
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the synthesis of biodegradable polyesters by sustainable polymerization: lipase-catalyzed polymerization
Ying Liua,
Lijie Songb,
Na Fengc,
Wei Jiang c,
Yongri Jina and
Xuwen Li*a
c,
Yongri Jina and
Xuwen Li*a
aCollege of Chemistry, Jilin University, No. 2699, Qianjin Road, Changchun, Jilin 130012, PR China. E-mail: xwli@jlu.edu.cn
bFirst Clinical Hospital, Jilin Province Academy of Traditional Chinese Medicine, Changchun, 130021, China
cDepartment of Molecular Pathology, Application Center for Precision Medicine, The Second Affiliated Hospital of Zhengzhou University, Academy of Medical Sciences, Zhengzhou, Henan 450052, China
First published on 1st October 2020
Abstract
Over the past three decades, enzymatic polymerization has dramatically developed and gradually broadened as a creative methodology in the construction of polymeric materials with tailor-made structures and properties. Compared with transition metal catalyst polymerizations, enzymatic polymerization is more attractive in the biomedicine field due to the metal-free residue, good biocompatibility, and few by-products. Meanwhile, enzymatic polymerization has far more activity towards macrolides. In this review, the synthesis of lipase-catalyzed polymer materials is systematically summarized, focusing on the synthesis of the complex and well-defined polymers. The enzymatic polyester synthesis was then discussed concerning the different reaction types, including ring-opening polymerization, polycondensation, a combination of ring-opening polymerization with polycondensation, and chemoenzymatic polymerization. Besides, exploration of novel biocatalysts and reaction media was also described, with particular emphasis on the enzymes obtained via immobilization or protein engineering strategies, green solvents, and reactors. Finally, recent developments in catalytic kinetics and mechanistic studies through the use of spectroscopy, mathematics, and computer techniques have been introduced. Besides, we addressed the remaining central issues in enzymatic polymerization and discussed current studies aimed at providing answers.
1. Introduction
With the development of the polymeric materials, the traditional chemical catalyst methods have been unable to meet the requirements of various new applications. In the last three decades, enzymatic polymerization, especially the lipase-catalyzed synthesis of aliphatic polyesters, has flourished as an important synthetic technique aimed specifically for biomedical applications, which has resulted in a diverse range of biodegradable polymers with advanced architecture.1–4 Compared with chemical routes, enzymatic polymerization has developed rapidly and become a creatively synthetic methodology for functional macromolecules,5–7 due to its unique characteristics of mild reaction conditions, high control of enantio-, chemo-, and regioselectivity, and few byproducts. Besides, enzymatic polymerization could also help to avoid problems related to trace residues of metallic catalysts, especially their unfavorable effects on the environment and toxicity in biomedical applications. More importantly, through combining with chemical methods, the diversity and complexity of macromolecules could be further increased, and chemoenzymatic polymerization could even be utilized to synthesize polymeric materials that are difficult to prepare via traditional routes. Meanwhile, lipase-catalyzed transesterification was also used to modify macromolecules by the tandem reaction to obtain new fictionalized macromolecules. Thus, enzymatic polymerization has been regarded as a potential substitute for traditional chemical polymerization, providing a good platform for green and complex polymer synthesis.Polyesters is one of the most fascinating polymeric materials and have gained huge attention because of their unique properties such as biocompatibility and biodegradability. Moreover, the polyesters have been extensive applicated in bioengineering, drug delivery systems and packaging. Several synthetic approaches have been explored in the preparation of polyesters with tailor structure and properties. Among the synthesis method of polyesters, two main modes of the lipase-catalyzed synthesis of aliphatic linear polyesters was included, one is the ring-opening polymerization of lactones, the other is polycondensation of hydroxyl acids or their activated esters (Fig. 1). In previous research of our lab, we successfully synthesized aliphatic polyester using thermophilic lipases and Novozym 435 as catalysts,8–15 more than that we also combined enzymatic ring-opening polymerization with living polymerization techniques for the preparation of well-defined functional copolymers.16–18 In the past five years, the enzymatic reactions have dramatically developed and transformed into newly functionalized macromolecules by using selective reaction characteristics for macromolecular synthesis and macromolecular modification.
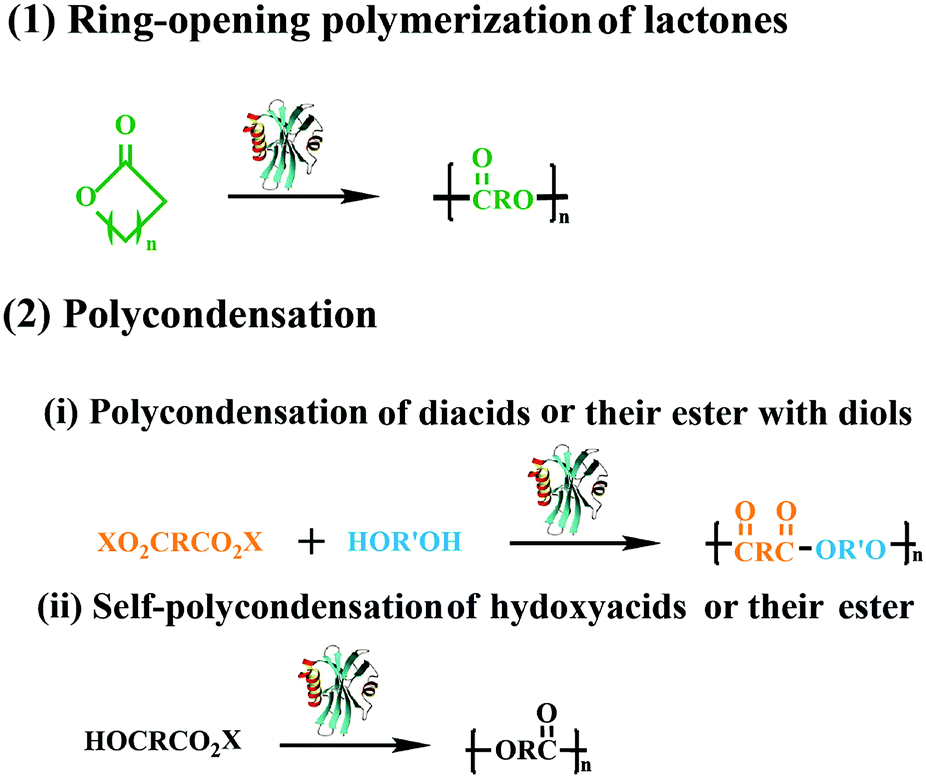 | ||
| Fig. 1 Schematic representation of two types lipase-catalyzed polymerization. | ||
In this review, we aimed at presenting systematically restate the history19 and comprehensively highlight the recent developments in lipase-catalyzed polyester, the exploration of biocatalysts, reaction media and monomers, even concurrent tandem polymerization (CTP) based on lipase-catalyzed polymerization will be presented as a primer to the latest developments. Also, the review provides an in-depth insight into the perspectives and future trends of enzymatic polymerization techniques. In the end, we would close with a future perspective towards the exploring of new monomers and polymerization method.
2. Exploration of the novel sustainable process
Except for commercially available enzyme or traditional organic solvent, new biocatalyst systems, e.g., biocatalysts and green reaction condition have been employed in lipase-catalyzed polyester.2.1 Exploration of biocatalysts
Since 1833, A. Payen and J. F. Persoz were first to discover diastase, which has aroused great interest to scientists in the function of enzymatic catalytic.20 To date, hydrolases, transferases, and oxidoreductases have been mainly used as biocatalysts in vitro enzymatic polymerizations. Lipase is an important member of the hydrolase family and is commonly used to catalyze the hydrolysis of fatty acid esters. It is also normally used as catalysts for esterification and transesterification reactions in an aqueous environment of living systems. So far, hydrolases, transferases, and oxidoreductases have been mainly used as biocatalysts for enzymatic polymerization in vitro. Lipase is an important member of the hydrolase family, which is usually used to catalyze the hydrolysis of fatty acid ester and can also be used as a catalyst for esterification and transesterification in the waterborne environment of life systems. This article focuses on the application of lipase as catalysts to the synthesis of polyester. Lipase-catalyzed polymerization mainly conducts in organic solvents, and it is necessary to optimize the enzyme characteristics in such solvents to achieve their biological function via immobilization or protein engineering. Among the lipases used in enzymatic polymerization, the most widely used enzyme for polyester synthesis is a physically immobilized form of C. Antarctica known as commercially available Novozym 435.21,22 To broaden the scope of biocatalysts, Ulrich Schwaneberg et al. constructed a physical entrapment immobilized Candida Antarctica Lipase B (CALB) using compartmentalized microgels based on poly(glycidol) as support and successfully applied it in the ring-opening polymerization of ε-caprolactone.23 Different with Novozym 435 and free CALB the equilibrium of esterification and hydrolysis depend on the solubility of the enzyme and water concentration rather than monomer concentration. Compared with the free enzyme and Novozym 435, the immobilized enzyme exhibited higher hydrolytic activity, and polymers with weight-average molecular weight (Mw) of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1 and a conversion to the polymeric product of 87 mol% was obtained. Further, to overcome the instability of the anchoring of the enzyme, Gardossi et al. immobilized CALB on an epoxy-functionalized methacrylic resin via covalently and used the enzyme for synthesizing novel bio-based functionalized polyesters by carrying out the solvent-free polycondensation of esters of diethyl adipate and 1,4-butanediol.24 Besides, the present methodology could recover different chemicals for special applications, for instance like emollient esters for cosmetic formulations, from the major issues related to the production on the industrial scale. Besides traditionally immobilized enzymes, an ionic liquid (IL) with different alkyl chain lengths coated Novozym 435 has also been used in polyester synthesis.25 Compared with Novozym 435, the coated catalyst exhibits much superior catalytic activity and cyclicity, and the IL with a longer alkyl chain was favorable for the synthesis of poly(δ-valerolactone) with a higher Mn and monomer conversion.
700 g mol−1 and a conversion to the polymeric product of 87 mol% was obtained. Further, to overcome the instability of the anchoring of the enzyme, Gardossi et al. immobilized CALB on an epoxy-functionalized methacrylic resin via covalently and used the enzyme for synthesizing novel bio-based functionalized polyesters by carrying out the solvent-free polycondensation of esters of diethyl adipate and 1,4-butanediol.24 Besides, the present methodology could recover different chemicals for special applications, for instance like emollient esters for cosmetic formulations, from the major issues related to the production on the industrial scale. Besides traditionally immobilized enzymes, an ionic liquid (IL) with different alkyl chain lengths coated Novozym 435 has also been used in polyester synthesis.25 Compared with Novozym 435, the coated catalyst exhibits much superior catalytic activity and cyclicity, and the IL with a longer alkyl chain was favorable for the synthesis of poly(δ-valerolactone) with a higher Mn and monomer conversion.
Very recently, a new concept named systems biocatalysis has emerged to construct an artificial metabolic pathway in vitro that can also be used to produce poly(ε-caprolactone) (PCL). Current examples are Bornscheuer et al. successfully synthesized poly(ε-caprolactone) by the combination of three different catalytic enzyme activities in a one-pot (Fig. 2).26,27 Beyond that, Liese designed a three-step ADH (alcohol dehydrogenase) – CHMO (cyclohexanone monooxygenase)-lipase cascade reaction for the synthesis of PCL, which was carried out in a fed-batch.28 As far as enzyme consumption is concerned, the fed-batch process makes the enzymatic polymerization more economical. Moreover, the process using covalently immobilized enzymes exhibited enhanced recyclability, favorable stability, and simple separation.
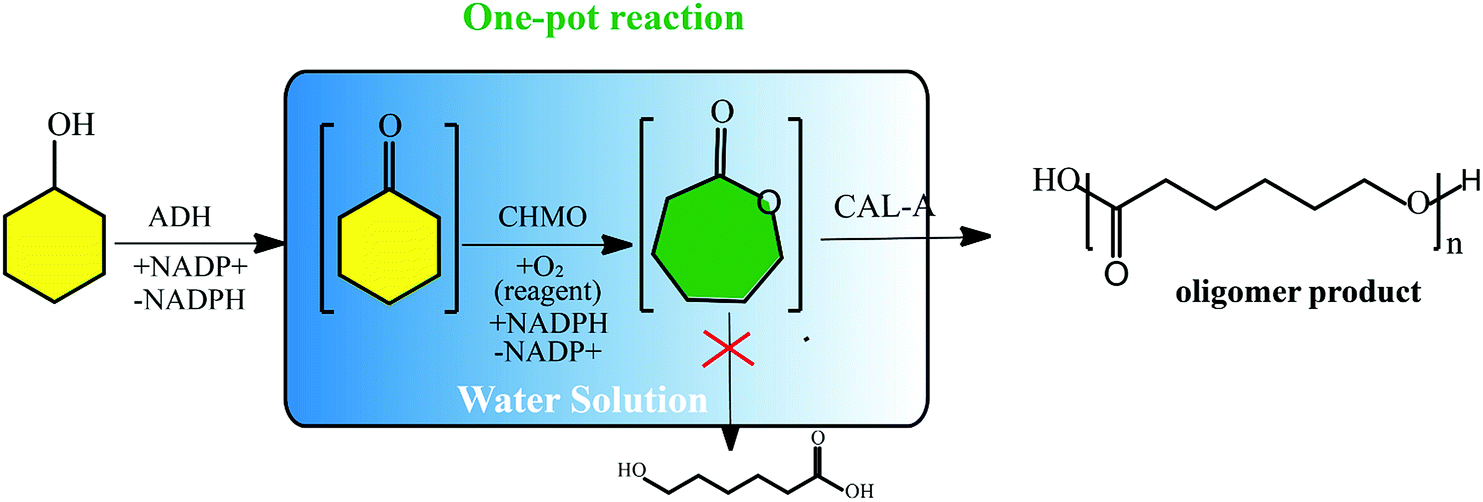 | ||
| Fig. 2 Schematic representation of the synthesis of the oligomer (ε-caprolactone) via an enzyme cascade.28 | ||
2.2 Exploration of green reaction conditions
The objectives of modern polymer chemistry could be partially met by developing a method to replace organic solvents with greener and more sustainable alternative reaction media, e.g., ionic liquids, supercritical carbon dioxide (scCO2) and hydrofluorocarbon solvents (HFCs). scCO2, a novel candidate solvent instead of traditional organic solvents, which shows many advantages in the enzymatic polymerization such as low-cost, environmentally friendly, secure and excellent transport performance. Howdle et al. have been successfully prepared novel green amphiphilic copolymers with terminal functionalization in azelaic acid, 1,6-hexanediol, and PEG in scCO2 by using C. Antarctica lipase B as a catalyst.29 Compared with an organic solvent, the development of scCO2 has many advantages, including mild reaction conditions, without pre-modification of the diacid, renewable diol, and excellent control over the polyester structure and properties. All the constructed green amphiphilic copolymers exhibited rather a low surface tension around 40 mN m−1 and their effect is similar to those of Tween 20 and Pluronic L121, demonstrating that this novel approach certainly provides an alternative method for preparing green biodegradable surfactant. In other words, well-defined structure poly(ω-pentadecalactone) and polycaprolactone eROP were easily synthesized using scCO2 as the reaction medium and Novozym 435 lipase as a catalyst in scCO2.30,31 Beyond that, ionic liquids also have been used as a novel and green reaction media for enzymatic transformation. Yuan et al. utilized Candida Antarctica to catalyst ROP of ε-CL in monocationic ionic liquid [Bmim]PF6 and systematically evaluated the effects of different variables on the enzymatic polymerization.32 The results showed that [Bmim]PF6 could efficiently promote the Mn and yield, as high to 5942 g mol−1 and 85%, respectively. Further, linear and hyperbranched poly-L-lactide with Mn values of above 28000 g mol−1 and 12000 g mol−1 was synthesized in a reaction medium composed of miscible compressed 1,1,1,2-tetrafluoroethane (R-134a) and ionic liquid [C4MIM] [PF6], which were much higher than those using ionic liquid as a medium.33
New sustainable batch reactors, variable-volume view reactors, and packed bed reactors have been designed to overcome the limitations of the traditional batch reactor, such as low operational stability, low product yield within a practical timescale, and complex operation process. For example, microwave energy (MWe) has been used as a cleaning synthetic tool to improve enzymatic polycondensation reactions and pre-polymer formation similar to conventional heating, while bulk phase polycondensations under intense microwave irradiation can quickly deactivate the enzyme.34 Poly(ω-pentadecalactone) has been prepared in a pressurized variable-volume view reactor using various solvents.35 Microreactor based continuous flow chemistry is a green reaction with many advantages such as improved energy efficiency, reaction speed, yield, safety, reliability, scalability, and on-site/on-demand production. Gross et al. constructed an aluminum microreactor in which microchannels were filled with Novozym 435.36 In this enzyme immobilized microreactor, ε-CL polymerization was initiated with benzyl alcohol using toluene as a solvent. The polymerization showed higher efficiency in the microreactor and the apparent rate constant (kapp = 0.027 s−1) was 27-fold larger than that of the batch reactor (kapp = 0.001 s−1).37 Compared with polyesters (Mn = 500–1000 g mol−1) synthesized in the batch system, higher molecular weight (Mn = 1000–2500 g mol−1) and benzyl end group fidelity (0.75 to >0.98) could be obtained in this microreactor.38 Under “dry” or “water-saturated” conditions, the recycling number of the enzyme could as high as 30 times. More importantly, simple and sustainable polymerization techniques could be extended to other enzymatic reaction systems.
3. Synthesis of tailor-made polymeric materials
To date, various polyester materials with tailor-made structures and properties have been successfully constructed through lipase-catalyzed polymerization, including ring-opening polymerization, polycondensation, and chemoenzymatic polymerization.3.1 Ring-opening polymerization
The lipase-catalyzed ROP, which was firstly reported by two independent groups in 1993, has developed rapidly and become an innovative approach to the synthesis of functional polyesters.39–41 Compared to polycondensation reactions, water or alcohol is not produced in the process of enzymatic ring-opening polymerization and thus favorable yields and product molecular weights can readily be obtained. The monomers of enzymatic ring-opening polymerization. However, are still mainly focused on the lactones, lactides, cyclic carbonates, and cyclic depsipeptides. Enzymatic ring-opening polymerization of six-membered carbonate monomer, 2-dimethylaminotrimethylene carbonate (DMATC) was first reported by Zhang et al. and then prepared a water-soluble aliphatic polycarbonate (PDMATC, Mn = 4900 g mol−1) with low cytotoxicity and good biodegradability,42 as shown in Fig. 3. Other experiments to the same effect have led to the extension of lipase-catalyzed ring-opening polymerization to cyclic monomers based on cholic acid, and the obtained functionalized polyesters showed significant lower residue Ru compare with the entropy-driven ring-opening metathesis polymerization (ED-ROMP) catalyzed solely by Grubbs' catalysts.43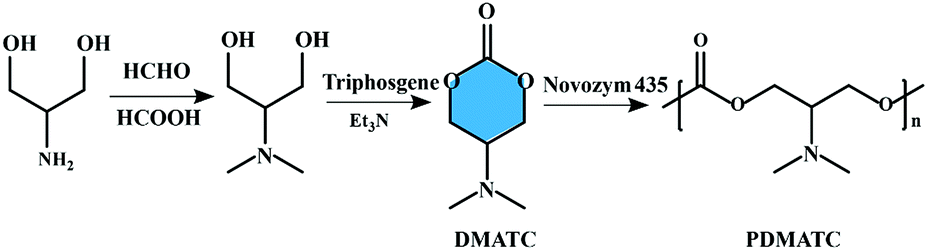 | ||
| Fig. 3 Schematic representation of the synthesis of six-membered carbonate monomer (DMATC) and water-soluble aliphatic polycarbonate (PDMATC).42 | ||
In enzymatic ring-opening polymerization, copolymerization of different monomers and construction of end-functionalized polyesters via the initiator method are two main methods available for altering the structure and further tuning the properties of polyesters. To decrease the crystallinity and hydrophobicity of poly(ω-pentadecalactone), ω-pentadecalactone was copolymerized with other monomers or substituted by macrolactones bearing double bonds, epoxide rings, and amide groups to regulate the poly(ω-pentadecalactone) of composition and properties.44–46 So far, many complex polyesters have been synthesized by enzymatic copolymerization, e.g., poly(b-thioether ester) and ε-caprolactone,47 D,L-lactides with ε-caprolactone,48 ε-caprolactone (ε-CL) and β-lactam,49 and ε-caprolactone (ε-CL) and ε-thiocaprolactone.50 Candida Antarctica lipase B was also used for ring-closure to prepare bile acid-containing macrocycles in a single step with 58% yield and then the products were polymerized using the same enzyme.51 Zhuo et al. first synthesized block copolymerization of cyclic carbonates utilized tetramethylene carbonate and amine-functionalized cyclocarbonate 6,14-dimethyl-1,3,9,11-tetraoxane-6,14-diaza-cyclohexadiene-2,10-dione [(ADMC)2] as monomer by enzymatically copolymerized in one-shot feed.52 Through the copolymerization of these two monomers, aliphatic polycarbonates with the amino group can be prepared. This method could be used to synthesize cationic polyesters with good biodegradability, biocompatibility and particularly pH-responsive that can spontaneously form micellar nanoparticles for use as the tumor-specific drug delivery. Very recently, Lang and co-workers designed a selenic cyclic carbonate dimer-monomer (MSe) through lipase CA as a catalyst for intermolecular cyclization in anhydrous toluene. Then living lipase-catalyzed homopolymerization and copolymerization could be executed easily via solution method using the same lipase CA as catalysts and new class polycarbonates with biodegradable selenide decorated, expected molecular weights and low molecular weight distribution, and tailor-made copolymer compositions were synthesized (Fig. 4). Furthermore, reaction kinetics was also determined in-depth to demonstrate the characteristics of this living ring-opening polymerization.53 The same group further demonstrated a one-pot two-step chemo-enzymatic ring-opening polymerization (ROP) approach for the construction of block copolyester poly(4-benzyl formate piperidine lactone-b-ω-pentadecalactone) (PNPIL-b-PPDL). Afterward, the PNPIL-b-PPDL were then carried out of hydrolysis reaction by acidic and obtained biocompatible cationic polymeric PPIL-b-PPDL with high antibacterial activity against Gram-negative E. coli and Gram-positive S. aureus. Moreover, the antibacterial property and biocompatible could be modulated by changing the ratio of PPIL content. This method could potentially be employed in the synthesis of medical polymeric carriers and implantation materials.54
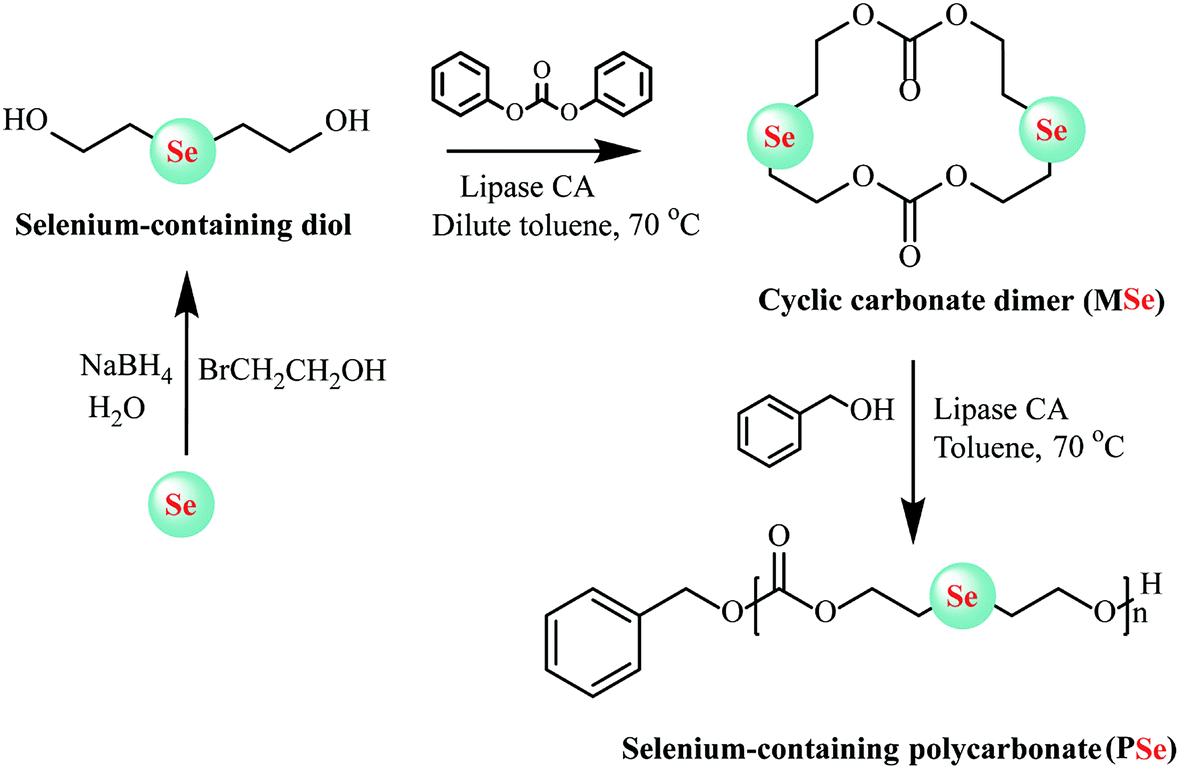 | ||
| Fig. 4 Schematic representation of selenium-containing polycarbonate (PSe) synthesized by enzymatic ring-opening polymerization.53 | ||
Another approaches to cope with the problem is to construct end-functionalized polyesters via the nucleophile (alcohol, amine or sugar), which acts as an initiator for enzymatic polymerization.55 In the past few years, the construction of end-functionalized polyesters through the use of initiators has sprung enzymatic polyester synthesis, examples include the use of the copolymer of poly(ethylene glycol) and δ-decalactone to induce the copolymerization of ω-pentadecalactone to prepare an amphiphilic block copolymer,56 propargyl alcohol, 6-mercapto-1-hexanol or 2-bromoisobutyrate to initiate the polymerization of ε-caprolactone,57,59 OH-terminated macroinitiator folate-conjugated poly(ethylene glycol) or methoxy poly(ethylene glycol) (mPEG) to initiate the copolymerization of dimethyl trimethylene carbonate and 5-methyl-5-allyloxycarbonyl-1,3-dioxane-2-one,60 OH-functional chain transfer agent dodecyl 4-(hydroxymethyl) benzyl carbonotrithioate to initiate the polymerization of ω-pentadecalactone, and 1-phenylethanol to initiate the polymerization of 3-methyl propiolactone.61
In the ring-opening polymerization of 3-methyl propiolactone with 1-phenylethanol as initiator, various catalytic systems have been compared and the in vitro enzyme catalysis approach showed the molar ratio of lactones to alcohols had the most significant effect on the reaction results, considering the over-consumption of lactones by the undesired polymerization so the lactones were excess compared with alcohols. Notably, Fels et al. first proved that CALB was used for enzyme-catalyzed ring-opening polymerization of β-lactam to obtain unbranched poly(β-alanine) and nylon 3.62
3.2 Polycondensation reaction
Enzymatic polycondensation plays pivotal roles in lipase-catalyzed polymerization, considering the more complex and structurally feasible monomers and is known as “enzyme-catalysis breathes new life into polyester condensation polymerizations”.63 According to the monomer, the enzymatic polycondensation reaction mainly classified into two types: (1) simple diacids with diols; (2) hydroxy acids (AB-type monomers). For the self-polycondensation of hydroxyl acids and their activated esters, monomers have so far been mainly limited to those previously reported, e.g., methyl 3-hydroxypropionate64 and methyl epoxyricinoleate.65 In contrast, it has been reported that various dicarboxylic acids and their activated esters are polymerized with diols such as ethyl glycolate with diethyl sebacate and 1,4-butanediol,66 diethyl carbonate with 1,8-octanediol and tris-(hydroxymethyl)ethane,67 dialkyl diester with diol and 2-azido-1,3-propanediol,68 sebacoyl chloride and N-methyldiethanolamine,46 itaconic acid and its esters with diol,69 diols (1,4-butanediol and 1,8-octanediol) and diacids or their derivatives (diethyl succinate, sebacic acid, 1,12-dodecanedioic acid, and 1,14-tetradecanedioic acid),70 cyclotetrasiloxane esters with diols,71 and L-glutamic acid dimethyl ester hydrochloride/2,2-(hydroxymethyl)propyl-tributyl phosphonium bromide with 1,4-butanediol.72 These copolymers have been successfully applied to control drug/gene delivery systems since their structure and characteristics can be readily tuned. Uhrich et al. synthesized a series of biodegradable polyester enzymatic copolymerization of malic acid sugar, ibuprofen and linear aliphatic diol (1,3-propanediol, 1,5-pentanediol, or 1,8-octanediol).73 These polymers all exhibited sustained ibuprofen release and release rate depending on the chain of hydrophobic diols. This technique could be further extended to other biomedical fields, such as amphiphilic micellization, targeted drug-delivery systems, and DNA transfection.The bio-based monomer has been encouraged over the past years as a potential alternative to partially meet future environmental requirements. Recently, various bio-based polyesters have been readily prepared by enzymatic polycondensation, e.g., succinate-based aliphatic polyesters,74 1,4:3,6-dianhydrohexitol-based polyesters,75 bio-based saturated aliphatic polyesters and itaconate-based unsaturated aliphatic polyesters,76 sorbitol-derived aliphatic polyesters,77 bio-based furan polyesters78,79 and sugar-derived diol-based polyesters, etc80,81. Martinelle et al. have utilized an epoxy-functional ω-hydroxy-fatty acid (EFA) extracted from birch bark for the synthesis of three different EFA-based telechelic oligomers with targeted molecular weight and high monomer conversion containing maleimide, methacrylate or oxetane as end-groups through enzymatic polycondensation.82 Avérous et al. synthesized fully bio-based poly(3-hydroxybutyrate-co-butylene succinate) via two different enzyme-catalyzed pathways (one and two-step processes) with poly[(R)-3-hydroxybutyrate] (PHB) based diol oligomers 1,4-butanediol and diethyl succinate (DES) (Fig. 5) and showed that the products obtained from the two-step process have higher high molar masses (Mn up to 18000 g mol−1).83 Also, the influence of the ester/hydroxyl functionality ratio, catalyst amount, PHB-diol oligomer chain length, hydroxybutyrate, and butylene succinate contents, and the nature of the solvent also were demonstrated. Polyamides, also known as nylons, are widely used polymers in daily and industrial applications like high-performance materials with many merits such as high mechanical strength, good dimensional stability, and superior processing characteristics. In recent years, polyamides have been successfully prepared base on bio-based monomer via enzymatic polycondensation. Loos et al. presented enzymatic polymerizations of polyamides by diethyl sebacate and aliphatic diamines (C4, C6, and C8).84 In the same group, the enzyme-catalyzed synthesis of furan aliphatic polyamides was also studied using dimethyl furan-2,5-dicarboxylate and 1,8-ODA as starting building blocks, and polymers with high Mw up to 54000 g mol−1 are much higher than those obtained from melt polycondensation at elevated temperatures.85,86 Accessible bio-monomers were performed for oligomerization using Novozym 435 as a catalyst to synthesized the furan-based hydrophilic and hydrophobic oligoesters.87 After then, the hydrophilic oligoesters were further heated through the intermolecular Diels–Alder reaction and the viscous polymer solution was obtained as well, more importantly, the self-assembly-assisted polymerization of oligoesters are formed a fibrous structure for the first time and extend a new method in the field of green polymer chemistry. Xia et al. used lipase catalysis to prepared a new class of chiral polyesters with high Tm and high crystallinity, D- and L-poly(aspartate-octanediol), containing a long-chain fatty alcohol backbone and readily available amino acids. Through DSC and XRD, it was confirmed that isotactic D- and L-poly(N-Cbz-Asp-octanediol) esters were constructed in such a way that the crystallinity and Tm value were increased by about 20 °C.88 The low polyester synthesized by Muthusamy et al. using furan-based bifunctional monomers monomer and Novozym 435 as biocatalyst and could be self-assemble into a viscous solution, which was further heated to form the polymer through the intermolecular Diels–Alder reaction. Hydrophilic oligomers were easy to self-assemble by hydrogen bond and van der Waals interaction, while hydrophobic oligomers presented amorphous structure which could be obtained by SAXD results. The morphology analysis of the polymer showed that the fiber structure was formed by rheology and SEM analysis.89
 | ||
| Fig. 5 Schematic of bio-based poly(3-hydroxybutyrate-co-butylene succinate) synthesized by enzymatic polycondensation via a one-step or two-step Process.83 | ||
3.3 Combination of ring-opening polymerization and polycondensation
Previous works have shown that lipase-catalyzed polycondensation and ring-opening polymerization are carried out through similar reaction intermediates. Therefore, it is possible to synthesize complex polyesters by a combination of eROP and polycondensation, which were difficult to obtain through any of them.90 Afterward, a series of monomers have been utilized in this tandem enzymatic polymerization (Fig. 6), e.g., diethyl succinate, 1,4-butanediol and p-dioxanone,91 L-lactide (LLA) with diesters and diols,92 ω-pentadecalactone (PDL) with diethyl carbonate (DEC), and 1,4-butanediol,93 lactone with disulfide-containing diester and diol.94 Among this synthesized polyester, a new biodegradable terpolymerization poly(amine-co-esters) formed synthesized by Jiang et al. through combined ring-opening and condensation copolymerization of the lactone with dialkyl diester and amino-substituted diols monomers particularly noteworthy.95–100 The terpolymers with minimal toxicity both in vitro and in vivo could be used for gene delivery and their high molecular weight and increased hydrophobicity could compensate for their low charge density. The same group synthesized a series of poly(amine-co-ester-co-orthoester) with acid sensitivity and redox-responsive, these polyesters have been proved to be gene delivery and nanocarriers for doxorubicin delivery.93,98,101 Similarly, Liu et al. developed the terpolymerization cationic poly(amine-co-ester) for DNA condensation with PEG shell for nanoparticle stabilization and poly(glutamic acid) and mTAT (a cell-penetrating peptide) for accelerating cellular uptake, and a nuclear localization signal peptide for enhanced intracellular transport of DNA to the nucleus.102 Such an approach presents a more sustainable and environmentally friendly synthesis and could also be used for controlled drug release and gene delivery.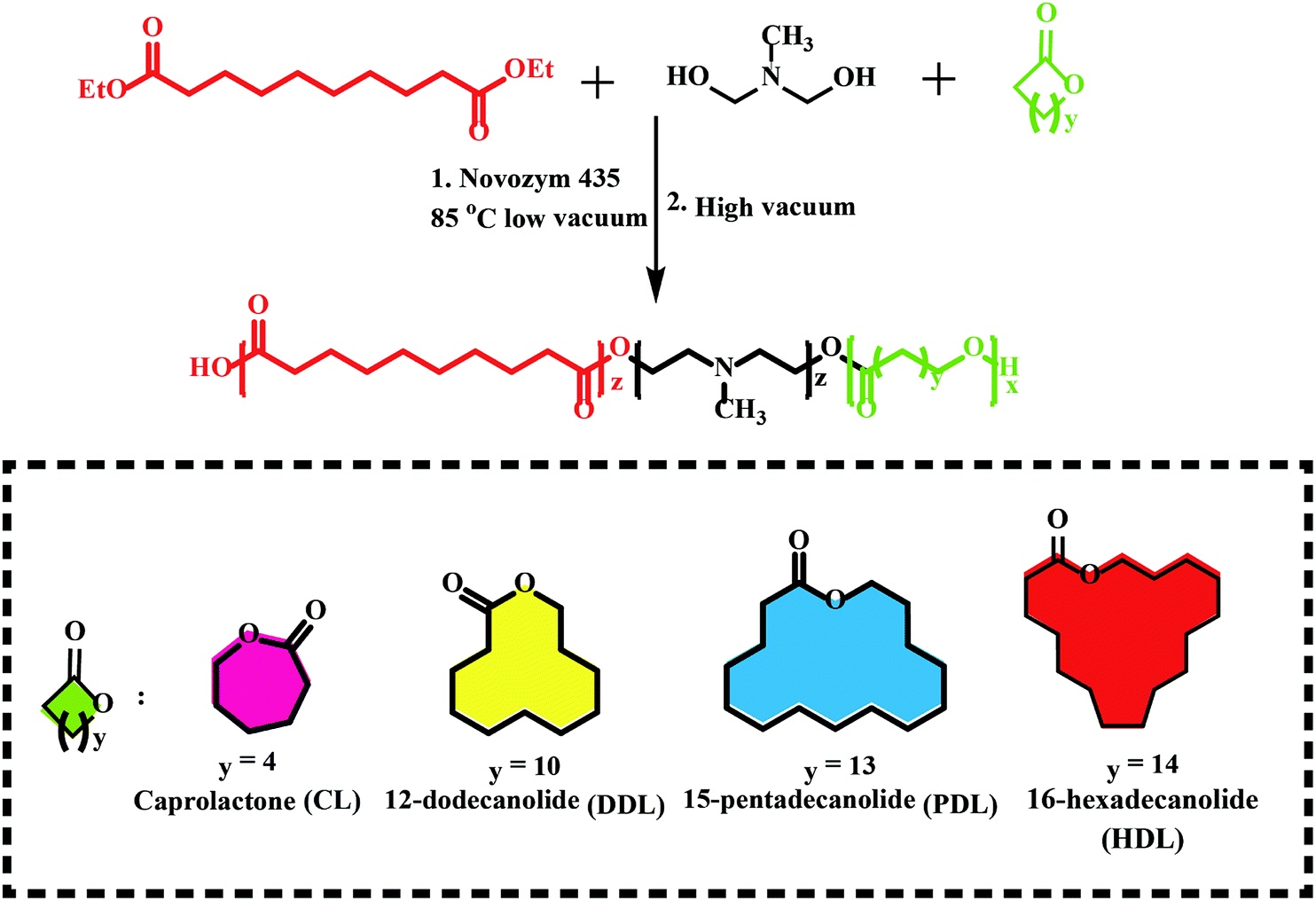 | ||
| Fig. 6 Schematic of a new biodegradable terpolymerization poly(amine-co-esters) synthesized by a combination of eROP and polycondensation.95 | ||
3.4 Chemoenzymatic polymerization
Furthermore, lipase-catalyzed polymerization is also used to tandem with chemical-mediate reactions and represented a new polymerization methodology based on multiple reactions sequentially or simultaneously in one-pot to achieve polymers with unique properties and functionalities represents a sophisticated green synthesis strategy for improving the synthetic efficiency and economic benefits. Recently, several research groups have constructed many multicomponent polymerization systems to prepare functional copolymers or optically active polymers using techniques such as click-chemoenzymatic-ATRP,103 chemoenzymatic monomer transformation with ATRP,104 light-controlled polymerization (LCP)105 or reversible addition–fragmentation chain transfer (RAFT),106–108 enzymatic ring-opening polymerization (eROP) with ATRP,58 enzymatic polycondensation with thiol–ene chemistry, enzymatic ring-opening polymerization (eROP).109 In our previous research, block copolymers were also synthesized through a one-pot polymerization by tandem enzymatic ring-opening polymerization (eROP) and ring-opening metathesis polymerization (ROMP) (Fig. 7). Moreover, a reasonable mechanism for the single-step tandem polymerization was also elucidated through kinetic analysis and structural analysis.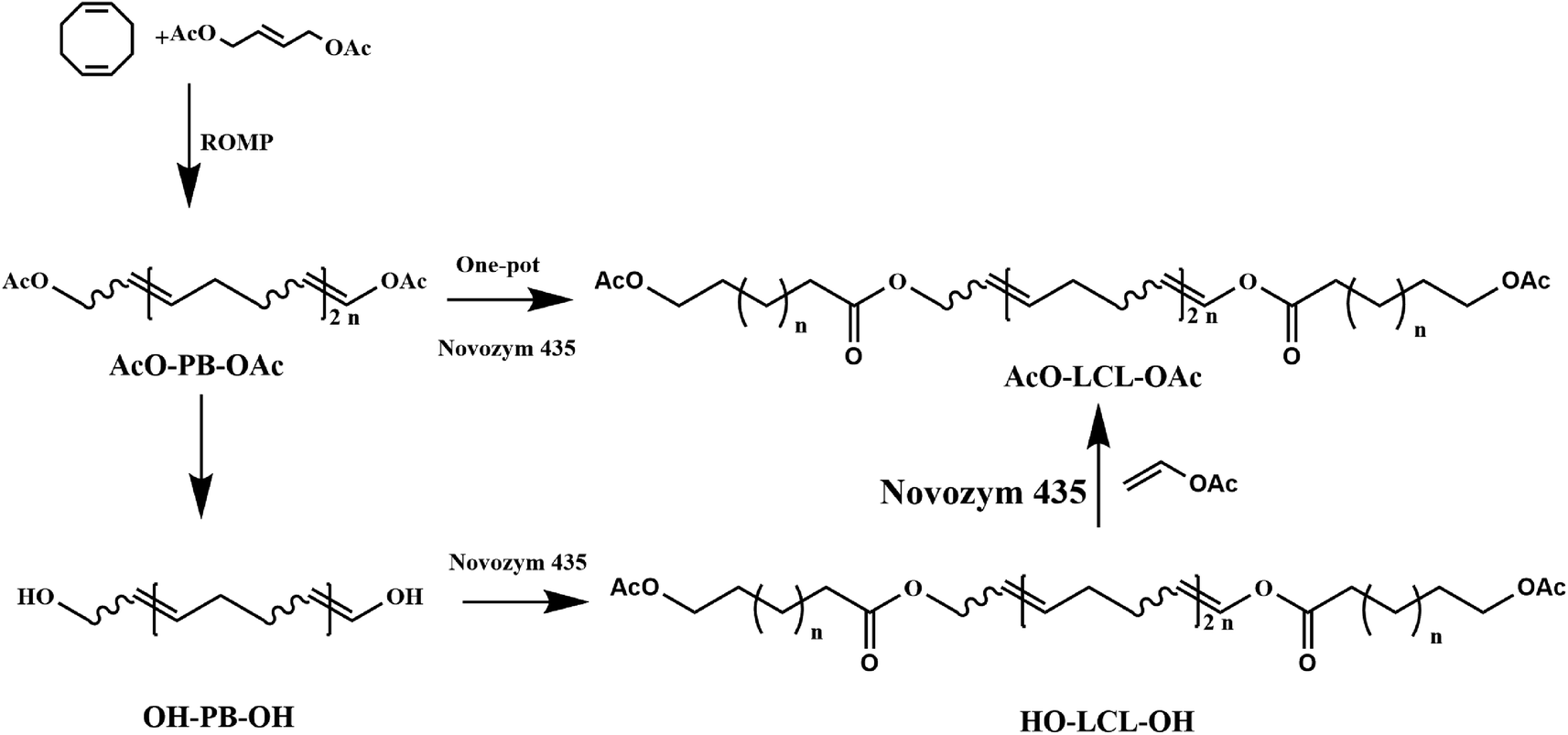 | ||
| Fig. 7 Synthesis of block copolymers by tandem ROMP and eROP. | ||
Besides eROP-based chemoenzymatic polymerization, Matt et al. reported a combination of highly regioselective biocatalytic approach and radical polymerization to constructed high-Tg polymer using isosorbide and monomethacrylated as monomers. Moreover, the experimental results showed that the position of methacrylate group had no significant influence on the polymerization yield and Mn value.110
In brief, the aforementioned examples demonstrate that these tandem polymerization systems could be used to construct polymer materials with desired properties and structures. More than that, the combination of chemical polymerization and enzymatic polymerization would help to dramatically improve the availability of complex macromolecules and even expand the range of polymers.
4. Reaction kinetics and mechanistic analysis
The kinetics of enzymatic polymerization has been studied in pioneering works and are generally expounded as a monomer-activated process.111 Although the enzymatic polymerization stage and the final product have been quantified in detail, most of the latest studies have only focused on the obvious reaction steps, and thus the reaction mechanism still needs to conduct an in-depth study. Profit from the using of novel kinetic analysis, computer-assisted molecular modeling,112,113 advanced spectroscopy technology114 and QM/MM methods e.g. ref. 115, the kinetics and mechanism of enzymatic polymerization have obtained a deeper insight. Puskas et al. conducted the lipase-catalyzed polycondensation of divinyl adipate (DVA) with tetraethylene glycol (TEG) and constructed both symmetric and asymmetric with narrow PDI via modulating of concentrations and reaction times (Fig. 8). Reaction sequences in the transesterification of DVA with TEG were detail analyzed by NMR spectra and MALDI-TOF mass spectrometry.116 The minimum conditions for the synthesis of symmetric and asymmetric telechelic TEGs are summarized in this report. In other words, at DVA/TEG = 1.5 molar ratio only symmetric telechelic TEGs could be detected after 20 minutes, at DVA/TEG = 3 only asymmetric telechelic TEGs were detected after 5 minutes. Zelisko et al. summarized the chain length selectivity for a series of acyl donors by lipase B from Candida Antarctica and CALB to accept aliphatic diesters of C4, C6, and C12 chain lengths equally.117 The introduction of a carbon–carbon double bond into the C4 esters dramatically lowered the rate constant associated with polymerization highlighting the role of geometry in catalysis; fumarate esters were polymerized at a reduced rate compared to the succinate esters, while the maleate esters were not polymerized above 5% for 24 h.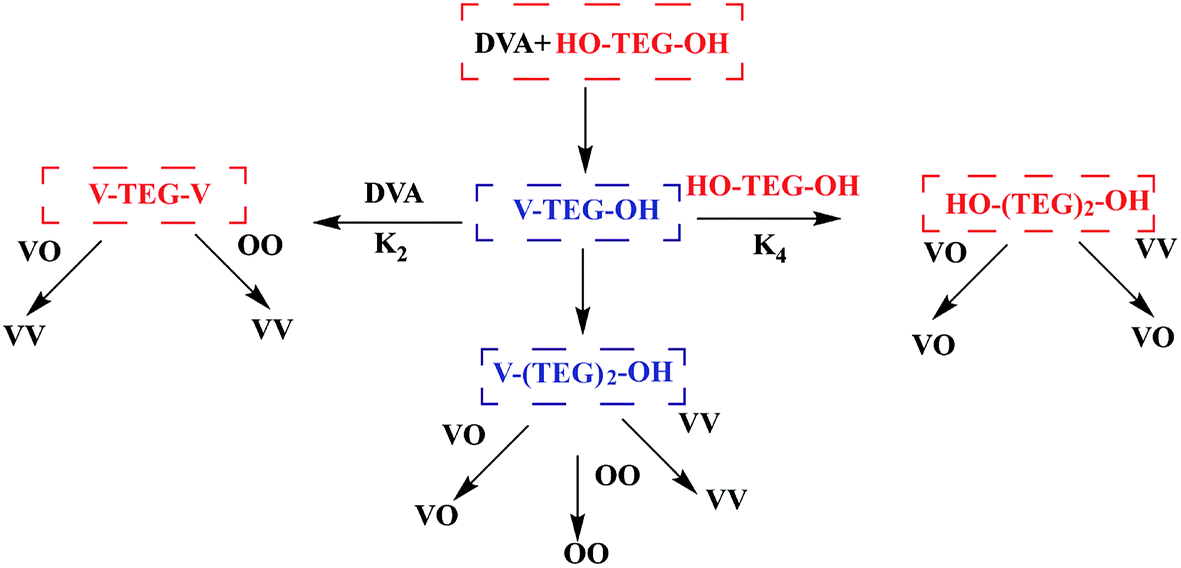 | ||
| Fig. 8 Scheme of reaction sequences in the transesterification of DVA with TEG.113 | ||
Avérous et al. demonstrated an aprotic amino TEA-assisted enzymatic ring-opening polymerization of lactide stereoisomers, indicating that this mixed system leads to a great improvement in the kinetics (five to six times faster reaction) and variation of slight molar mass.118 Furthermore, they proposed five possible mechanisms for the eROP activation by TEA: (1) a nucleophile activation of lactide, (2) basic activation of lipases, (3) basic activation of oligomers chain-end, (4) the epimerization of lactide and (5) nucleophile activation of esters bond in the oligomer backbone. Results seem to attest for the occurrence of more than three of these activation ways: the epimerization, the chain-end activation and the activation of the ester bond in the backbone of the chain.
Gardossi et al. analyzed the feasibility of enzymatic polycondensation of diethyl itaconate for the first time and provided a comprehensive solution embracing both the formulation of the biocatalyst, the reaction conditions and the choice of the comonomers.69 Computational docking was used to disclose the structural factors responsible for the low reactivity of dimethyl itaconate and to identify possible solutions. The computational and experimental data show that the bulky and rigid CHDM is a promising candidate for achieving a better elongation of dimethyl itaconate (DMI) polyesters.
Immobilized Candida antarctica lipase B (CALB) also was used to synthesis of renewable polyamides with high molecular weights and amine end-capped oligoamides.119 Based on the Yet Another Scientific Artificial Reality Application (YASARA), Finnveden et al. built a model to confirm the binding position of the two MO-cys molecules in the enzyme. Polymeric materials obtained by lipase-catalyzed ROPs of benzyl malolactonate (MLABe) play an important role in the biomedical applications. Casajus et al. conducted lipase-catalyzed ROPs of MLABe and obtained monobenzyl fumarate group terminated poly(benzyl malate) (PMLABe) in presence of the lipase/acyltransferase CpLip2 and its serine knockout. To further understanding the enzyme–substrate interactions and mechanism of lipase-catalyzed MLABe, Casajus et al. constructed a molecular modeling with a DFR quantum-chemical calculations, indicating that the catalytic histidine plays an important role in the polymerization of cyclic lactones.120
We firmly believe that continuously updated theories and technologies in the near future could provide reliable tools for lipase-catalyzed polymerization, mainly used to produce polymer materials with specific structures and properties, and provides a greener strategy for polyester synthesis.
5. Conclusion
Overall, we systematically summarize the main contributions of lipase-catalyzed polyester synthesis during the past five years, the technique has been largely studied for a wide range of applications and has made significant progress from the exploration of biocatalysts, green and sustainable reaction conditions, novel polymerization method to more in-depth catalytic kinetics and mechanism research. Concerning the limitations of cost and catalytic activity of enzymes, enzymatic polymerization is still mainly used in laboratory scale and only a small part was employed for industrial production so far. Recent methods have focused on overcoming the problems by developing novel biocatalysts with tailor-made catalytic profiles. More than that, with the development of enzymatic synthetic biology and protein engineering techniques, it has made an important contribution to the fields of lipase-catalyzed polymerization, even and green polymer synthesis.121 We have reasons to believe that promising methodologies and technologies will establish enzymatic polymerization as a versatile tool for constructing customized special structures and polymer properties.Funding
This research was funded by National Science Foundation of China (Grant Nos. 81901882).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful to Prof. Quanshun Li for guidance in the investigation, validation, and writing-original draft preparation.References
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353, DOI:10.1021/cr900165z.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413, DOI:10.1021/acs.chemrev.5b00472.
- E. Champagne, S. Strandman and X.-X. Zhu, Macromol. Rapid Commun., 2016, 37, 1986–2004, DOI:10.1002/marc.201600494.
- B. Yeniad, H. Naik and A. Heise, Lipases in Polymer Chemistry, Biofunctionalization of Polymers and their Applications, ed. G. S. Nyanhongo, W. Steiner and G. Gübitz, Springer Berlin Heidelberg, 2010, pp. 69–95, DOI:10.1007/10_2010_90.
- S. Kobayashi, Macromol. Rapid Commun., 2009, 30, 237–266, DOI:10.1002/marc.200800690.
- Y. Yang, Y. Yu, Y. Zhang, C. Liu, W. Shi and Q. Li, Process Biochem., 2011, 46, 1900–1908, DOI:10.1016/j.procbio.2011.07.016.
- J. Zhang, H. Shi, D. Wu, Z. Xing, A. Zhang, Y. Yang and Q. Li, Process Biochem., 2014, 49, 797–806, DOI:10.1016/j.procbio.2014.02.006.
- J. Ma, Q. Li, B. Song, D. Liu, B. Zheng, Z. Zhang and Y. Feng, J. Mol. Catal. B: Enzym., 2009, 56, 151–157, DOI:10.1016/j.molcatb.2008.03.012.
- Q. Li, G. Li, S. Yu, Z. Zhang, F. Ma and Y. Feng, Process Biochem., 2011, 46, 253–257, DOI:10.1016/j.procbio.2010.08.019.
- Q. Li, G. Li, F. Ma, Z. Zhang, B. Zheng and Y. Feng, Process Biochem., 2011, 46, 477–481, DOI:10.1016/j.procbio.2010.09.020.
- Q. Li, G. Li and W. Shi, J. Controlled Release, 2011, 152, 221–223, DOI:10.1016/j.jconrel.2011.09.023.
- G. Li and Q. Li, Biotechnol. Bioprocess Eng., 2011, 16, 1201–1207, DOI:10.1007/s12257-011-0260-y.
- H. Cao, H. Han, G. Li, J. Yang, L. Zhang, Y. Yang, X. Fang and Q. Li, Int. J. Mol. Sci., 2012, 13, 12232–12241, DOI:10.3390/ijms131012232.
- Y. Sun, Y. Yang, C. Wang, J. Liu, W. Shi, X. Zhu, L. Lu and Q. Li, Appl. Biochem. Biotechnol., 2013, 170, 399–405, DOI:10.1007/s12010-013-0152-z.
- Y. Yu, D. Wu, C. Liu, Z. Zhao, Y. Yang and Q. Li, Process Biochem., 2012, 47, 1027–1036, DOI:10.1016/j.procbio.2012.04.006.
- S. Xiang, Q. Zhang, G. Zhang, W. Jiang, Y. Wang, H. Zhou, Q. Li and J. Tang, Biomacromolecules, 2014, 15, 3112–3118, DOI:10.1021/bm500723k.
- W. Jiang, N. An, Q. Zhang, Z. Bai, H. Han, X. Li, Q. Li and J. Tang, Macromol. Chem. Phys., 2015, 216, 2107–2114, DOI:10.1002/macp.201500313.
- J. Chen, W. Jiang, H. Han, J. Yang, W. Chen, Y. Wang, J. Tang and Q. Li, ACS Macro Lett., 2017, 6, 523–528, DOI:10.1021/acsmacrolett.7b00211.
- A. Idris and A. Bukhari, Biotechnol. Adv., 2012, 30, 550–563, DOI:10.1016/j.biotechadv.2011.10.002.
- A. Payen and J. F. Persoz, Ann. Chim. Phys., 1833, 53, 73–92 Search PubMed.
- N. Miletić, A. Nastasović and K. Loos, Bioresour. Technol., 2012, 115, 126–135, DOI:10.1016/j.biortech.2011.11.054.
- L. Mespouille, O. Coulembier, M. Kawalec, A. P. Dove and P. Dubois, Prog. Polym. Sci., 2014, 39, 1144–1164, DOI:10.1016/j.progpolymsci.2014.02.003.
- S. Engel, H. Höck, M. Bocola, H. Keul, U. Schwaneberg and M. Möller, Polymers, 2016, 8, 372, DOI:10.3390/polym8100372.
- A. Pellis, L. Corici, L. Sinigoi, N. D'Amelio, D. Fattor, V. Ferrario, C. Ebert and L. Gardossi, Green Chem., 2015, 17, 1756–1766, 10.1039/C4GC02289K.
- Y. He, J. J. Li, Y. K. Luo, F. Song, X. L. Wang and Y. Z. Wang, RSC Adv., 2015, 5, 68276–68282, 10.1039/C5RA10227H.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Gröger, A. Liese, H.-G. Herz and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2015, 54, 2784–2787, DOI:10.1002/anie.201410633.
- S. Schmidt, H. C. Büchsenschütz, C. Scherkus, A. Liese, H. Gröger and U. T. Bornscheuer, ChemCatChem, 2015, 7, 3951–3955, DOI:10.1002/cctc.201500823.
- C. Scherkus, S. Schmidt, U. T. Bornscheuerm, H. Gröger, S. Kara and A. Liese, ChemCatChem, 2016, 8, 3446–3452, DOI:10.1002/anie.201410633.
- S. Curia and S. M. Howdle, Polym. Chem., 2016, 7, 2130–2142, 10.1039/C6PY00066E.
- A. E. Polloni, J. G. Veneral, E. A. Rebelatto, D. de Oliveira, J. V. Oliveira, P. H. H. Araújo and C. Sayer, J. Supercrit. Fluids, 2017, 119, 221–228, DOI:10.1016/j.supflu.2016.09.019.
- S. R. R. Comim, J. G. Veneral, D. de Oliveira, S. R. S. Ferreira and J. V. Oliveira, J. Supercrit. Fluids, 2015, 96, 334–348, DOI:10.1016/j.supflu.2014.07.004.
- J. Yuan, Y. Dai, Y. Yu, P. Wang, Q. Wang and X. Fan, Eng. Life Sci., 2016, 16, 371–378, DOI:10.1002/elsc.201500081.
- M. Mena, K. Shirai, A. Tecante, E. Bárzana and M. Gimeno, J. Supercrit. Fluids, 2015, 103, 77–82, DOI:10.1016/j.supflu.2015.04.024.
- A. Pellis, G. M. Guebitz and T. J. Farmer, Molecules, 2016, 21, 1245, DOI:10.3390/molecules21091245.
- A. E. Polloni, E. A. Rebelatto, J. G. Veneral, D. de Oliveira, J. V. Oliveira, P. H. H. Araújo and C. Sayer, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1219–1227, DOI:10.1002/pola.28486.
- S. Kundu, A. S. Bhangale, W. E. Wallace, K. M. Flynn, C. M. Guttman, R. A. Gross and K. L. Beers, J. Am. Chem. Soc., 2011, 133, 6006–6011, DOI:10.1021/ja111346c.
- A. S. Bhangale, K. L. Beers and R. A. Gross, Macromolecules, 2012, 45, 7000–7008, DOI:10.1021/ma301178k.
- K. J. Thurecht, A. Heise, M. de Geus, S. Villarroya, J. Zhou, M. F. Wyatt and S. M. Howdle, Macromolecules, 2006, 39, 7967, DOI:10.1021/ma061310q.
- H. Uyama and S. Kobayashi, Chem. Lett., 1993, 22, 1149–1150, DOI:10.1246/cl.1993.1149.
- H. Uyama, K. Takeya and S. Kobayashi, Proc. Jpn. Acad., Ser. B, 1993, 69, 203–207 CrossRef CAS.
- D. Knani, A. L. Gutman and D. H. Kohn, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1221–1232, DOI:10.1002/pola.1993.080310518.
- X. Zhang, M. Cai, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2012, 33, 693–697, DOI:10.1002/marc.201100765.
- S. Strandman, I. H. Tsai, R. Lortie and X. X. Zhu, Polym. Chem., 2013, 4, 4312–4316, 10.1039/c3py00651d.
- J. Liu, Z. Jiang, S. Zhang, C. Liu, R. A. Gross, T. R. Kyriakides and W. M. Saltzman, Biomaterials, 2011, 32, 6646–6654, DOI:10.1016/j.biomaterials.2011.05.046.
- I. van der Meulen, Y. Li, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 837–843, DOI:10.1021/bm200084y.
- C. Vaida, H. Keul and M. Moeller, Green Chem., 2011, 13, 889–899, 10.1039/C1GC15044H.
- W. X. Wu, L. Qu, B. Y. Liu, W. W. Zhang, N. Wang and X. Q. Yu, Polymer, 2015, 59, 187–193, DOI:10.1016/j.polymer.2015.01.002.
- H. Ö. Düşkünkorur, A. Bégué, E. Pollet, V. Phalip, Y. Güvenilir and L. Avérous, J. Mol. Catal. B: Enzym., 2015, 115, 20–28, DOI:10.1016/j.molcatb.2015.01.011.
- E. Stavila, G. O. R. Alberda van Ekenstein, A. J. J. Woortman and K. Loos, Biomacromolecules, 2013, 15, 234–241, DOI:10.1021/bm401514k.
- S. W. Duchiron, E. Pollet, S. Givry and L. Avérous, Eur. Polym. J., 2017, 87, 147–158, DOI:10.1016/j.eurpolymj.2016.12.024.
- É. Champagne, N. Lévaray and X. X. Zhu, ACS Sustainable Chem. Eng., 2016, 5, 689–695, DOI:10.1021/acssuschemeng.6b02043.
- H. F. Wang, H. Z. Jia, Y. F. Chu, J. Feng, X. Z. Zhang and R. X. Zhuo, Macromol. Biosci., 2014, 14, 526–536, DOI:10.1002/mabi.201300414.
- C. Wei, Y. Xu, B. Yan, J. Hou, Z. Du and M. Lang, ACS Macro Lett., 2018, 7, 336–340, DOI:10.1021/acsmacrolett.8b00039.
- Y. Xiao, J. Pan, D. Wang, A. Heise and M. Lang, Biomacromolecules, 2018, 19, 2673–2681, DOI:10.1021/acs.biomac.8b00296.
- H. Uyama, S. Suda and S. Kobayashi, Acta Polym., 1998, 49, 700–703 CrossRef CAS.
- K. K. Bansal, D. Kakde, L. Purdie, D. J. Irvine, S. M. Howdle, G. Mantovani and C. Alexander, Polym. Chem., 2015, 6, 7196–7210, 10.1039/C5PY01203A.
- M. Castano, J. Zheng, J. E. Puskas and M. L. Becker, Polym. Chem., 2014, 5, 1891–1896, 10.1039/C3PY01536J.
- N. Zhu, Z. L. Zhang, W. He, X. C. Geng, Z. Fang, X. Li, Z. J. Li and K. Guo, Chin. Chem. Lett., 2015, 26, 361–364, DOI:10.1016/j.cclet.2014.11.016.
- H. Zhou, W. Jiang, N. An, Q. Zhang, S. Xiang, L. Wang and J. Tang, RSC Adv., 2015, 5, 42728–42735, 10.1039/C5RA06548H.
- Y. Lv, B. Yang, T. Jiang, Y. M. Li, F. He and R. X. Zhuo, Colloids Surf., B, 2014, 115, 253–259, DOI:10.1016/j.colsurfb.2013.11.049.
- B. Xia, Y. Li, G. Cheng, X. Lin and Q. Wu, ChemCatChem, 2014, 6, 3448–3454, DOI:10.1002/cctc.201402672.
- I. Baum, B. Elsässer, L. W. Schwab, K. Loos and F. Gregor, ACS Catal., 2011, 1, 323–336, DOI:10.1021/cs1000398.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443, DOI:10.1016/j.tibtech.2010.05.004.
- S. Hao, Z. Yongjie, Y. Zhang, W. Kong and C. Xia, Chin. J. Catal., 2012, 33, 432–438, DOI:10.1016/S1872-2067(11)60336-7.
- A. Kazariya and S. Matsumura, Polymers, 2012, 4, 486–500, DOI:10.3390/polym4010486.
- Z. Yang, X. Zhang, X. Luo, Q. Jiang, J. Liu and Z. Jiang, Macromolecules, 2013, 46, 1743–1753, DOI:10.1021/ma302433x.
- C. Liu, Z. Jiang, J. Decatur, W. Xie and R. A. Gross, Macromolecules, 2011, 44, 1471–1479, DOI:10.1021/ma102899c.
- W. X. Wu, N. Wang, B. Y. Liu, Q. F. Deng and X. Q. Yu, Soft Matter, 2014, 10, 1199–1213, 10.1039/c3sm52496e.
- L. Corici, A. Pellis, V. Ferrario, C. Ebert, S. Cantone and L. Gardossi, Adv. Synth. Catal., 2015, 357, 1763–1774, DOI:10.1002/adsc.201500182.
- M. Kanelli, A. Douka, S. Vouyiouka, C. D. Papaspyrides, E. Topakas, L. M. Papaspyridi and P. Christakopoulos, J. Appl. Polym. Sci., 2014, 131(19), 40820, DOI:10.1002/app.40820.
- M. B. Frampton, T. R. B. Jones and P. M. Zelisko, RSC Adv., 2015, 5, 1999–2008, 10.1039/C4RA14828B.
- M. Bautista, A. M. de Ilarduya, A. Alla, M. Vives, J. Morató and S. Muñoz-Guerra, Eur. Polym. J., 2016, 75, 329–342, DOI:10.1016/j.eurpolymj.2015.12.012.
- N. D. Stebbins, W. Yu and K. E. Uhrich, Macromol. Biosci., 2015, 15, 1115–1124, DOI:10.1002/mabi.201500030.
- Y. Jiang, G. O. R. van Ekenstein, A. J. J. Woortman and K. Loos, Macromol. Chem. Phys., 2014, 215, 2185–2197, DOI:10.1002/macp.201400164.
- A. F. Naves, H. T. C. Fernandes, A. P. S. Immich and L. H. Catalani, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(18), 3881–3891, DOI:10.1002/pola.26789.
- Y. Jiang, A. J. J. Woortman, G. O. R. A. van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5451–5463, 10.1039/C5PY00660K.
- L. Gustini, C. Lavilla, L. Finzel, B. A. J. Noordover, M. M. R. M. Hendrix and C. E. Koning, Polym. Chem., 2016, 7, 6586–6597, 10.1039/C6PY01339B.
- Y. Jiang, A. J. J. Woortman, G. O. R. van Ekenstein, D. M. Petrović and K. Loos, Biomacromolecules, 2014, 15, 2482–2493, DOI:10.1021/bm500340w.
- Y. Jiang, A. J. J. Woortman, G. O. R. A. van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5198–5211, 10.1039/C5PY00629E.
- L. Gustini, B. A. J. Noordover, C. Gehrels, C. Dietz and C. E. Koning, Eur. Polym. J., 2015, 67, 459–475, DOI:10.1016/j.eurpolymj.2014.12.025.
- C. Japu, M. A. de Ilarduya, A. Alla, Y. Jiang, K. Loos and S. Muñoz-Guerra, Biomacromolecules, 2015, 16, 868–879, DOI:10.1021/bm501771e.
- S. Semlitsch, S. Torron, M. Johansson and M. Martinelle, Green Chem., 2016, 18, 1923–1929, 10.1039/C5GC02597D.
- T. Debuissy, E. Pollet and L. Avérous, Biomacromolecules, 2016, 17, 4054–4063, DOI:10.1021/acs.biomac.6b01494.
- E. Stavila, R. Z. Arsyi, D. M. Petrovic and K. Loos, Eur. Polym. J., 2013, 49, 834–842, DOI:10.1016/j.eurpolymj.2012.12.010.
- Y. Jiang, D. Maniar, A. J. J. Woortman, G. O. R. A. van Ekenstein and K. Loos, Biomacromolecules, 2015, 16, 3674–3685, DOI:10.1021/acs.biomac.5b01172.
- Y. Jiang, D. Maniar, A. J. J. Woortman and K. Loos, RSC Adv., 2016, 6, 67941–67953, 10.1039/C6RA14585J.
- K. Muthusamy, K. Lalitha, Y. S. Prasad, A. Thamizhanban, V. Sridharan, C. U. Maheswari and S. Nagarajan, ChemSusChem, 2018, 11, 2453–2463, DOI:10.1002/cssc.201800446.
- B. Xia, Y. Zhang, Q. Zhu, X. Lin and Q. Wu, Biomacromolecules, 2019, 20, 3584–3591, DOI:10.1021/acs.biomac.9b00918.
- K. Muthusamy, K. Lalitha, Y. S. Prasad, A. Thamizhanban, V. Sridharan, U. Maheswaria and S. Nagarajan, ChemSusChem, 2018, 11, 2453–2463, DOI:10.1002/cssc.201800446.
- S. Namekawa, H. Uyama and S. Kobayashi, Biomacromolecules, 2000, 1, 335–338, DOI:10.1021/bm000030u.
- W. C. Nie, H. C. Dang, X. L. Wang, F. Song and Y. Z. Wang, Polymer, 2017, 111, 107–114, DOI:10.1016/j.polymer.2017.01.055.
- Z. Jiang and J. Zhang, Polymer, 2013, 54, 6105–6113, DOI:10.1016/j.polymer.2013.09.005.
- Z. Jiang, Biomacromolecules, 2011, 12, 1912–1919, DOI:10.1021/bm2002522.
- B. Liu, X. Zhang, Y. Chen, Z. Yao, Z. Yang, D. Gao, Q. Jiang, J. Liu and Z. Jiang, Polym. Chem., 2015, 6, 1997–2010, 10.1039/C4PY01321B.
- J. Liu, Z. Jiang, J. Zhou, S. Zhang and W. M. Saltzman, J. Biomed. Mater. Res., Part A, 2011, 96, 456–465, DOI:10.1002/jbm.a.32994.
- Z. Jiang, Biomacromolecules, 2010, 11, 1089–1093, DOI:10.1021/bm1000586.
- J. Zhou, J. Liu, C. J. Cheng, T. R. Patel, C. E. Weller, J. M. Piepmeier, Z. Jiang and W. M. Saltzman, Nat. Mater., 2012, 11, 82–90, DOI:10.1038/nmat3187.
- L. Martino, M. Scandola and Z. Jiang, Polymer, 2012, 53, 1839–1848, DOI:10.1016/j.polymer.2012.03.005.
- X. Zhang, B. Liu, Z. Yang, C. Zhang, H. Li, X. Luo, H. Luo, D. Gao, Q. Jiang, J. Liu and Z. Jiang, Colloids Surf., B, 2014, 115, 349–358, DOI:10.1016/j.colsurfb.2013.12.029.
- Y. Chen, Y. Li, J. Gao, Z. Cao, Q. Jiang, J. Liu and Z. Jiang, ACS Appl. Mater. Interfaces, 2015, 8, 490–501, DOI:10.1021/acsami.5b09437.
- J. Zhang, J. Cui, Y. Deng, Z. Jiang and W. M. Saltzman, ACS Biomater. Sci. Eng., 2016, 2, 2080–2089, DOI:10.1021/acsbiomaterials.6b00502.
- Z. Yang, Z. Jiang, Z. Cao, C. Zhang, D. Gao, X. Luo, X. Zhang, H. Luo, Q. Jiang and J. Liu, Nanoscale, 2014, 6, 10193–10206, 10.1039/C4NR02395A.
- Y. Zhang, C. Fu, C. Zhu, S. Wang, L. Tao and Y. Wei, Polym. Chem., 2013, 4, 466–469, 10.1039/C2PY21039H.
- C. Fu, L. Tao, Y. Zhang, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9062–9064, 10.1039/c2cc34633h.
- E. Hrsic, H. Keul and M. Möller, Macromol. Rapid Commun., 2015, 36, 2092–2096, DOI:10.1002/marc.201300512.
- J. Lu, C. Fu, S. Wang, L. Tao, L. Yan, D. M. Haddleton, G. Chen and Y. Wei, Macromolecules, 2014, 47, 4676–4683, DOI:10.1021/ma500664u.
- C. Fu, A. Bongers, K. Wang, B. Yang, Y. Zhao, H. Wu, Y. Wei, H. T. T. Duong, Z. Wang and L. Tao, Polym. Chem., 2016, 7, 546–552, 10.1039/C5PY01652E.
- Z. Huang, C. Fu, S. Wang, B. Yang, X. Wang, Q. Zhang, J. Yuan, L. Tao and Y. Wei, Macromol. Chem. Phys., 2015, 216, 1483–1489, DOI:10.1002/macp.201500106.
- M. Finnveden, S. Nameer, M. Johansson and M. Martinelle, Macromol. Chem. Phys., 2016, 217, 1335–1341, DOI:10.1002/macp.201500490.
- M. Livia, P. Jaan, P. Omar, P. Tõnis, T. H. Pham, L. Ilme, L. Vares and P. Jannasch, ACS Sustainable Chem. Eng., 2018, 6, 17382–17390, DOI:10.1021/acssuschemeng.8b05074.
- S. Namekawa, S. Suda, H. Uyama and S. Kobayashi, Int. J. Biol. Macromol., 1999, 25, 145–151, DOI:10.1016/S0141-8130(99)00028-8.
- L. Van der Mee, F. Helmich, R. de Bruijn, J. A. J. M. Vekemans, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2006, 39, 5021–5027, DOI:10.1021/ma060668j.
- M. A. J. Veld, L. Fransson, A. R. A. Palmans, E. W. Meijer and K. Hult, Chembiochem, 2009, 10, 1330–1334, DOI:10.1002/cbic.200900128.
- J. Cai, B. S. Hsiao and R. A. Gross, Macromolecules, 2011, 44, 3874–3883, DOI:10.1021/ma102949h.
- B. Elsasser, I. Schoenen and G. Fels, ACS Catal., 2013, 3, 1397–1405, DOI:10.1021/cs3008297.
- M. Castano, K. S. Seo, K. Guo, M. L. Becker, C. Wesdemiotis and J. E. Puskas, Polym. Chem., 2015, 6, 1137–1142, 10.1039/C4PY01223B.
- M. B. Frampton and P. M. Zelisko, Enzyme Microb. Technol., 2014, 58, 87–92, DOI:10.1016/j.enzmictec.2014.03.006.
- S. W. Duchiron, E. Pollet, S. Givry and L. Avérous, RSC Adv., 2015, 5, 84627–84635, 10.1039/C5RA18954C.
- M. Finnveden, P. Hendil-Forssell, M. Claudino, M. Johansson and M. Martinelle, Polymers, 2019, 11, 1730, DOI:10.3390/polym11111730.
- H. Casajus, E. Dubreucq, S. Tranchimand, V. Perrier, C. Nugier-Chauvin and S. Cammas-Marion, Biomacromolecules, 2020, 21, 2874–2883, DOI:10.1021/acs.biomac.0c00593.
- J. Yang, Y. Liu, X. Liang, Y. Yang and Q. Li, Macromol. Biosci., 2018, 18, 1800131, DOI:10.1002/mabi.201800131.
| This journal is © The Royal Society of Chemistry 2020 |