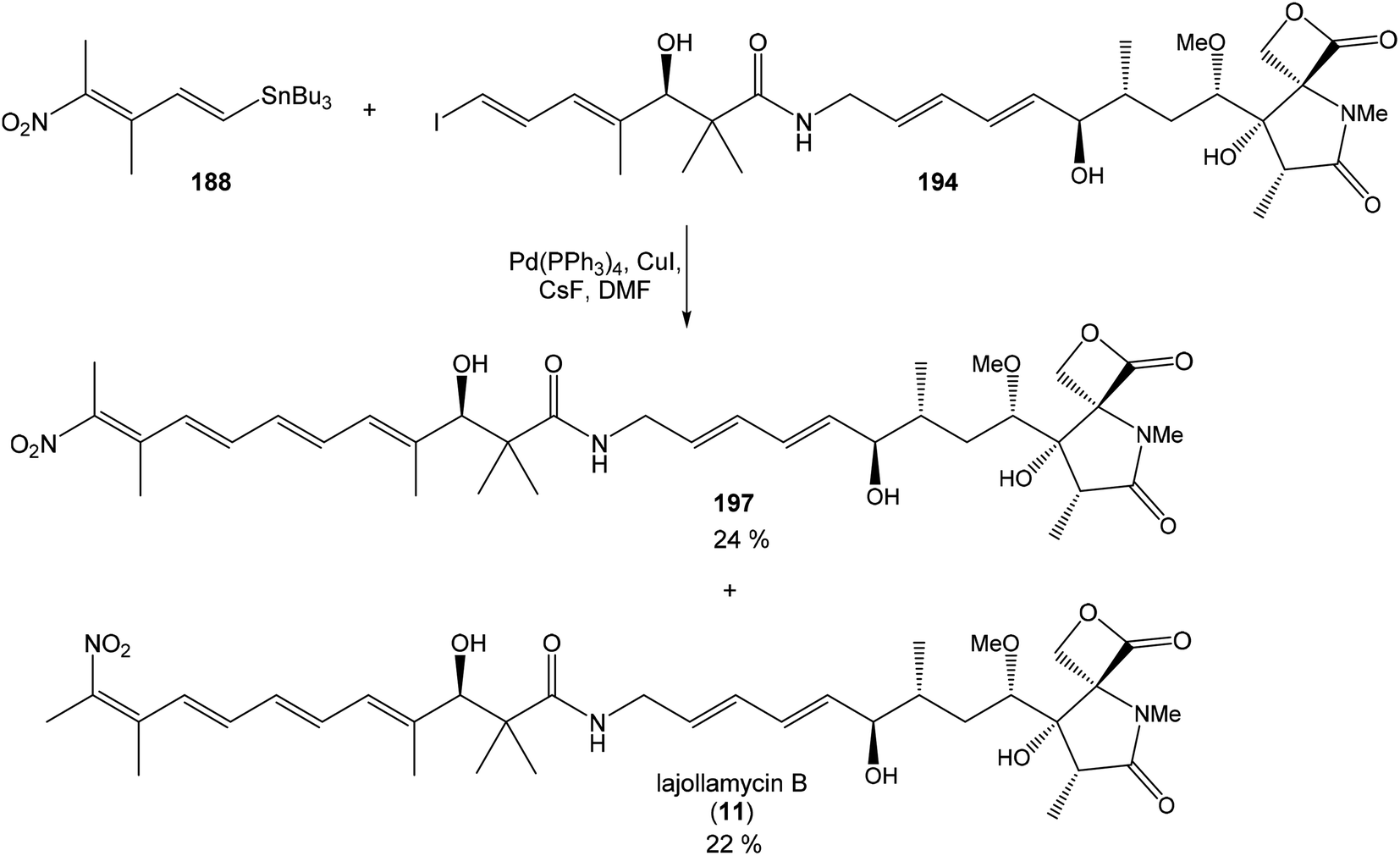 | ||
| Scheme 20 Finalization of the total synthesis of lajollamycin B (11) by Hatakeyama. | ||
Surprisingly, the spectroscopic comparison (1H and 13C NMR) of unexpected (Z)-configurated compound 11 found it to be identical with those reported9 as natural lajollamycin B, in contrary to expected (E)-configurated compound 197. Furthermore, Hatakeyama and co-workers carefully reinvestigated COSY, HSQC, HMBC and ROESY spectra of lajollamycin B reported by Oh et al.9 and found that key ROESY correlation between H9′ and Me-11′, confirming (E)-configuration was not observed.9,15 Hatakeyama et al. unambiguously assigned stereostructures of prepared products 11 and 197 based on their NOESY spectra (Fig. 29).15 Moreover, Hatakeyama and co-workers observed, that the nitrotetraene domain of 197 when compared to 196 and spectra of 11 when compared to 195 exhibited close similarities in 1H and 13C NMR.15 These findings allowed Hatakeyama et al. to revise the initially assigned (E)-geometry of the terminal nitro-containing tetrasubstituted alkene in lajollamycin B to (Z)-geometry.
 | ||
| Fig. 29 NOESY correlations reported by Hatakeyama et al. for final products in the total synthesis of lajollamycin B. | ||
Hatakeyama et al. prepared the first total synthesis of lajollamycin B (11) and its (10′E)-isomer 197. The synthesis involved conjugation of key intermediates as the right-hand fragment 181 and the middle fragment 191, which was developed for synthesis of other members of oxazolomycin family.12,16,43,131 This synthetical strategy represents applicable methodology for both lajollamycins and oxazolomycins. And importantly, the stereochemistry of natural lajollamycin B was revised and corrected.15
5 Formal synthesis of oxazolomycin family member
5.1 Formal synthesis of (+)-neooxazolomycin by Taylor
In 2011 Taylor et al. reported first formal synthesis of (+)-neooxazolomycin (2).18 Talyor's formal synthesis of neooxazolomycin included the same late stage amide formation between left-hand fragment 71 (Scheme 25) and already combined right-hand and middle fragment 91, which were prepared in both previously reported total syntheses by Kende and Hatakeyama.13,16 The left-hand fragment 71 of neooxazolomycin, was readily available as an intermediate in formerly reported synthesis of inthomycin A.115,132 Synthesis of the compound 91, which represents combined right-hand and middle fragment of neooxazolomycin is described herein.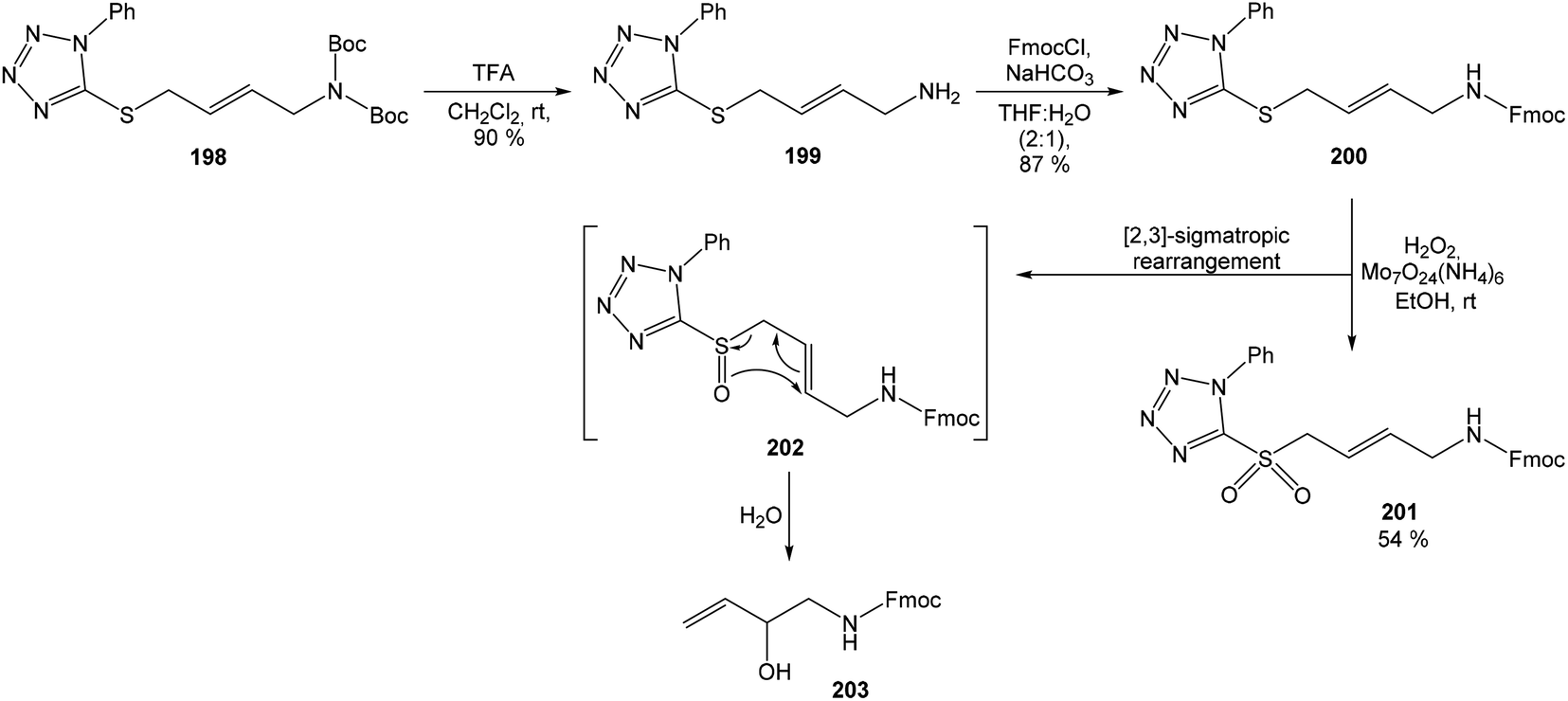 | ||
| Scheme 21 Synthesis of the middle fragment derivative 201 of neooxazolomycin by Taylor. | ||
:1. N-Acetylation with acid chloride 205 with pyridine in CH2Cl2 provided N-acylated oxazolidine 206 in 92% yield as the mixture of diastereomers in ratio 1:1 (stereocenter on malonate moiety). Dieckmann condensation promoted by t-BuOK in t-BuOH led to regioselective cyclisation to desired bicyclic oxazolidine 207 in 93% yield. Compound 207 was prepared as a single diastereomer, albeit as a mixture of 9:1 enol/keto tautomers. Next reaction with Tf2O and i-Pr2NEt in CH2Cl2 at −50 °C gave corresponding triflate 208 in 70% yield (Scheme 22), which represents one of two partners for Stille cross coupling.
 | ||
| Scheme 22 Synthesis of bicyclic precursor for right-hand fragment 208 of neooxazolomycin by Taylor. | ||
(Trimethylsilyl)propargyl alcohol was in 2 reaction steps transformed to required (E)-trisubstituted alkene 209 according to the procedure of Denmark and co-workers.137 Sharpless epoxidation of alkene with Ti(OiPr)4, (+)-DIPT and TBHP in CH2Cl2 provided epoxide 210 in 85% as an enantiomeric mixture in ratio 95:5. Enantioselectivity of prepared epoxide 210 was determined by formation of Mosher's esters with both (R)- and (S)-MTPA-Cl. Corresponding esters 211 and 212 were determined by 1H NMR analysis. Further, primary hydroxyl group in epoxide 210 was protected via bezylation with BnBr/NaH in THF to fully protected epoxide 213 in 85% yield. Epoxide 214 was obtained in 85% yield after desilylation under standard conditions with TBAF in THF. Lewis acid conditions with the alanate reagent derived from lithium trimethylsilylacetylide and AlMe3138 allowed regioselective epoxide opening at the C3 position, to produce desired alcohol 215 in 97% yield, as a mixture 9:1 of regioisomers (in favor of desired regioisomer). It was observed that the control of temperature in epoxide opening reaction was essential to ensure high regioselectivity. Standard conditions of desilylation with TBAF in THF provided alkyne 216 in 86% yield. Finally, radical promoted hydrostannylation with AIBN and n-Bu3SnH led to stannane 217 in 67% yield as a 10:1 mixture of (E/Z)-isomers (Scheme 23). Desired (E)-configurated stannane 217 represents a second partner for Stille cross coupling.
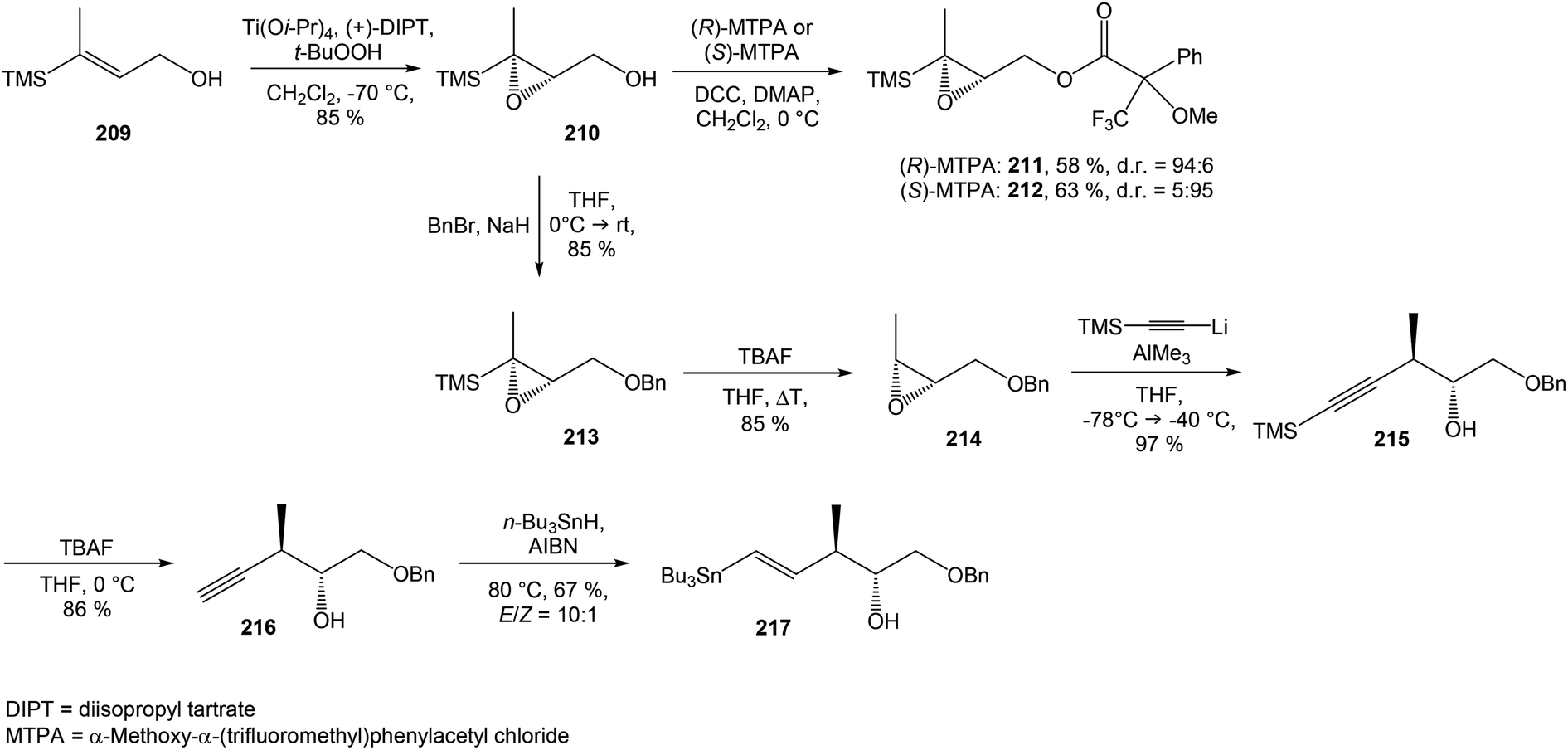 | ||
| Scheme 23 Synthesis of Stille cross coupling partner 217 for right-hand fragment of neooxazolomycin by Taylor. | ||
The combination of the above-mentioned two fragments, 208 and 217, was performed under Stille cross coupling conditions with Pd2(dba)3 and P(2-fur)3 in DMF at 50 °C for 6 h. In this Stille reaction, control of the temperature was essential to afford desired geometric isomer. When the reaction was done at higher temperature, higher formation of unwanted isomer was observed. Under mentioned Stille conditions, diene 218 was obtained with the same crude ratio 10:1 of (E/Z)-isomers as was presented for the stannane precursor 217. After chromatographic separation, 87% yield of desired (E)-218 and 4% yield of undesired (Z)-218 were obtained. Disubstituted alkene 218 was selectively reduced with in situ generated diimide from p-TsNHNH2 and KOAc. Not surprisingly, the reduction of (E)-218 proved to be much easier than the reduction of more steric hindered (Z)-218 and alkene 219 was prepared in 89% yield (Scheme 24).
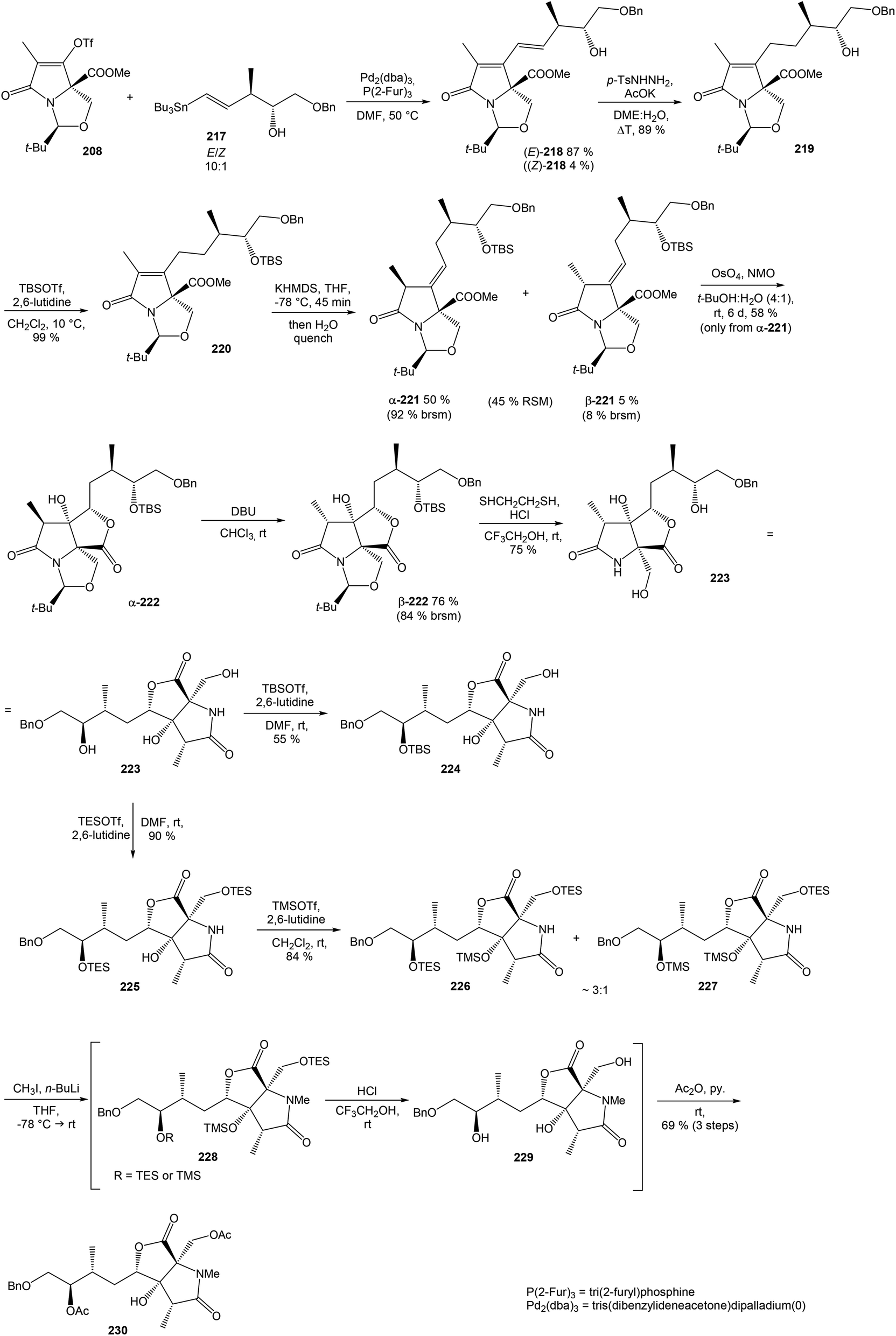 | ||
| Scheme 24 Synthesis of the right-hand fragment derivative 230 of neooxazolomycin by Taylor. | ||
Silylation of secondary hydroxyl group with TBSOTf and 2,6-lutidine in CH2Cl2 provided protected derivative 220 in 99% yield. For the purpose of necessary dihydroxylation, the deconjugation of α,β-unsaturated lactam 220 was required. Unfortunately, the applied conditions of deconjugation model studies to unsaturated lactam 220 led only to recovery of starting material. After a series of experiments, appropriate reaction conditions were identified with KHMDS in THF followed with rapid quench by pouring into stirred water. These conditions provided 10:1 mixture of α-221 in 50% yield, β-221 in 5% yield and the rest of mixture was recovered as the starting material (Scheme 24). The stereoconfiguration of the α-centre, as well as the (E)-configuration of alkene were assigned by NOE studies.18 Interestingly, if a longer reaction time or more basic conditions were applied, none deconjugation product was obtained.
Following dihydroxylation of deconjugated double bond was found to be more difficult than expected as the result of the crowded steric environment around the trisubstituted olefin. Epimer β-221 with correct configuration at the C2 position proved to be inert under any conditions of dihydroxylation and solely starting material was recovered. In contrast, epimer α-221 displayed reduced reactivity, but it was able to provide product of dihydroxylation with concomitant lactonization as a tricyclic lactone α-222 in 58% yield. The dihydroxylation was carried out with OsO4 and NMO in t-BuOH/H2O for 6 days and provided only single oxidation product derived from attack on concave face of alkene α-221 (Fig. 30).18 The obtained tricyclic lactone α-222 necessitated inversion of configuration on the stereocenter adjacent to the carbonyl group of lactam core. For this purpose, Taylor and co-workers found that a catalytic amount of DBU (10 mol%) is suitable for desired epimerisation. The process of isomerisation was studied by Taylor and co-workers by 1H NMR spectroscopy.18 It was found that equilibrium process led to >95% conversion over 24 h. Synthetically, epimerisation of α-222 was accomplished with DBU (10 mol%) in CHCl3 and provided β-222 in 76% yield with 10% of recovered starting material α-222 (Scheme 24).
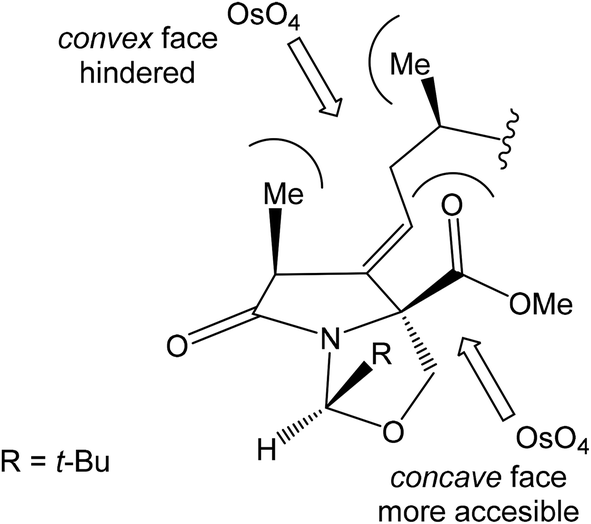 | ||
| Fig. 30 Model for dihydroxylation reported by Taylor et al. | ||
Further, acidic deprotection of the oxazolidine moiety under standard Corey conditions139 with ethane-1,2-dithiol and HCl in CF3CH2OH gave a bicyclic triol 223 in 75% yield. Taylor and co-workers focused their attention on the lactam core N-methylation without the need for protection of some or all hydroxyl groups. It was found that the most suitable strategy involved the preparation of tris-silyl derivative. Triple silylation in a single step shown to be complicated by poor solubility of triol 223 in common solvents with exception of MeOH and DMF. Protection with TBSOTf and 2,6-lutidine in DMF afforded only monosilyl derivative 224 in 55% yield, presumably due to steric hindrance. Silylation was accomplished with less hindered TESOTf and 2,6-lutidine in DMF and provided double silylated product 225 in 90% yield. TMSOTf and 2,6-lutidine in CH2Cl2 for 24 h were used for a tertiary alcohol protection. As the result of this silylation, the mixture 3:1 of compounds 226 and 227 in 84% yield was obtained. An inevitable displacement of one of the TES groups with a TMS group led to the mixture of silyl isomers, but chromatographic separation of compounds 226 and 227 was found to be not necessary. The lactam N-methylation with CH3I and n-BuLi in THF at −78 °C of both silyl isomers was done. The subsequent total desilylation with HCl in CF3CH2OH and finally acetylation under standard conditions with Ac2O in pyridine was accomplished. The desired diacetate 230 was prepared in 69% yield after 3 steps (Scheme 24).
:1 mixture of (4E,6E):(4Z,6E)-isomers. Using chromatographic separation, (4E,6E)-91 was isolated in 63% yield along with 14% of its isomer (4Z,6E)-91 (Scheme 25). The obtained data were in accordance with those reported by Kende et al.13
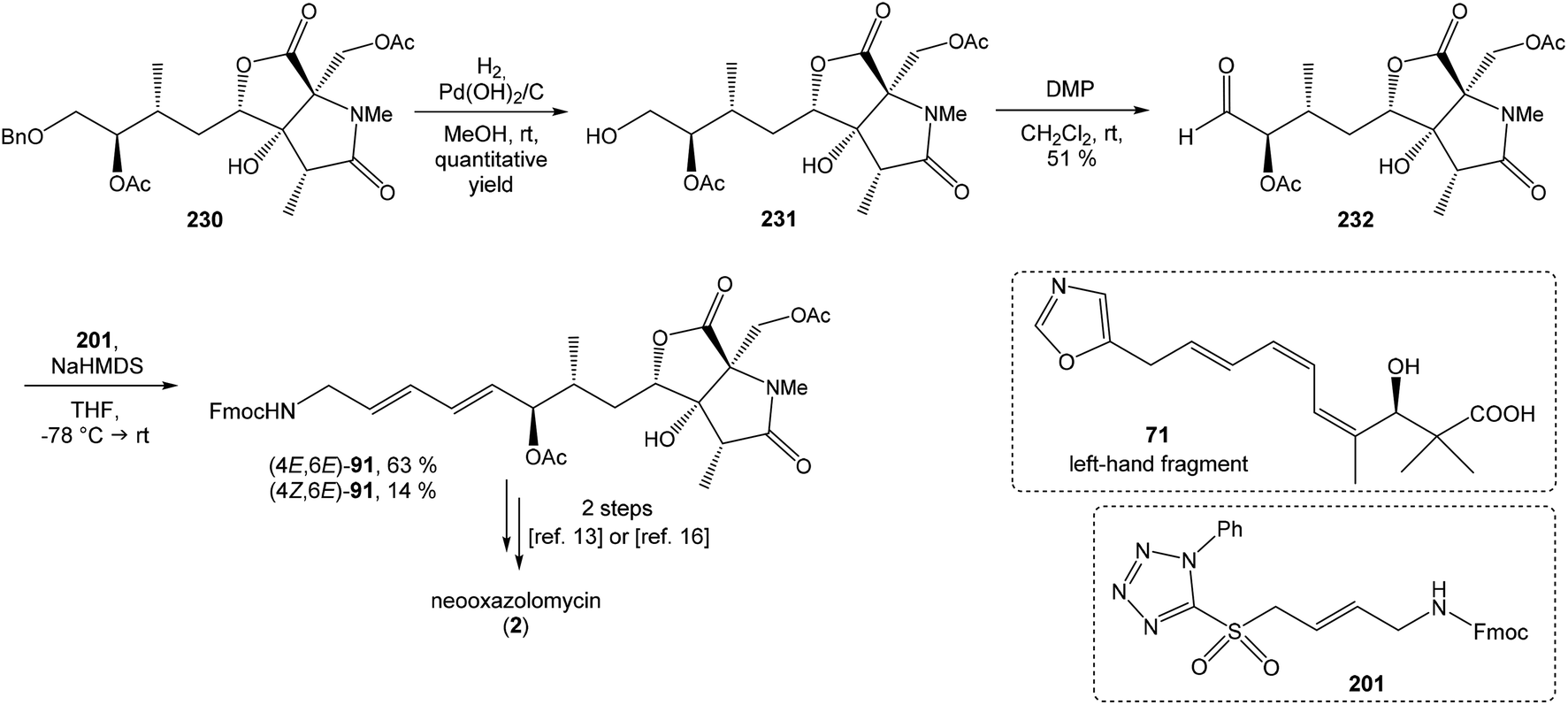 | ||
| Scheme 25 Finalization of the formal synthesis of neooxazolomycin (2) by Taylor. | ||
To conclude, Taylor and co-workers completed a formal synthesis of neooxazolomycin in 23 steps with the preparation of Kende's intermediate 91. In this approach a Moloney's synthetic strategy136 was used toward key lactam core preparation. The Stille coupling reaction and Julia–Kocienski olefination were used to achieve combination of three key segments. The stereoselective construction of the bicyclic core was achieved with the preparation of Stille adduct via strategic deconjugation and dihydroxylation.
6 Discussion
Detailed biosynthesis of oxazolomycin A (1) has been investigated in Streptomyces albus JA3453 using labeling experiments20 and gene analysis25 of the oxazolomycin A gene cluster. These data suggest, that a whole structure of 1 is constructed from six building blocks: N-formyl-tetrahydrofolate, glycine, L-methionine, L-serine, malonyl-ACP and methoxymalonyl-ACP (Fig. 7). Zhao et al. further proposed disquisitional model for biosynthesis of 1 in S. albus (Fig. 6).25 Despite a precise biosynthetic analysis of 1, some details remain still unclear to date, e.g. domains responsible for cyclizations to afford the oxazole or γ-lactam ring.As mentioned above, the main biological effects of oxazolomycins are attributed to their right-hand pyroglutamate cores, due the structural similarity with other bioactive pyroglutamates (Fig. 8). The results from biological assays suggest, that the unique spiro-β-lactone-γ-lactam moiety of right-hand fragment in some oxazolomycin members plays a significant role in the bioactivity. For example, oxazolomycin A (1) displays a selective antibacterial activity against Agrobacterium tumefaciens, while neooxazolomycin (2) with bicyclic γ-lactam-γ-lactone core is inactive.60 Similarly, 1 showed more potent antibacterial activities against: Bacillus subtilis ATCC 6633, Escherichia coli NIHJ, Kocuria rhizophila ATCC 9341, Staphylococcus aureus ATCC 6538p and Xanthomonas campestris pv. oryzae KB 88 at all tested concentrations compared to its hydrolyzed natural member oxazolomycin A2 (14). Moreover, the cytotoxicity of 1 against human leukemia HL60 cells growth was considerably higher compared to 14.10 These results could suggest higher activity of 1 due the angle strain of the β-lactone moiety. Bagwell et al. evaluated the influence of structural subunit 45 localized close to the middle fragment of oxazolomycins.55 Thorpe–Ingold effect of gem-dimethyl amide motif causes a structural U-shaped conformation of oxazolomycins, which is assumed to be beneficial for final biological activity. Additionally, suitable substituted derivatives of subunit 45 exhibit biological activity itself. Further studies revealed the importance of left-hand fragment on the biological activity of oxazolomycins. For example, 1 showed antibacterial activity against Agrobacterium rhizogenes IFO 13257, Agrobacterium tumefaciens EHA 101 and Agrobacterium tumefaciens IFO 13263, while its geometric isomers oxazolomycin B (3) and C (4) were inactive.6 Even the phytotoxic activity is likely associated with left-hand fragment geometry. For the inhibition of Medicago sativa seeds germination and potato tuber disc necrosis 1 was the most potent geometric isomer, followed by 3 and then by 4.6,71 However, left-hand fragment geometric isomers 1, 3 and 4 inhibited the crown gall formation at the same doses.6 The biological activity comparison hand in hand with a structural diversity of single fragments could bring a better understanding in oxazolomycin's mechanism of action, thus further bioassays in this field are needed.
The aggregated structures of oxazolomycins as the combination of particular fragments result in unique skeletons with remarkable bioactivity. A widespread antibacterial activity of oxazolomycins against both Gram-positive and Gram-negative bacterial strains was reported. Besides the selective activity of oxazolomycin A (1) against Agrobacterium tumefaciens,60 growth inhibition of several other bacteria including Agrobacterium rhizogenes,6 Escherichia coli10 and Pseudomonas putida60 were found. Curromycin A (6) and B (7) affect against bacterial strains Bacillus subtilis,61 Micrococcus luteus4 and Staphylococcus aureus.4 Lajollamycin (10) showed significant antibacterial properties against Enterococcus faecalis, Staphylococcus aureus, Streptococcus pneumoniae and Escherichia coli.8 Bisoxazolomycin (15), oxazolomycin A2 (14) and 1 were active against Staphylococcus aureus and Xanthomonas campestris.10 These data approved that diverse oxazolomycin members possess a broad antibacterial activity. Moreover, the antiviral activity of some oxazolomycins provided interesting results. The bioactivity of 1 was reported against herpes simplex type 1, influenza A and vaccinia viruses.63 Further study revealed anti-HIV activity of curromycins 6 and 7, with indication that curromycins affect in the virus replication cycle in later steps.64 Similarly, the cytotoxic activity bioassays of oxazolomycins resulted in notable findings. The activity against Ehrlich ascites carcinoma was reported for 1,1 as well as for neooxazolomycin (2).2 Significant cytotoxicity against mouse leukemia P388 cells was reported for 1,1 16-methyloxazolomycin (5)5 and for both curromycins 6 and 7.3 KSM-2690 B (8) and C (9) exhibited the inhibition activity against human bladder carcinoma T24 cells.7 Despite none cytotoxic activity of lajollamycins (10–13) was observed against selected cell lines,9 10 showed potential against mouse melanoma B16–F10 cells growth.8 Human leukemia HL60 cell line growth was inhibited by monocyclic oxazolomycin 14, dimeric structure 15, but the most potent activity was reported for 1.10 As seen, structural diverse oxazolomycins have a strong potential in growth inhibition of different carcinoma cell lines. Further considerable bioactivity of oxazolomycins was observed toward the inhibition of crown gall formation. The highest inhibition activity was reported for 1, oxazolomycins B (3) and C (4),6 although the curromycins 6 and 7 also showed inhibitory potential.62 The phytotoxic activity was observed for 1, 3, 4, 6 and 7 against Medicago sativa seeds germination. Similarly, these five oxazolomycin members induced the necrosis of potato tuber disc.6,62,71 The broad range of biological activities exhibited by oxazolomycin family members or their fragments indicates their potential research or application in medicine, especially as antibacterial, antiviral, cytotoxic or phytotoxic compounds. Therefore, it is not surprising that different research groups developed many synthetic strategies toward oxazolomycins.
Kende et al.13 achieved first total synthesis of neooxazolomycin (2) in 1990, just five years since its discovery by Uemura et al.2 in 1985. Kende's synthesis involved diastereoselective Reformatsky-type condensation, Schöllkopf condensation and Stille coupling. Kende's synthesis started from anhydrogalactoside 73 (Fig. 31), which was also utilized as a chiral source. Longest linear sequence form 73 involves 22 steps with overall yield 1.9%. For the next 17 years it was the only total synthesis of oxazolomycin family member. Second total synthesis of neooxazolomycin was reported by Hatakeyama et al. in 2007.16 Hatakeyama's synthesis of 2 involves Stille coupling, Tamao hydrosilylation, Pd-catalyzed enolate alkenylation, dihydroxylation accompanied by lactonization and a Nozaki–Hiyama–Kishi reaction. Hatakeyama's total synthesis of neooxazolomycin starting from commercially available methyl (S)-3-hydroxy-2-methylpropionate (100; Fig. 31), which can be convert to neooxazolomycin in 27 steps with 2.2% overall yield (based on the same preparation methodology of 103, as reported in Hatakeyama's total synthesis of oxazolomycin A12). Third total synthesis of neooxazolomycin (2) was reported by Kim et al. in 2019.17 Kim's synthesis of 2 using a minimum number of chiral sources. Several chirality propagation processes were employed, including memory of chirality, dynamic kinetic resolution and substrate-controlled asymmetric inductions. Kim's synthesis used as the starting material as well as a source of chirality D-serine (Fig. 31), which was in 24 steps of longest linear sequence transformed to neooxazolomycin in 1.7% overall yield. Additionally, Taylor et al. reported in 2011 formal synthesis of neooxazolomycin.18 Taylor's synthesis utilized Stille cross coupling, a base-promoted enone deconjugation and Julia–Kocienski methodology for the preparation of Kende's key intermediate 91. The longest linear sequence of Taylor's formal synthesis starting from alkene 209 (Fig. 31) and led to neooxazolomycin in 23 steps, in formal overall yield 0.6% (based on the yields of conversion Kende's key intermediate 91 to neooxazolomycin in Kende's total synthesis13).
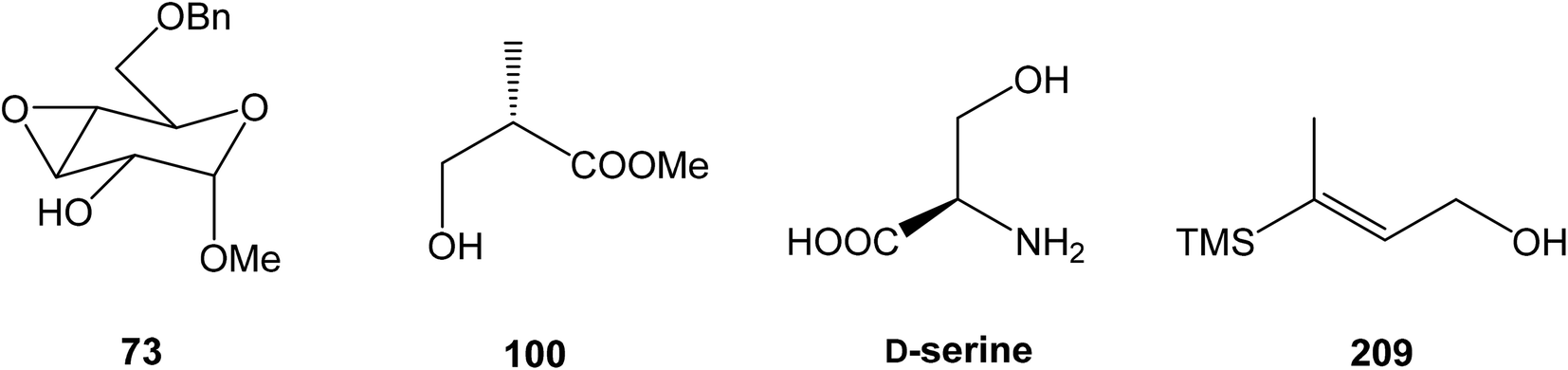 | ||
| Fig. 31 Used starting materials for total and formal syntheses of oxazolomycin family members. | ||
The first total synthesis of oxazolomycin A (1) was reported by Hatakeyama et al. in 2011.12 Hatakeyama's synthesis of 1 included stereo controlled construction of the right-hand fragment by taking advantage of an In(III)-catalyzed Conia-ene type cyclization and the asymmetric synthesis of the left-hand fragment with Cinchona alkaloid-catalyzed cyclocondensation. Hatakeyama's synthesis of oxazolomycin A started from commercially available (S)-Roche ester 100, which after 34 steps of longest linear sequence provided oxazolomycin A in 1.4% overall yield. Notably, Hatakeyama's synthesis of oxazolomycin A involved synthesis of later discovered oxazolomycin A2 (14),10 which represented the penultimate compound in the synthesis. Based on Hatakeyama's synthesis, oxazolomycin A2 could be prepared according to reported procedure12 from 100 in 33 steps, in 1.4-3.5% range of yield. Thus, Hatakeyama's synthesis of oxazolomycin A is to date the only total synthesis, which involves preparation of two different natural members of oxazolomycin family.
Total synthesis of lajollamycin B (11) was reported by Hatakeyama et al. in 2019.15 Hatakeyama's synthesis of 11 involved the construction of nitrodienylstannane and its coupling under Stille conditions with compound prepared form ω-iodoheptadienoic acid and the right-hand fragment 181, previously utilized for synthesis of oxazolomycin A. The longest linear sequence of Hatakeyama's synthesis of lajollamycin B begun from (S)-Roche ester 100 and involved 35 steps, which give to lajollamycin B in 0.3% overall yield (based on the same preparation methodology of 181, as reported in Hatakeyama's total synthesis of oxazolomycin A12). Moreover, Hatakeyama et al. revised the structure of lajollamycin B from initially assigned (10′E)-configuration to (10′Z)-geometry. Hatakeyama's synthesis of lajollamycin B involved synthesis of its (10′E)-isomer, too. The results of above-mentioned syntheses are summarized in following table (Table 10).
| Synthesis of | Longest linear sequence | Overall yield | Published in year | Reported by | Used starting material |
|---|---|---|---|---|---|
| a Based on the same preparation methodology of 103, as reported in Hatakeyama's synthesis of 1.12b ormal synthesis.c Based on the yields of conversion Kende's key intermediate 91 to 2 in Kende's total synthesis of 2.13d Product was not isolated, used as a crude mixture to a following reaction.12e Expected range of overall yield, according to reported procedure.12f Based on the same preparation methodology of 181, as reported in Hatakeyama's synthesis of 1.12 | |||||
| Neooxazolomycin (2) | 22 steps | 1.9% | 1990 | Kende et al.88 | 73 |
| Neooxazolomycin (2) | 27 steps | 2.2%a | 2007 | Hatakeyama et al.16 | 100 |
| Neooxazolomycin (2)b | 23 steps | 0.6%c | 2011 | Taylor et al.18 | 209 |
| Neooxazolomycin (2) | 24 steps | 1.7% | 2019 | Kim et al.17 | D-Serine |
| Oxazolomycin A (1) | 34 steps | 1.4% | 2011 | Hatakeyama et al.12 | 100 |
| Oxazolomycin A2 (14)d | 33 steps | 1.4–3.5%e | 2011 | Hatakeyama et al.12 | 100 |
| Lajollamycin B (11) | 35 steps | 0.3%f | 2019 | Hatakeyama et al.15 | 100 |
Further in 2018, Hatakeyama et al. reported application of γ-lactone-γ-lactam core 113,43 which was formerly utilized in Hatakeyama's synthesis of neooxazolomycin16 and oxazolomycin A12 as a common intermediate for the construction of the right-hand fragment of oxazolomycins bearing methyl group at C16 position, with (16R)- or (16S)-configurated center. Thus, Hatakeyama et al. developed methodology toward total syntheses of 16-methylated members of oxazolomycin family: KSM-2690 B (8), KSM-2690 C (9), lajollamycin (10), lajollamycin C (12) and lajollamycin D (13).43
7 Summary and outlook
This review summarizes previously reported structures and biological properties of oxazolomycin family members. Oxazolomycin's family includes 15 different extraordinary structures usually divided into three fragments (left-hand, middle and right-hand fragment). Left-hand fragment represents either oxazole-triene-amide or tetraene-nitro-hydroxy-amide. Middle fragment is identical for all oxazolomycins and it can be described as pentadienylamine. Right-hand fragment of oxazolomycins represents in general pyroglutamate core, which forms different structural scaffolds including spiro-β-lactone-γ-lactam, bicyclic γ-lactone-γ-lactam or monocyclic γ-lactam. Unique pyroglutamate core is presented in the dimeric structure of bisoxazolomycin (15). The pyroglutamate core of oxazolomycins shows structural similarities with pyroglutamate core of known proteasome inhibitors such as omuralide (27) and salinosporamide A (28). These findings support hypothesis of the similar bioactivity for oxazolomycins with β-lactone-γ-lactam core. In addition, all members of oxazolomycin's family were already tested toward some biological targets. The bioassays revealed widespread biological activity including antibacterial, antiviral, cytotoxic activity and ability of enzyme inhibition. These remarkable biological activities shown, that oxazolomycin members have promising medicinal properties, although a better understanding of the mechanism of action as well as further bioassays are needed. Combination of attractive biological properties with a unique structural arrangement of oxazolomycins represents synthetic challenge for organic chemists. Therefore, different research groups developed the synthetic strategies toward oxazolomycins. In summary, total syntheses of four different oxazolomycin members were achieved. Specifically, four syntheses of neooxazolomycin (2), one synthesis of oxazolomycin A (1) including oxazolomycin A2 (14) and one synthesis of lajollamycin B (11) were reported to date.In future years we assume, that novel oxazolomycin members will be discovered and encompassed to the family. We expect further studies of biological activity, which will prove exact oxazolomycin's mechanism of action for particular targets. The beneficial data could be obtained from comparison studies between bioactivity of oxazolomycin's fragments and bioactivity of the whole structures. Analogously, bioactivity comparison of known pyroglutamate core proteasome inhibitors with oxazolomycins would give better insight to biological effect of oxazolomycins. We suppose there would be increasing number of total syntheses toward oxazolomycins, which have not yet been synthetized. These syntheses will allow faster access to oxazolomycin's preparation in higher yields. It seems likely, that oxazolomycins will attract more attention on fields of organic chemistry and biochemistry.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The present work was supported by Slovak Grant Agency VEGA (no. 1/0375/19), Slovak Research and Development Agency (no. APVV-14-0883) and University of Hradec Kralove (Faculty of Science, no. VT2019-2021).References
- T. Mori, K. Takahashi, M. Kashiwabara, D. Uemura, C. Katayama, S. Iwadare, Y. Shizuri, R. Mitomo, F. Nakano and A. Matsuzaki, Tetrahedron Lett., 1985, 26, 1073–1076 CrossRef CAS.
- K. Takahashi, M. Kawabata, D. Uemura, S. Iwadare, R. Mitomo, F. Nakano and A. Matsuzaki, Tetrahedron Lett., 1985, 26, 1077–1078 CrossRef CAS.
- M. Ogura, H. Nakayama, K. Furihata, A. Shimazu, H. Seto and N. Otake, J. Antibiot., 1985, 38, 669–673 CrossRef CAS.
- Y. Ikeda, S. Kondo, H. Naganawa, S. Hattori, M. Hamada and T. Takeuchi, J. Antibiot., 1991, 44, 453–455 CrossRef CAS.
- G. Ryu, S. Hwang and S.-K. Kim, J. Antibiot., 1997, 50, 1064–1066 CrossRef CAS.
- H. Kanzaki, K. Wada, T. Nitoda and K. Kawazu, Biosci., Biotechnol., Biochem., 1998, 62, 438–442 CrossRef CAS.
- T. Otani, K.-I. Yoshida, H. Kubota, S. Kawai, S. Ito, H. Hori, T. Ishiyama and T. Oki, J. Antibiot., 2000, 53, 1397–1400 CrossRef CAS.
- R. R. Manam, S. Teisan, D. J. White, B. Nicholson, J. Grodberg, S. T. C. Neuteboom, K. S. Lam, D. A. Mosca, G. K. Lloyd and B. C. M. Potts, J. Nat. Prod., 2005, 68, 240–243 CrossRef CAS.
- K. Ko, S.-H. Lee, S.-H. Kim, E.-H. Kim, K.-B. Oh, J. Shin and D.-C. Oh, J. Nat. Prod., 2014, 77, 2099–2104 CrossRef CAS.
- W. Koomsiri, Y. Inahashi, T. Kimura, K. Shiomi, Y. Takahashi, S. Ōmura, A. Thamchaipenet and T. Nakashima, J. Antibiot., 2017, 70, 1142–1145 CrossRef CAS.
- H. M. Jacobs, B. A. Burke and A. Brossi, The Alkaloids: Chemistry and Pharmacology, 1989, ch. 6, vol. 35 Search PubMed.
- K. Eto, M. Yoshino, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Lett., 2011, 13, 5398–5401 CrossRef CAS.
- A. S. Kende, K. Kawamura and R. J. DeVita, J. Am. Chem. Soc., 1990, 112, 4070–4072 CrossRef CAS.
- G. Ryu and S.-K. Kim, J. Antibiot., 1999, 52, 193–197 CrossRef CAS.
- T. Nishimaru, K. Eto, K. Komine, J. Ishihara and S. Hatakeyama, Chem. - Eur. J., 2019, 25, 7927–7934 CrossRef CAS.
- E. O. Onyango, J. Tsurumoto, N. Imai, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2007, 46, 6703–6705 CrossRef CAS.
- J. H. Kim, I. Kim, Y. Song, M. J. Kim and S. Kim, Angew. Chem., Int. Ed., 2019, 58, 11018–11022 CrossRef CAS.
- R. Bastin, J. W. Dale, M. G. Edwards, J. P. N. Papillon, M. R. Webb and R. J. K. Taylor, Tetrahedron, 2011, 67, 10026–10044 CrossRef CAS.
- M. Yoshino, K. Eto, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Biomol. Chem., 2012, 10, 8164–8174 RSC.
- U. Gräfe, H. Kluge and R. Thiericke, Liebigs Ann. Chem., 1992, 1992, 429–432 CrossRef.
- S. C. Wenzel, R. M. Williamson, C. Grünanger, J. Xu, K. Gerth, R. A. Martinez, S. J. Moss, B. J. Carroll, S. Grond, C. J. Unkefer, R. Müller and H. G. Floss, J. Am. Chem. Soc., 2006, 128, 14325–14336 CrossRef CAS.
- P. C. Dorrestein, S. G. Van Lanen, W. Li, C. Zhao, Z. Deng, B. Shen and N. L. Kelleher, J. Am. Chem. Soc., 2006, 128, 10386–10387 CrossRef CAS.
- Y. A. Chan, M. T. Boyne, A. M. Podevels, A. K. Klimowicz, J. Handelsman, N. L. Kelleher and M. G. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14349–14354 CrossRef CAS.
- B. J. Carroll, S. J. Moss, L. Bai, Y. Kato, S. Toelzer, T.-W. Yu and H. G. Floss, J. Am. Chem. Soc., 2002, 124, 4176–4177 CrossRef CAS.
- C. Zhao, J. M. Coughlin, J. Ju, D. Zhu, E. Wendt-Pienkowski, X. Zhou, Z. Wang, B. Shen and Z. Deng, J. Biol. Chem., 2010, 285, 20097–20108 CrossRef CAS.
- C. Zhao, J. Ju, S. D. Christenson, W. C. Smith, D. Song, X. Zhou, B. Shen and Z. Deng, J. Bacteriol., 2006, 188, 4142–4147 CrossRef CAS.
- D. A. Hopwood, T. Kieser, M. Bibb, M. Buttner and K. Chater, Practical Streptomyces Genetics, John Innes Foundation, 2000 Search PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- G. Schoenafinger, N. Schracke, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 7406–7407 CrossRef CAS.
- L. Rouhiainen, L. Paulin, S. Suomalainen, H. Hyytiäinen, W. Buikema, R. Haselkorn and K. Sivonen, Mol. Microbiol., 2000, 37, 156–167 CrossRef CAS.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS.
- A. Tanovic, S. A. Samel, L.-O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS.
- M. He, M. Varoglu and D. H. Sherman, J. Bacteriol., 2000, 182, 2619–2623 CrossRef CAS.
- J. G. Olsen, A. Kadziola, P. von Wettstein-Knowles, M. Siggaard-Andersen, Y. Lindquist and S. Larsen, FEBS Lett., 1999, 460, 46–52 CrossRef CAS.
- D. Song, J. Coughlin, J. Ju, X. Zhou, B. Shen, C. Zhao and Z. Deng, Acta Biochim. Biophys. Sin., 2008, 40, 319–326 CrossRef CAS.
- R. M. Kagan and S. Clarke, Arch. Biochem. Biophys., 1994, 310, 417–427 CrossRef CAS.
- M. Vrljic, H. Sahm and L. Eggeling, Mol. Microbiol., 1996, 22, 815–826 CrossRef CAS.
- H. Savage, G. Montoya, C. Svensson, J. D. Schwenn and I. Sinning, Structure, 1997, 5, 895–906 CrossRef CAS.
- S. Lin, S. G. V. Lanen and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4183–4188 CrossRef CAS.
- K. Zaleta-Rivera, C. Xu, F. Yu, R. A. E. Butchko, R. H. Proctor, M. E. Hidalgo-Lara, A. Raza, P. H. Dussault and L. Du, Biochemistry, 2006, 45, 2561–2569 CrossRef CAS.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- R. Finking and M. A. Marahiel, Annu. Rev. Microbiol., 2004, 58, 453–488 CrossRef CAS.
- K. Eto, J. Ishihara and S. Hatakeyama, Tetrahedron, 2018, 74, 711–719 CrossRef CAS.
- B. C. Potts and K. S. Lam, Mar. Drugs, 2010, 8, 835–880 CrossRef CAS.
- M. Groll, R. Huber and B. C. M. Potts, J. Am. Chem. Soc., 2006, 128, 5136–5141 CrossRef CAS.
- L. R. Dick, A. A. Cruikshank, L. Grenier, F. D. Melandri, S. L. Nunes and R. L. Stein, J. Biol. Chem., 1996, 271, 7273–7276 CrossRef CAS.
- M. Groll, L. Ditzel, J. Löwe, D. Stock, M. Bochtler, H. D. Bartunik and R. Huber, Nature, 1997, 386, 463–471 CrossRef CAS.
- N. Chandan and M. G. Moloney, Org. Biomol. Chem., 2008, 6, 3664–3666 RSC.
- T. J. Hill, P. Kocis and M. G. Moloney, Tetrahedron Lett., 2006, 47, 1461–1463 CrossRef CAS.
- Y.-C. Jeong and M. Moloney, Synlett, 2009, 2009, 2487 CrossRef.
- C. L. Bagwell, M. G. Moloney and M. Yaqoob, Bioorg. Med. Chem. Lett., 2010, 20, 2090–2094 CrossRef CAS.
- C. A. Holloway, C. J. Matthews, Y.-C. Jeong, M. G. Moloney, C. F. Roberts and M. Yaqoob, Chem. Biol. Drug Des., 2011, 78, 229–235 CrossRef CAS.
- M. Anwar and M. G. Moloney, Tetrahedron Lett., 2007, 48, 7259–7262 CrossRef CAS.
- P. Angelov, Y. K. S. Chau, P. J. Fryer, M. G. Moloney, A. L. Thompson and P. C. Trippier, Org. Biomol. Chem., 2012, 10, 3472–3485 RSC.
- C. L. Bagwell, M. G. Moloney and A. L. Thompson, Bioorg. Med. Chem. Lett., 2008, 18, 4081–4086 CrossRef CAS.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC.
- S. Omura, Y. Tanaka, I. Kanaya, M. Shinose and Y. Takahashi, J. Antibiot., 1990, 43, 1034–1036 CrossRef CAS.
- Y. Tanaka, I. Kanaya, Y. Takahashi, M. Shinose, H. Tanaka and S. Omura, J. Antibiot., 1993, 46, 1208–1213 CrossRef CAS.
- M. Kawada, H. Inoue, I. Usami and D. Ikeda, Cancer Sci., 2009, 100, 150–157 CrossRef CAS.
- S. Kawai, G. Kawabata, A. Kobayashi and K. Kawazu, Agric. Biol. Chem., 1989, 53, 1127–1133 CAS.
- M. Ogura, H. Nakayama, K. Furihata, A. Shimazu, H. Seto and N. Otake, Agric. Biol. Chem., 1985, 49, 1909–1910 CAS.
- H. Kanzaki, T. Ichioka, A. Kobayashi and K. Kawazu, Biosci., Biotechnol., Biochem., 1996, 60, 1535–1537 CrossRef CAS.
- E. Tonew, M. Tonew, U. Gräfe and P. Zöpel, Acta Virol., 1992, 36, 166–172 CAS.
- M. Nakamura, H. Honma, M. Kamada, T. Ohno, S. Kunimoto, Y. Ikeda, S. Kondo and T. Takeuchi, J. Antibiot., 1994, 47, 616–618 CrossRef CAS.
- R. L. Willey, D. H. Smith, L. A. Lasky, T. S. Theodore, P. L. Earl, B. Moss, D. J. Capon and M. A. Martin, J. Virol., 1988, 62, 139–147 CrossRef CAS.
- J. Carmichael, W. G. DeGraff, A. F. Gazdar, J. D. Minna and J. B. Mitchell, Cancer Res., 1987, 47, 936–942 CAS.
- A. J. Petit, F. G. Terpstra and F. Miedema, J. Clin. Invest., 1987, 79, 1883–1889 CrossRef CAS.
- Y. Hayakawa, M. Akimoto, A. Ishikawa, M. Izawa and K. Shin-ya, J. Antibiot., 2016, 69, 187–188 CrossRef CAS.
- M. C. Lorenz and G. R. Fink, Nature, 2001, 412, 83–86 CrossRef CAS.
- H.-R. Park, A. Tomida, S. Sato, Y. Tsukumo, J. Yun, T. Yamori, Y. Hayakawa, T. Tsuruo and K. Shin-ya, J. Natl. Cancer Inst., 2004, 96, 1300–1310 CrossRef CAS.
- K. Kawazu, H. Kanzaki, G. Kawabata, S. Kawai and A. Kobayashi, Biosci., Biotechnol., Biochem., 1992, 56, 1382–1385 CrossRef CAS.
- W. Fischetti, K. T. Mak, F. G. Stakem, J. Kim, A. L. Rheingold and R. F. Heck, J. Org. Chem., 1983, 48, 948–955 CrossRef CAS.
- R. D. Stephens and C. E. Castro, J. Org. Chem., 1963, 28, 3313–3315 CrossRef CAS.
- T. Harada and T. Mukaiyama, Chem. Lett., 1982, 11, 161–164 CrossRef.
- A. S. Kende, K. Kawamura and M. J. Orwat, Tetrahedron Lett., 1989, 30, 5821–5824 CrossRef CAS.
- D. A. Evans, T. C. Britton and J. A. Ellman, Tetrahedron Lett., 1987, 28, 6141–6144 CrossRef CAS.
- M. Ohno and M. Okamoto, Org. Synth., 1973, 281 Search PubMed.
- U. Schöllkopf and R. Schröder, Angew. Chem., Int. Ed. Engl., 1971, 10, 333 CrossRef.
- W. J. Scott, G. T. Crisp and J. K. Stille, J. Am. Chem. Soc., 1984, 106, 4630–4632 CrossRef CAS.
- J. K. Stille and B. L. Groh, J. Am. Chem. Soc., 1987, 109, 813–817 CrossRef CAS.
- J. G. Buchanan and R. Fletcher, J. Chem. Soc., 1965, 6316–6323 RSC.
- K. A. Parker and R. E. Babine, Tetrahedron Lett., 1982, 23, 1763–1766 CrossRef CAS.
- J. R. Rasmussen, C. J. Slinger, R. J. Kordish and D. D. Newman-Evans, J. Org. Chem., 1981, 46, 4843–4846 CrossRef CAS.
- F. Dasgupta, P. P. Singh and H. C. Srivastava, Indian J. Chem., 1980, 19, 1056–1059 Search PubMed.
- A. S. Kende and R. J. DeVita, Tetrahedron Lett., 1988, 29, 2521–2524 CrossRef CAS.
- T. Fujisawa, T. Mori and T. Sato, Chem. Lett., 1983, 12, 835–838 CrossRef.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408–7410 CrossRef CAS.
- A. S. Kende and R. J. DeVita, Tetrahedron Lett., 1990, 31, 307–310 CrossRef CAS.
- J. Cabré and A. L. Palomo, Synthesis, 1984, 1984, 413–417 CrossRef.
- Y. Watanabe, WO2003006442A1, 2003.
- C. M. Shafer and T. F. Molinski, J. Org. Chem., 1998, 63, 551–555 CrossRef CAS.
- J. P. Marino and H. N. Nguyen, Tetrahedron Lett., 2003, 44, 7395–7398 CrossRef CAS.
- B. M. Trost and J. P. N. Papillon, J. Am. Chem. Soc., 2004, 126, 13618–13619 CrossRef CAS.
- A. Abiko, O. Moriya, S. A. Filla and S. Masamune, Angew. Chem., 1995, 107, 869–871 CrossRef.
- A. Abiko, O. Moriya, S. A. Filla and S. Masamune, Angew. Chem., Int. Ed. Engl., 1995, 34, 793–795 CrossRef CAS.
- S. E. Denmark and W. Pan, Org. Lett., 2001, 3, 61–64 CrossRef CAS.
- R. Heckendorn, Helv. Chim. Acta, 1990, 73, 1700–1718 CrossRef CAS.
- X. Liu, J. R. Deschamp and J. M. Cook, Org. Lett., 2002, 4, 3339–3342 CrossRef CAS.
- J. S. Panek and P. Liu, J. Am. Chem. Soc., 2000, 122, 11090–11097 CrossRef CAS.
- K. J. Hale, Z. Xiong, L. Wang, S. Manaviazar and R. Mackle, Org. Lett., 2015, 17, 198–201 CrossRef CAS.
- T. N. T. Huynh, P. Retailleau, C. Denhez, K. P. P. Nguyen and D. Guillaume, Org. Biomol. Chem., 2014, 12, 5098–5101 RSC.
- H. C. Brown, J. Chandrasekharan and P. V. Ramachandran, J. Am. Chem. Soc., 1988, 110, 1539–1546 CrossRef CAS.
- J. Uenishi, R. Kawahama, O. Yonemitsu and J. Tsuji, J. Org. Chem., 1998, 63, 8965–8975 CrossRef CAS.
- K. J. Hale, M. Grabski, S. Manaviazar and M. Maczka, Org. Lett., 2014, 16, 1164–1167 CrossRef CAS.
- M. Falorni, S. Conti, G. Giacomelli, S. Cossu and F. Soccolini, Tetrahedron: Asymmetry, 1995, 6, 287–294 CrossRef CAS.
- K. Takeda, A. Akiyama, H. Nakamura, S. Takizawa, Y. Mizuno, H. Takayanagi and Y. Harigaya, Synthesis, 1994, 1994, 1063–1066 CrossRef.
- D. J. Kempf, J. Org. Chem., 1986, 51, 3921–3926 CrossRef CAS.
- A. G. M. Barrett, R. A. E. Carr, S. V. Attwood, G. Richardson and N. D. A. Walshe, J. Org. Chem., 1986, 51, 4840–4856 CrossRef CAS.
- S. Zhang, T. Govender, T. Norström and P. I. Arvidsson, J. Org. Chem., 2005, 70, 6918–6920 CrossRef CAS.
- G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 2001, 1731–1736 CrossRef.
- G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945–7946 CrossRef CAS.
- C. Zhu, X. Shen and S. G. Nelson, J. Am. Chem. Soc., 2004, 126, 5352–5353 CrossRef CAS.
- J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 6358–6360 CrossRef CAS.
- D. Seebach, J. Aebi and D. Wasmuth, Org. Synth., 1985, 63, 109 CrossRef CAS.
- M. R. Webb, M. S. Addie, C. M. Crawforth, J. W. Dale, X. Franci, M. Pizzonero, C. Donald and R. J. K. Taylor, Tetrahedron, 2008, 64, 4778–4791 CrossRef CAS.
- S. P. H. Mee, V. Lee and J. E. Baldwin, Chem. - Eur. J., 2005, 11, 3294–3308 CrossRef CAS.
- K. Takahashi, M. Midori, K. Kawano, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2008, 47, 6244–6246 CrossRef CAS.
- J. García-Fortanet, J. R. Debergh and J. K. De Brabander, Org. Lett., 2005, 7, 685–688 CrossRef.
- G. V. M. Sharma, K. L. Reddy, P. Sree Lakshmi and P. Radha Krishna, Tetrahedron Lett., 2004, 45, 9229–9232 CrossRef CAS.
- D. Sawada and Y. Ito, Tetrahedron Lett., 2001, 42, 2501–2504 CrossRef CAS.
- K. Nishide, S. Ohsugi, T. Miyamoto, K. Kumar and M. Node, Monatsh. Chem., 2004, 135, 189–200 CrossRef CAS.
- J. Cossy, S. BouzBouz and A. H. Hoveyda, J. Organomet. Chem., 2001, 634, 216–221 CrossRef CAS.
- H. Jin, J. Uenishi, W. J. Christ and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 5644–5646 CrossRef CAS.
- K. Takai, M. Tagashira, T. Kuroda, K. Oshima, K. Utimoto and H. Nozaki, J. Am. Chem. Soc., 1986, 108, 6048–6050 CrossRef CAS.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS.
- H. Oda, T. Kobayashi, M. Kosugi and T. Migita, Tetrahedron, 1995, 51, 695–702 CrossRef CAS.
- K. C. Nicolaou, G. Bellavance, M. Buchman and K. K. Pulukuri, J. Am. Chem. Soc., 2017, 139, 15636–15639 CrossRef CAS.
- S. E. Denmark and L. Gomez, Org. Lett., 2001, 3, 2907–2910 CrossRef CAS.
- S. E. Denmark and L. Gomez, J. Org. Chem., 2003, 68, 8015–8024 CrossRef CAS.
- S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed., 2004, 43, 1132–1136 CrossRef CAS.
- J. Ishihara and S. Hatakeyama, Chem. Rec., 2014, 14, 663–677 CrossRef CAS.
- B. K. Senapati, L. Gao, S. I. Lee, G.-S. Hwang and D. H. Ryu, Org. Lett., 2010, 12, 5088–5091 CrossRef CAS.
- R. Bastin, M. Liron and R. Taylor, Synlett, 2008, 2008, 2183–2187 CrossRef.
- P. Bickart, F. W. Carson, J. Jacobus, E. G. Miller and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4869–4876 CrossRef CAS.
- H. S. Schultz, H. B. Freyermuth and S. R. Buc, J. Org. Chem., 1963, 28, 1140–1142 CrossRef CAS.
- M. D. Andrews, A. G. Brewster, K. M. Crapnell, A. J. Ibbett, T. Jones, M. G. Moloney, K. Prout and D. Watkin, J. Chem. Soc., Perkin Trans. 1, 1998, 223–236 RSC.
- S. E. Denmark, K. L. Habermas and G. A. Hite, Helv. Chim. Acta, 1988, 71, 168–194 CrossRef CAS.
- T. Skrydstrup, M. Bénéchie and F. Khuong-Huu, Tetrahedron Lett., 1990, 31, 7145–7148 CrossRef CAS.
- E. J. Corey and G. A. Reichard, J. Am. Chem. Soc., 1992, 114, 10677–10678 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |