DOI:
10.1039/C9RA10263A
(Review Article)
RSC Adv., 2020,
10, 2313-2326
Asymmetric catalysis in direct nitromethane-free Henry reactions
Received
7th December 2019
, Accepted 6th January 2020
First published on 13th January 2020
Abstract
A great number of reports have described asymmetric catalytic Henry reactions using nitromethanes as pronucleophiles, but far more challenging is diastereoselective catalytic Henry reactions using substituted higher nitroalkanes instead of nitromethane to generate chiral β-nitro alcohol scaffolds with four adjacent stereogenic centers in a one-pot operation. This review summarizes the current state and applications of such reactions involving complex nitroalkane coupling with various carbonyl compounds for resolving double chiral centers with high enantio- and diastereoselectivities.
 Lin Dong | Lin Dong obtained her PhD at Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences, supervised by Prof. Liu-Zhu Gong in 2006. After her postdoctoral studies at the University of Queensland under the guidance of Prof Craig. M. Williams in 2009, she worked as a Research Scientist under the guidance of Prof. K. C. Nicolaou and Prof. David Yu-Kai Chen at the Chemical Synthesis Laboratory (CSL) @ Biopolis, Singapore until 2011. Since 2011, she has relocated her research operation to Sichuan University, China, where she was appointed as a professor. Her research interests are focused on organometallic chemistry and metal-catalyzed organic reactions, and drug synthesis. |
 Fen-Er Chen | Fen-Er Chen received a M. S. in Pharmacy and a PhD in organic chemistry from Sichuan University. He joined Wuhan Institute of Technology in 1988 and was promoted to professor in 1996. In 1998, he moved to Fudan University as a full professor. His current research focuses on the development of new asymmetric catalysts, asymmetric total synthesis of natural products, process chemistry and computer assisted mechanism-based drug design (CADD). Prof. Fen-Er Chen has been a visiting professor at numerous prestigious universities, including Washington University, King's College London, etc. He is currently a member of Academician of Chinese Academy of Engineering. |
1. Introduction
Over the past two decades, asymmetric catalytic Henry reactions have been established as an integral part of asymmetric catalysis.1,2 During this period, a lot of reviews using nitromethanes as pronucleophiles have been reported. Despite the significant developments of the high enantioselectivity of these single chiral center products, many synthetic challenges arising from the demand for complex nitroalkanes instead of simply using nitromethanes as pronucleophiles have inspired chemists to construct chiral β-nitro alcohol scaffolds with four adjacent stereogenic centers in a one-pot operation. In particular, applications of these nitromethane-free reactions in the synthesis of chiral β-nitro alcohols have created new possibilities for the asymmetric preparation of natural products, pharmaceutical drugs, and bioactive molecules in academic and industrial settings.3
Since the seminal work reported by Shibasaki using metal/chiral ligand complexes in catalytic asymmetric nitroaldol reactions in 1992,4 chemists have overcome myriad synthetic challenges by developing various efficient catalytic systems, particularly chiral metal catalysts;1,2,5 chiral ligands such as Schiff bases, tetrahydrosalens, amino alcohols, and diamines;1 and small organic molecules such as guanidine, cinchona alkaloid-derived organocatalysts and quaternary ammonium salts.2 2D materials are also a powerful platform to construct efficient catalysts for various reactions, such as asymmetric catalysis, CO2 reduction, CO oxidation.6
The present review aims to describe the impressive growth in this rapidly expanding field. It examines the current state of direct asymmetric catalytic Henry reactions involving higher nitroalkanes rather than simple nitromethanes coupling with various carbonyl compounds that control the stereochemistry at double chiral centers. The review will also present perspectives on the use of these reactions in the syntheses of natural products or bioactive molecules. The discussion is divided in two based on the type of catalyst: metal/chiral ligand complex-based reactions and organocatalytic reactions.
2. Rare earth-catalyzed asymmetric diastereoselective Henry reaction
Lanthanides seldom show simple, predictable coordination chemistry because of their variable coordination number and geometry.7 Their coordination modes depend largely on ligand structure. Only lanthanum and neodymium have been applied so far in asymmetric diastereoselective Henry reactions.
2.1. Lanthanum-based catalysis
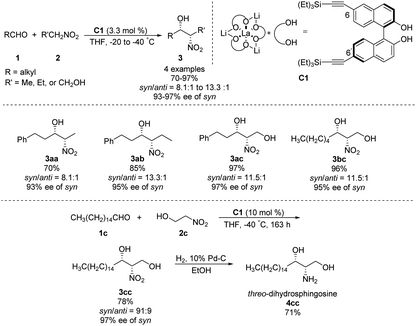
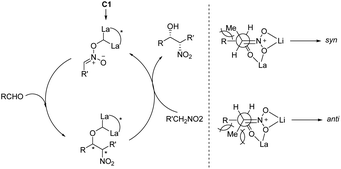
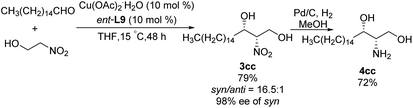
In 1992, Shibasaki reported the first transition metal-catalyzed asymmetric Henry reaction.4 The optimal catalyst was a lanthanum–alkoxide complex C1, in which bulky TES groups at the 6,6′-positions of BINOL led to β-nitroalcohols in yields of 70–96%, a syn/anti ratio up to 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, and enantioselectivity of 93–97% ee (Scheme 1).8 Catalyst C1 also worked efficiently with the nitroethanol 2c bearing an alkyl aldehyde to afford the corresponding propylene glycol 3cc in good yields and a syn/anti ratio of 91:9. Subsequent hydrogenation reduced the nitro group to give threo-dihydrosphingosine 4cc in 71% yield. Mechanistic studies on the catalyst system indicated that the first step of the reaction might undergo the ligand exchange between the binaphthol and nitromethane (Scheme 2). The model proposed to explain that the syn-selectivity is most favorable due to steric hindrance in the bicyclic transition state via chelate formation which can be seen in Newman projections.
:8, and enantioselectivity of 93–97% ee (Scheme 1).8 Catalyst C1 also worked efficiently with the nitroethanol 2c bearing an alkyl aldehyde to afford the corresponding propylene glycol 3cc in good yields and a syn/anti ratio of 91:9. Subsequent hydrogenation reduced the nitro group to give threo-dihydrosphingosine 4cc in 71% yield. Mechanistic studies on the catalyst system indicated that the first step of the reaction might undergo the ligand exchange between the binaphthol and nitromethane (Scheme 2). The model proposed to explain that the syn-selectivity is most favorable due to steric hindrance in the bicyclic transition state via chelate formation which can be seen in Newman projections.
 |
| | Scheme 1 La–alkoxide complex C1 catalyzed asymmetric Henry reaction. | |
 |
| | Scheme 2 The mechanism of C1 catalyzed asymmetric Henry reaction. | |
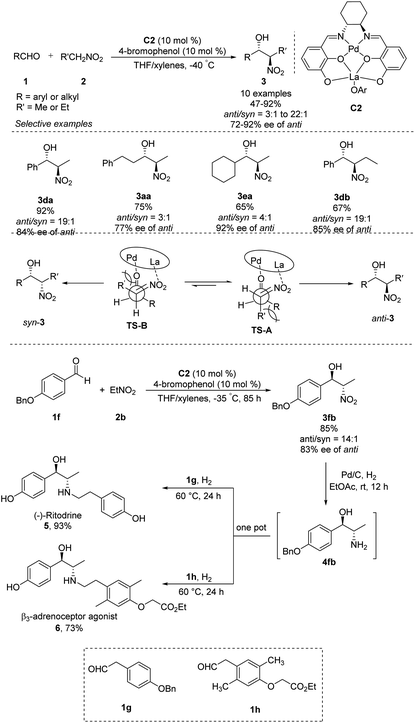
Shibasaki's group expanded the usefulness of this reaction for pharmaceutical syntheses by developing heterobimetallic Pd/La/C2 complexes that can catalyze anti-selective asymmetric Henry reactions of various aldehydes with nitroethane or nitropropane. In the presence of catalytic amounts of 4-bromophenol additive, products were generated in yields of 65–92% with high anti/syn ratios of 22:1–3:1 and excellent enantioselectivities of 72–92% ee (Scheme 3).9 The La–OAr moiety in the catalyst acts as a Brønsted base to generate a La–nitronate. Then La–nitronate reacts with the aldehyde, which is coordinated to the Pd metal center to favorably form TS-A than TS-B to avoid steric repulsion between the R′ group and the Pd/La catalyst, preferentially giving anti-adducts (Scheme 3).10 This approach generated anti-nitroaldol adduct 3fb, which was converted in a one-pot reaction into β-adrenoceptor agonists 5 and 6.
 |
| | Scheme 3 Asymmetric anti-selective Henry reactions catalyzed by heterobimetallic catalyst C2. | |
2.2. Catalysis by a neodymium–sodium heterobimetallic complex
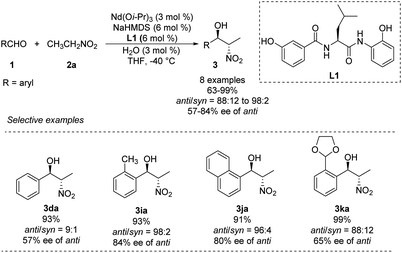
In 2007, Shibasaki and Kumagai designed a novel lanthanum/amide complex that mimics an enzyme's structure to support asymmetric amination.11 Their Nd/Na/amide heterobimetallic catalyst smoothly generated anti-1,2-nitro alkanols with good enantioselectivities and excellent diastereoselectivities (Scheme 4).12 Benzaldehydes afforded the corresponding products in high yields with anti-selectivity, albeit only moderate enantioselectivity. Reactions of aromatic aldehydes bearing o-alkyl substituents proceeded smoothly with 3 mol% catalyst loading, yielding products with good diastereo- and enantioselectivities. No aliphatic aldehydes have been studied.
 |
| | Scheme 4 Catalytic asymmetric anti-selective Henry reactions with Nd/Na/L1 heterobimetallic complex. | |
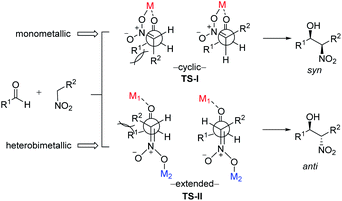
To account for the extremely high catalytic diastereoselectivities in the presence of heterobimetallic catalyst, Shibasaki and Kumagai proposed a transition state model for metal-catalyzed nitroaldol reactions (Scheme 5).13 In this mechanism, the monometallic catalyst forms the cyclic transition state TS-I when metal and oxidant chelate each other, and this transition state affords syn diastereomers. In contrast, chelation between a heterobimetallic catalyst and chiral amide ligand generates transition state TS-II, which prefers an extended conformation in which each metal cation works independently as a Lewis acid to activate the aldehyde and as a Brønsted base to form metal nitronate (Scheme 5). This transition state affords predominantly anti diastereomers, overriding the undesirable chelate formation.
 |
| | Scheme 5 Transition state models of metal-catalyzed Henry reaction. | |
Kumagai and Shibasaki improved the anti-selectivity and enantioselectivity of catalytic asymmetric Henry reactions by using a fluorine-substituted chiral amide ligand L2 with their Nd/Na heterobimetallic complex (Scheme 6).14 The corresponding products formed with nearly perfect anti-selectivity, which the researchers attributed to (1) a C–F⋯H–N intramolecular hydrogen bond in the o-fluorobenzamide, which may restrict rotation of the C–C bond; (2) the influence of the fluorine substituent on the electronic properties of the aminophenol moiety (Scheme 7). Regardless of the reasons, the researchers found that aliphatic aldehydes led to much lower anti-selectivity than aromatic aldehydes.
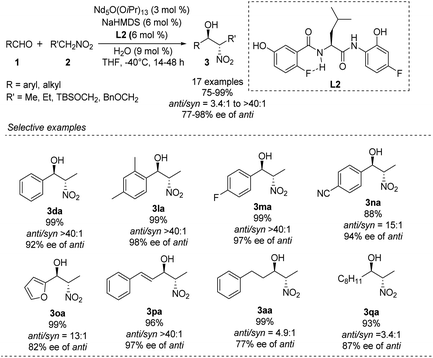 |
| | Scheme 6 o-Fluorobenzamide L2 as chiral ligand of the asymmetric anti-selective Henry reaction. | |
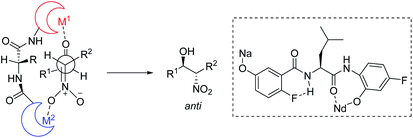 |
| | Scheme 7 Amide backbone as a platform for bimetallic complex. | |
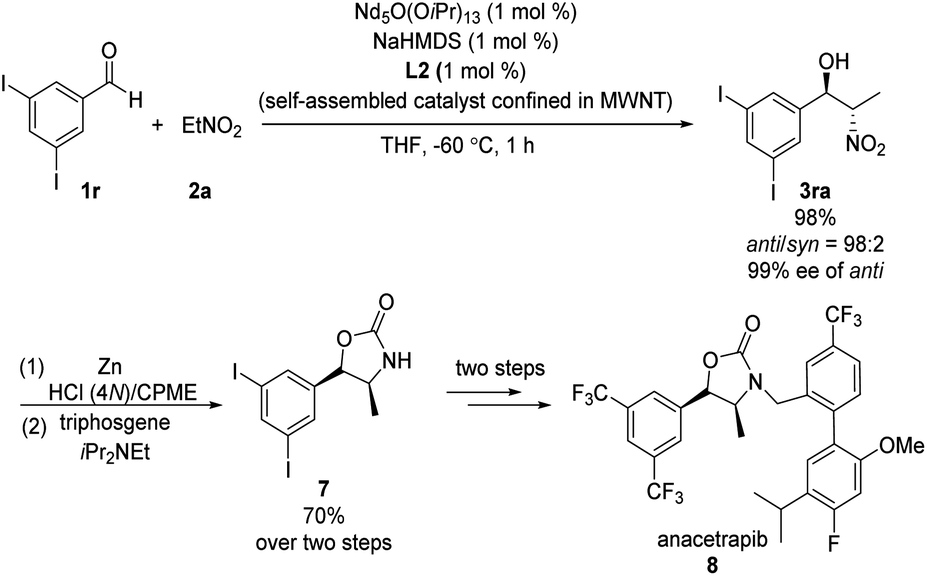
Kumagai and Shibasaki found that immobilizing their Nd/Na heterobimetallic catalyst on an entangled multiwalled carbon nanotube substantially increased its efficiency and facilitated its reuse.15 Using this self-assembling catalyst system, they concisely prepared anacetrapib (8) enantioselectively (Scheme 8). The catalyst promoted the reaction of diiodobenzaldehyde 1r with nitroethane 2a, providing anti-β-nitroethanol 3ra in excellent yield as well as excellent diastero- and enantioselectivity. A further four steps completed the synthesis of 8. In later work, Shibasaki replaced NdO1/5(OiPr)13/5/NaHMDS with bench-stable, inexpensive NdCl3·6H2O/NaOtBu.16
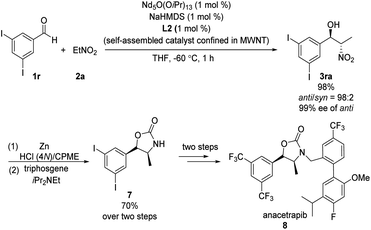 |
| | Scheme 8 Enantioselective synthesis of anacetrapib. | |
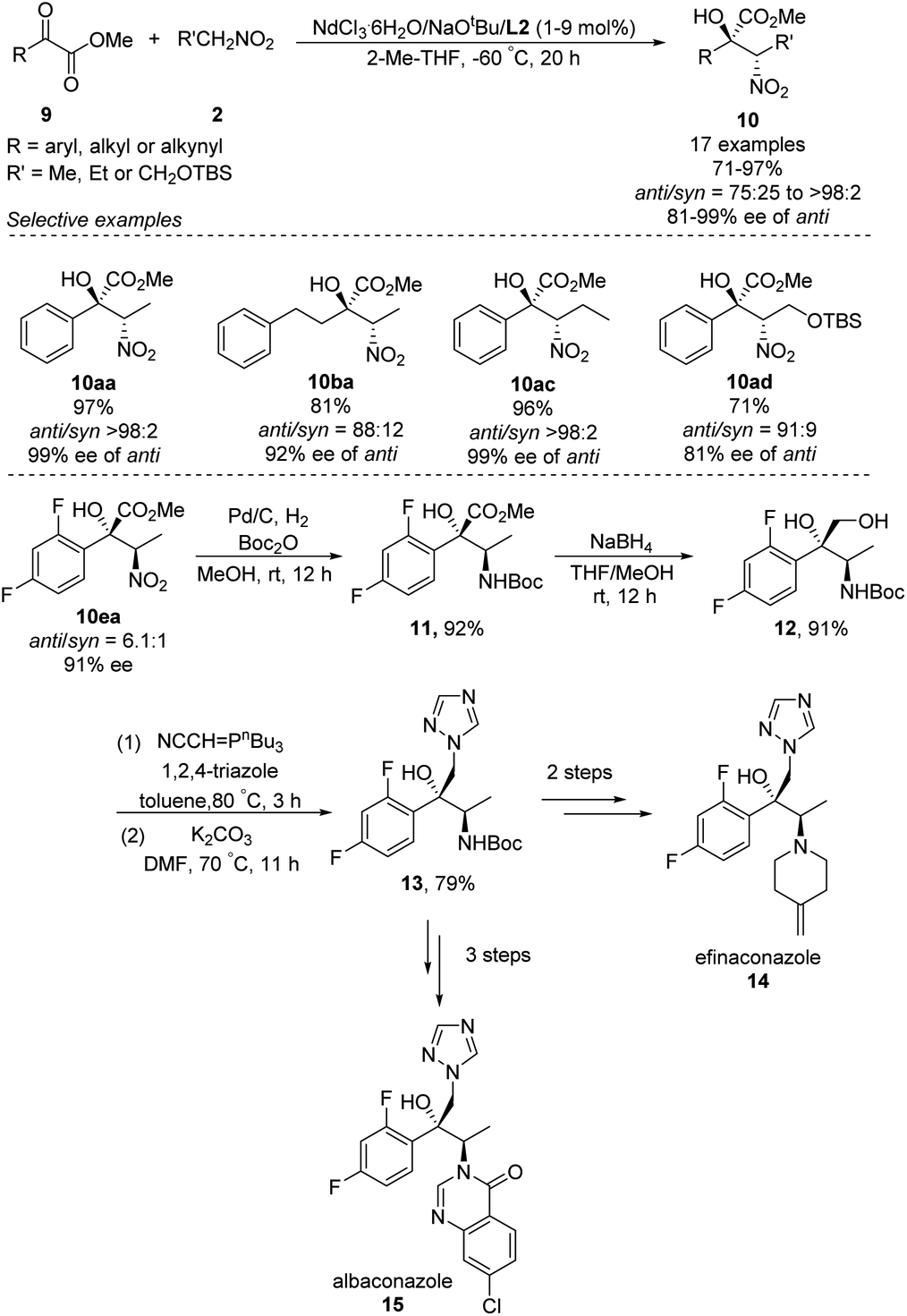
In 2018, Kumagai and Shibasaki extended Nd/Na heterobimetallic catalysts to reactions between α-keto esters and nitroalkanes (Scheme 9).17 In this approach, a range of aryl α-keto esters afforded α-nitro tertiary alcohols in good to excellent yields as well as diastero- and enantioselectivities. The solvent 2-Me-THF gave better stereoselectivity than THF. The reaction also tolerated alkyl and alkynyl α-keto esters, albeit with modest diastereo- and enantioselectivities. The researchers exploited the anti-selectivity of their asymmetric Henry reaction to streamline the stereoselective synthesis of the commercial antifungal agents efinaconazole (14) and albaconazole (15). In the shared starting pathway, the nitro group in α-keto esters 10ea was reduced and the compound protected with a Boc group, giving methyl ester 11 in 92% yield. Reduction with NaBH4 gave diol 12, and introduction of 1,2,4-triazole afforded the key intermediate 13, from which two further steps generated efinaconazole (14) or three further steps generated albaconazole (15).
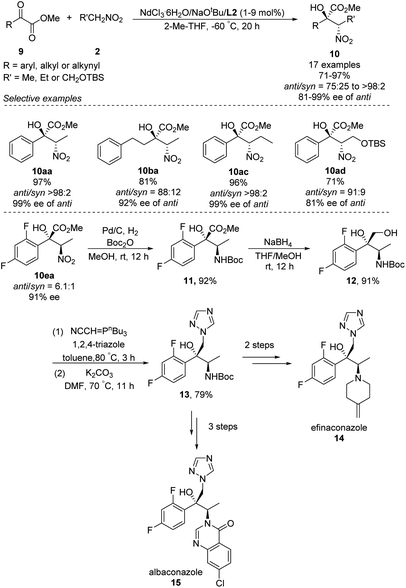 |
| | Scheme 9 Anti-selective asymmetric Henry reaction of α-keto esters. | |
Highly anti-selective Henry reactions of various trifluoromethyl ketones with nitroethane and 1-nitropropane were also achieved using heterobimetallic Nd–Na–L2 or Pr–Na–L2 complex (Scheme 10).18 Nitroethane led to CF3-appended vic-nitroalkanols in anti/syn ratios up to 98:2 and 95% ee. Nitropropane, in contrast, led to much lower enantioselectivity of 70% ee. This reaction was also able to generate CF3-appended ephedrine 19.
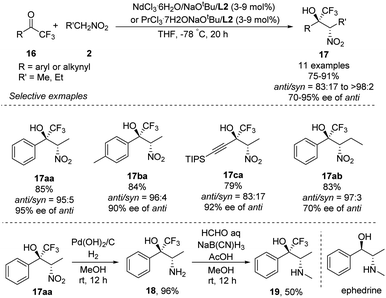 |
| | Scheme 10 Asymmetric Henry reactions of trifluoromethyl ketones. | |
While these impressive results highlight the ability of rare earth metals to support efficient enantio- and diastereoselective Henry reactions, such metals are scarce and expensive. This has led researchers to search for more accessible and inexpensive metal catalysts.
3. Copper-catalyzed asymmetric diastereoselective Henry reaction
Copper is abundant, shows low toxicity and can form stable chiral metal complexes with ligands containing nitrogen- and oxygen.19 It is no surprise, then, that since Jørgensen's groundbreaking work in 2001,20 numerous asymmetric diastereoselective Henry reactions have been developed using chiral copper-based catalysts and various chiral ligands such as imidazolines, Schiff bases, tetrahydrosalens, amino alcohols and diamines.
3.1. Chiral imidazoline ligands
In 2007, You replaced the oxygen atom of oxazoline with nitrogen to generate tridentate imidazoline ligand L3.21 The complex of Cu(OTf)2–L3 supported enantioselective Henry reactions in the presence of catalytic amounts of Et3N. In reactions using nitroethane as nucleophile, this catalyst demonstrated good synthetic potential, that aromatic, aliphatic and even heterocyclic aldehydes are well tolerated (Scheme 11).22 Surprisingly, using N-methylmorpholine as base generated the desired adducts with a syn/anti ratio up to 50:1 and enantioselectivity up to 99% ee. In this process, TS-I is most favoured and results in syn product, because of the repulsion between the methyl group of nitroethane and the isopropyl group of the catalyst in Ts-II, which will lead to the anti product (Scheme 11).
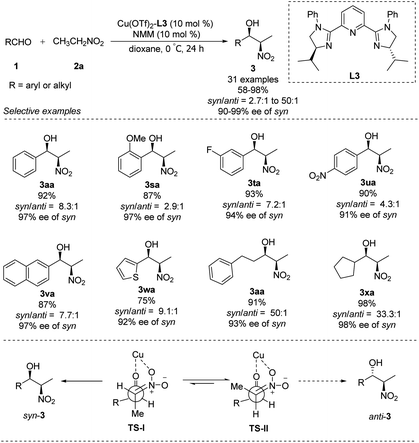 |
| | Scheme 11 You's asymmetric diastereoselective Henry reaction. | |
3.2. Chiral Schiff-base ligands
A chiral Schiff-base ligand L4, derived from cinchona alkaloid, supported smooth asymmetric Henry reactions between various aldehydes and nitroethane.23 The corresponding products were obtained in yields around 70% with enantioselectivities up to 99% ee, but anti/syn ratios up to only 2.7:1 (Scheme 12).
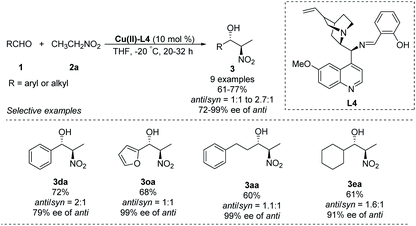 |
| | Scheme 12 Cu–cinchona alkaloid complex catalyzed asymmetric Henry reaction. | |
3.3. Chiral tetrahydrosalen ligands
Chiral salen-type ligands have proven useful in a variety of asymmetric metal-catalyzed reactions.24 In Henry reactions, chiral tetrahydrosalen ([H4]salen) ligands produce strong asymmetry by increasing the basicity and framework flexibility of the nitrogen atom.25
In 2012, White synthesized chiral [H4]salen ligand L5 from cis-2,5-diaminobicyclo[2.2.2]octane (Scheme 13)26 and used it to conduct a highly enantio- and diastereoselective copper(I)-catalyzed Henry reaction. Reaction of benzaldehyde and 1-naphthaldehyde with nitropropane in the presence of Cu(I) and L5 strongly favoring the syn product 3 which was formed in high enantiomeric excess. A transition state TS-I rationalizing this outcome is proposed. In this model, N–H hydrogen bond with the nitronate leads to high enantioselectivity. In addition, the copper complexed nitronate of nitropropane in TS-I has a (Z) configuration with attack occurring at the si face of the aldehyde carbonyl (Scheme 13). While the applicability of this approach is limited by the expense of the metal and complicated preparation of ligand L5.
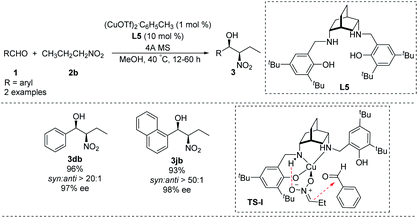 |
| | Scheme 13 Asymmetric syn-selective Henry reaction catalyzed by copper(I)–[H4] salen complex. | |
Kureshy developed Cu–L6 complexes to catalyze diastereoselective Henry reactions (Scheme 14).27 Benzaldehyde reacted with nitroethanol to give the desired product in 82% yield with a syn/anti ratio of 92:8. However, the reaction did not work well with aliphatic and aromatic aldehydes bearing electron-withdrawing substitutions. The researchers were able to use the catalyst more than 8 times without significant loss of performance.
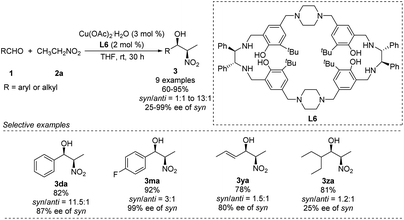 |
| | Scheme 14 Asymmetric syn-selective Henry reaction catalyzed by Cu(II)–L6 complex. | |
3.4. Chiral amino-alcohol ligands
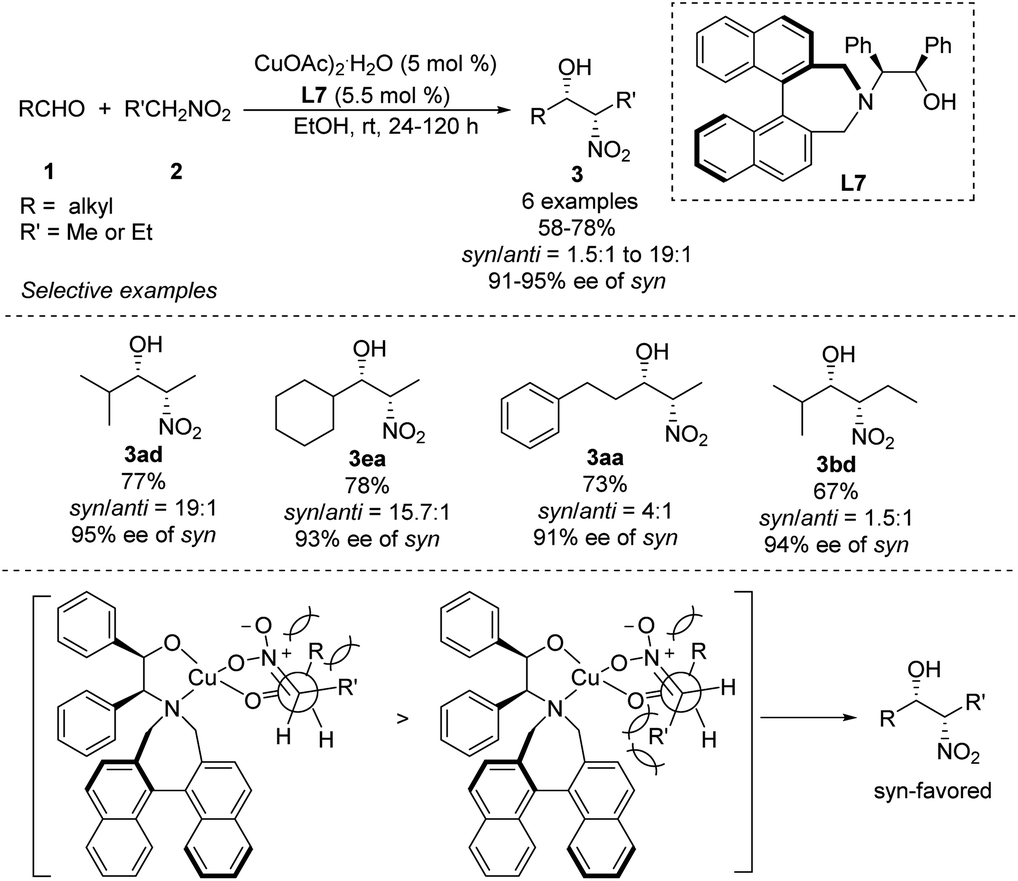
Early in 2011, Lu derived amino alcohol L7 from 1,1-binaphthylazepine and demonstrated its efficiency as a chiral ligand in Cu-catalyzed asymmetric Henry reactions (Scheme 15).28 In this catalytic system, aliphatic aldehydes showed better enantio- and diastereoselectivities than aromatic aldehydes. For example, isobutyraldehyde reacted with nitroethane to give the corresponding product in 77% yield with a syn/anti ratio of 95:5, and the syn-adduct showed enantioselectivity up to 95%. Under the same conditions, aldehydes reacted with nitropropanes to give products in syn/anti ratios of only 61:39. The reaction model proposed to explain the steric hindrance of binaphthylazepine in the L7 could result in higher stereocontrol.
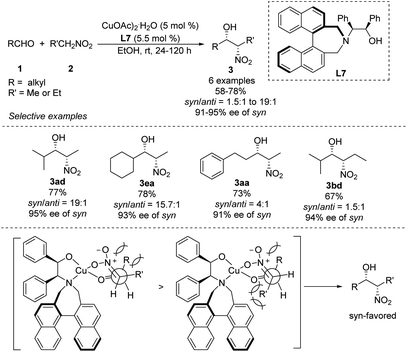 |
| | Scheme 15 Asymmetric syn-selective Henry reaction catalyzed by amino alcohol ligand L7. | |
Systematic screening of a library of C1-symmetrical amino-alcohol compounds showed that those bearing a pyridine group and a phenol substituted with a bulky alkyl group were the best ligands in diastereoselective Henry reactions.29 In the presence of 5 mol% of Cu(OAc)2–L8 as catalyst and 5 mol% diisopropylethylamine as base, the Henry reaction of 3-phenylpropionaldehyde 1a and methyl 4-nitrobutyrate 2e furnished the desired products with a syn/anti ratio of 85:15 (Scheme 16). Using aromatic aldehydes substantially reduced the syn/anti ratio to 1.7:1.
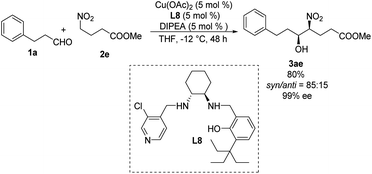 |
| | Scheme 16 Asymmetric syn-selective Henry reaction catalyzed by C1-symmetrical amino-alcohol ligand L8. | |
Chen developed an amino alcohol copper(II) catalyst (Cu–L9) for syn- and enantioselective Henry reactions of aliphatic aldehydes with nitroethane (Scheme 17).30 The desired products were obtained with very good enantioselectivity and syn/anti ratios up to 18.6:1. In contrast, 4-chlorobenzaldehyde generated product in a syn/anti ratio of only 2.9:1 under optimal conditions. Using 2-nitroethanol as nucleophile increased the syn/anti ratio, presumably because the hydroxyl group formed additional intermolecular hydrogen bonds and thereby stabilized transition states. This approach allowed the preparation of safingol (4cc) in only two steps with 57% overall yield (Scheme 18).31
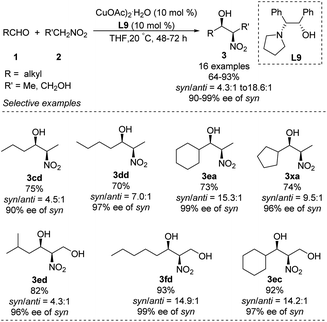 |
| | Scheme 17 Syn- and enantioselective Henry reactions of aliphatic aldehydes. | |
 |
| | Scheme 18 Synthesis of safingol. | |
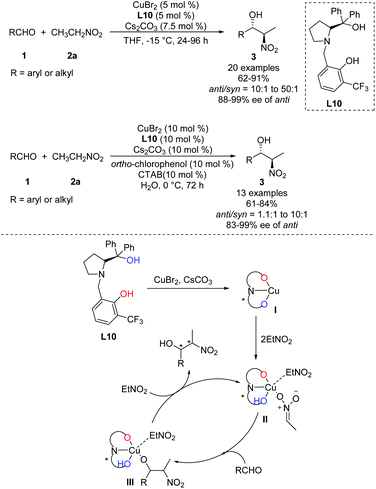
In 2011, Wang achieved one of the few monometal-catalyzed anti-selective asymmetric Henry reactions ever reported. They conducted the reaction in organic solvents and water using the catalyst Cu–L10 and phase-transfer catalyst Bu4NBr.32 They achieved good anti-selectivity and excellent enantioselectivities, even 99% ee in water (Scheme 19).33 The transition metal model exhibited that copper complex was bonded to one molecule of nitronate and coordinated with one molecule of EtNO2 in the intermediate II, then only metal–nitronate worked with aldehyde to give anti-selectivity Henry reactions. Therefore, this reaction requires excess EtNO2(Scheme 19). Further analysis showed that diastereoselectivity did not depend on 4-tert-butylphenol or Bu4NBr, and that phenol may facilitate proton transfer by functioning as a weak acid.34
 |
| | Scheme 19 Anti-selective asymmetric Henry reactions in the presence of water. | |
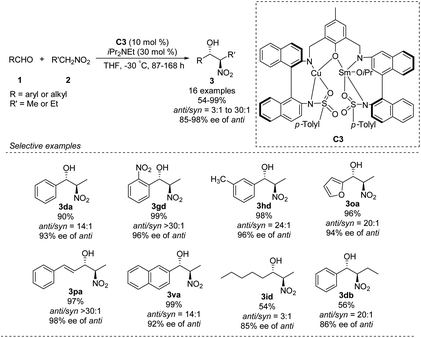
In 2016, Zhou prepared the novel heterobimetallic Cu/Sm/aminophenol sulfonamide complex C3 in one pot and used it to achieve an anti-selective asymmetric Henry reaction (Scheme 20).35 Aryl aldehydes substituted with electron-donating or -withdrawing substituents afforded the products in up to 99% yield with anti/syn ratios > 30:1 and enantioselectivity of 98% ee. The reaction with benzaldehyde also proceeded with 1-nitropropane as nucleophile. As showed in the report, aromatic aldehydes led to higher diastereoselectivity.
 |
| | Scheme 20 Anti-selective asymmetric Henry reaction catalyzed by heterobimetallic Cu/Sm/aminophenol sulfonamide complex. | |
3.5. Chiral diamines ligands
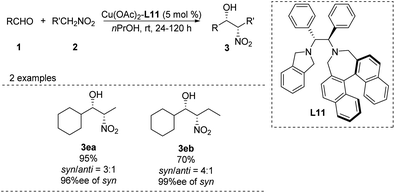
In 2006, Arai catalyzed the Henry reaction using a C2-symmetric diamine catalyst,36 whose usefulness is limited by the air sensitivity of Cu(I) and the hygroscopic cyclohexyl-1,2-diamine ligand. To overcome these drawbacks, the researchers replaced the binaphthyl azepine ring in the ligand with a simple isoindoline to generate ligand L11.37 In the presence of 5 mol% of Cu(OAc)2–L11 complex, the adduct was obtained in >99% yield with 98% ee at room temperature (Scheme 21). The same catalyst supported other syn-selective Henry reactions, which afforded both diastereomers with excellent enantiomeric excess.
 |
| | Scheme 21 Arai's asymmetric Henry reaction. | |
Kanger reported an asymmetric Henry reaction using L12 as the chiral ligand (Scheme 22).38 The desired Henry adducts were efficiently obtained at low temperature after reasonable reaction times with enantioselectivities up to 96%. Using other nitroalkyl compounds as ligands gave lower anti/syn ratios from 2.6:1 to 4.5:1.
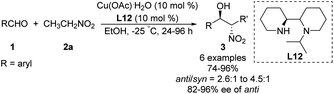 |
| | Scheme 22 Cu(II)–L12 catalyzed Henry reactions. | |
Zhang and Guo developed the C1-symmetric chiral diamine L13 as an efficient ligand for the copper-catalyzed asymmetric nitroaldol reaction.39 Their catalyst system supported the reaction of nitroethane or 1-nitropropane with 2-nitroethylbenzene to afford products with excellent enantioselectivities and moderate to good diastereoselectivities (Scheme 23). Due to its stability, ligand L13 was recovered in good yield without loss of catalytic performance via simple aqueous acid/base workup.
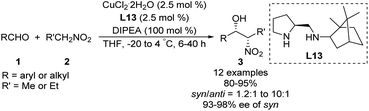 |
| | Scheme 23 Syn-selective asymmetric Henry reaction catalyzed by L13. | |
Using a chiral bis(sulfonamide)-diamine skeleton and nitromethane as nucleophile, Wan achieved copper-catalyzed enantioselective Henry reactions giving good yields and high enantioselectivities.40 A scalable version of the reaction gave adducts in up to 99% yield with a syn/anti ratio of 32.3:1, and 97% ee of the syn adduct (Scheme 24).41 This syn-selectivity appears to depend on pyridine, and base additives increase catalyst reactivity.42 Future work should examine whether other complex nitroalkanes can support high diastereo- and enantioselectivities in this reaction.
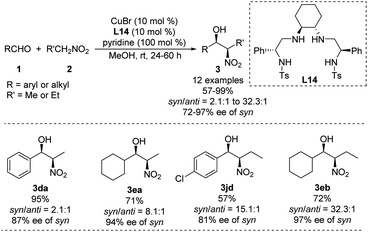 |
| | Scheme 24 Syn-selective asymmetric Henry reaction catalyzed by Cu–L14. | |
Some of the few monometal-catalyzed anti-selective asymmetric Henry reactions were achieved using Gou's chiral N-monoalkyl cyclohexane-1,2-diamine ligand L1543 and Breuning's chiral bispidine ligand L16.44 These reactions gave products with anti/syn ratios of 6.1:1 and enantioselectivity above 90% ee (Scheme 25).
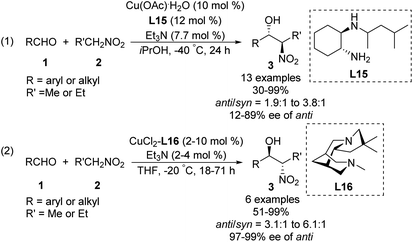 |
| | Scheme 25 Anti-selective asymmetric Henry reaction catalyzed by Cu–L15 and Cu–L16. | |
3.6. Other type of chiral ligands
N,N-dioxide/metal complexes have been used to catalyze a number of enantioselective reactions.45 In 2007, Feng reported enantioselective Henry reactions catalyzed by N,N′-dioxide-Cu(I).46 These ligands also proved efficient for asymmetric anti-selective Henry reactions of nitroethane with aromatic aldehydes, which generated the corresponding products in good yields with moderate to excellent dr values (Scheme 26).47 However, these results required low reaction temperatures and long reaction times. Naphthaldehyde, α,β-unsaturated aldehyde and heteroaromatic aldehyde proceeded well to afford the nitroaldol products in good yields but with poor anti/syn ratio. In addition, the poor reactivity of 1-nitropropane was observed via its steric hindrance.
 |
| | Scheme 26 N,N-dioxide ligands applied to the Cu-catalyzed asymmetric anti-selective Henry reactions. | |
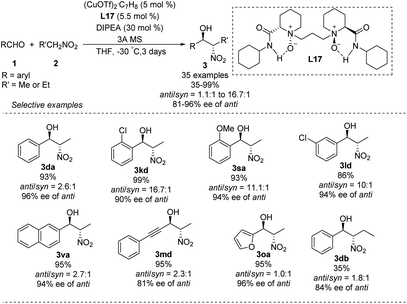
In 2008, Blay and Pedro reduced the imine bond of iminopyridine to synthesize a more flexible chiral aminopyridine ligand L18.48 In the presence of 5 mol% of this ligand, 1.0 equiv. of DIPEA and 5 mol% of Cu(OAc)2·H2O, various aldehydes reacted smoothly with nitroethane and bromonitromethane49 to give the expected products in yields up to 99%, enantioselectivities up to 98% ee and diastereoselectivities up to 82:18 (Scheme 27).
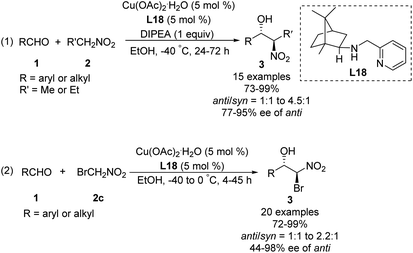 |
| | Scheme 27 Chiral amino-pyridine ligand L18 applied to the Cu-catalyzed asymmetric Henry reactions. | |
4. Cobalt-catalyzed asymmetric diastereoselective Henry reaction
Optically active ketoiminatocobalt complexes were originally employed as chiral Lewis acid catalysts in enantioselective hetero Diels–Alder reactions50 and carbonyl-ene reactions.51
In 2008, Hong revealed that self-assembled dinuclear cobalt(II)–salen catalyst C4 promoted cobalt-catalyzed asymmetric anti-selective Henry reactions (Scheme 28).52 Dimers self-assembled from 2-pyridone and aminopyridine as the hydrogen-bonding pair.
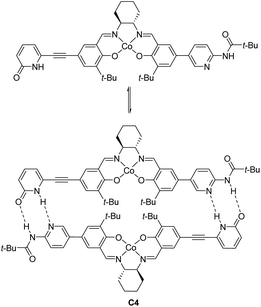 |
| | Scheme 28 Hong's self-assembled catalyst. | |
Hong went on to develop second-generation catalysts that self-assembled through urea–urea hydrogen bonding,53 such as the [(bisurea–salen)Co] catalyst C5.54 The NH moiety of urea was critical for yield and stereoselectivity: both parameters decreased when the catalyst was replaced by unfunctionalized [(salen)CoIII] catalyst C6 or methyl-functionalized catalyst C7 (Table 1). The author proposed that the [(bisurea–salen)Co] catalyst might enable the antiparallel transition state for the Henry reaction, either by bimetallic dual activation (TS-I) or by H-bond/metal bifunctional activation (TS-II) (Table 1).
Table 1 Self-assembled [(bisurea–salen)Co] catalyzed asymmetric Henry reaction
High anti-selectivity was obtained using methyl tert-butyl ether as solvent and N-ethylpiperidine as base, but this selectivity fell to an anti/syn ratio around 2:1 when the substrate was benzaldehyde without an ortho substitution (Scheme 29). The catalyst 5-promoted Henry reaction was applied to the synthesis of (1R,2S)-methoxamine hydrochloride 4ha, an α1-adrenergic receptor agonist.
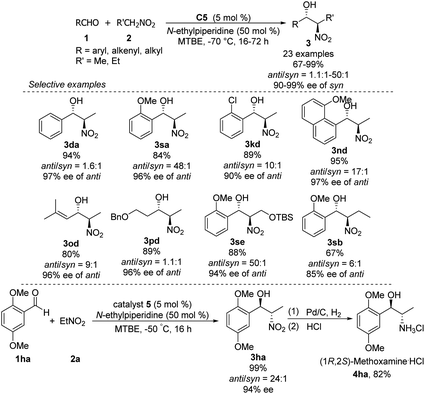 |
| | Scheme 29 Hong's Synthesis of (1R,2S)-methoxamine hydrochloride. | |
5. Organocatalytic asymmetric diastereoselective Henry reactions
5.1. Guanidine derived organocatalysts
The Nagasawa group achieved the first organocatalytic asymmetric diastereoselective Henry reaction in 2006.55 The guanidine–thiourea bifunctional organocatalyst C8 catalyzed reaction between various aliphatic aldehydes and nitroalkanes (Scheme 30). Yields were moderate to good, and syn-selective products were obtained with high enantioselectivity. The inorganic salt KI proved crucial for inhibiting the retro-nitroaldol reaction and for improving enantioselectivity. The same organocatalyst C8 was extended to the reaction of nitroalkanes with different α-keto esters, giving products in moderate yields with moderate enantioselectivities and high syn selectivity.56 Transition state of the Henry reaction catalyzed by C8 was based on the chemoselective dual activation concept.57
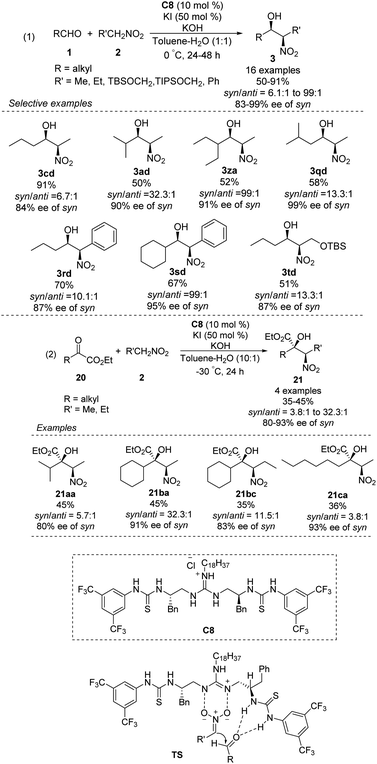 |
| | Scheme 30 Guanidine-thiourea C8 catalyzed Henry reaction. | |
Then, the Terada group58 and the Herrera group59 independently reported the diastereo- and enantioselective Henry reactions of nitroalkanes with aldehydes using axially chiral guanidine C9 and C10 as the catalyst. However, optically active products were obtained in moderate yield and poor enantio- and diastereoselectivities (Scheme 31).
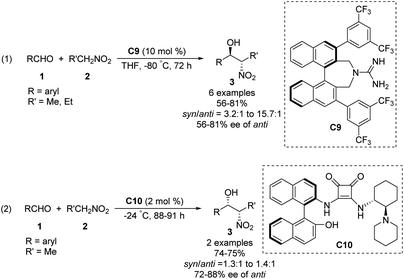 |
| | Scheme 31 Axially chiral guanidine catalysed Henry reactions. | |
5.2. Tetraaminophosphonium salt derived organocatalysts
Asymmetric phosphine catalysis has emerged as a remarkable and powerful strategy for constructing chiral molecules,60 but few such reactions involve quaternary phosphonium salts. In 2007, Ooi used chiral P-spirocyclic tetra-aminophosphonium salts to promote asymmetric diastereoselective Henry reactions.61 In reactions of aromatic aldehydes with nitroethane or nitropropane, the catalysts provided enantioselectivity up to 99% ee and anti/syn ratios up to 19:1 (Scheme 32). Using aliphatic aldehydes resulted in moderate yields, enantioselectivities and diastereoselectivities. The reaction has been proposed to proceed via an ion pair complex 22 that forms when nitronate anions hydrogen-bond to the secondary amino group of catalyst C11.
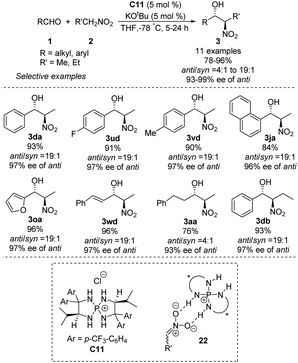 |
| | Scheme 32 Tetra-aminophosphonium salt-catalyzed Henry reactions of aldehydes. | |
Ooi extended the scope of this approach to ynals, a relatively unexplored substrate in asymmetric Henry chemistry (Scheme 33).62 Adding N,N-dimethylformamide as a co-solvent suppressed decomposition of the aminophosphonium alkoxide intermediate. Aromatic and aliphatic ynals were suitable substrates, affording the corresponding propargylic alcohols in excellent yields, enantioselectivities, and diastereoselectivities. Through this approach, (2S,3R)-(+)-xestoaminol C (24) and (−)-codonopsinine 27 were synthesized concisely (Scheme 34).
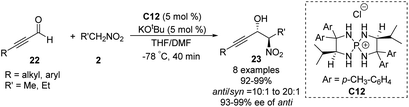 |
| | Scheme 33 Catalytic asymmetric direct Henry reaction of ynals. | |
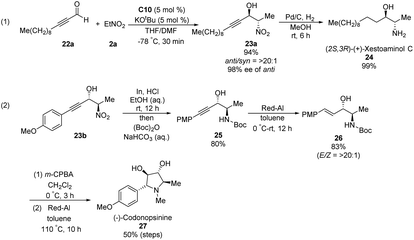 |
| | Scheme 34 Short syntheses of (2S,3R)-(+)-xestoaminol C and (−)-codonopsinines. | |
5.3. Cyclodextrins derived organocatalysts
In 2010, the Pitchumani group used per-6-amino-β-cyclodextrin C13 to catalyze the highly syn-selective Henry reaction of nitroethane with different aldehydes (Scheme 35).63 Syn-products were formed in good yields and enantioselectivities. The catalyst was easily recovered by simple filtration, and reused without loss of activity. So far, however, catalyst C13 has not been applied to other nitroalkanes.
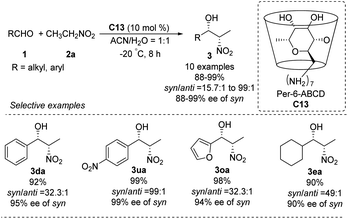 |
| | Scheme 35 Per-6-amino-β-cyclodextrin C13 catalyzed Henry reactions. | |
5.4. Cinchona alkalloid derived organocatalysts
The He group reported the use of a new family of cinchona alkaloid-thiourea catalysts in anti-selective asymmetric Henry reactions.64 Catalyst C14 led to isomers in yields up to 95%, an anti/syn ratio of 91:9 and enantioselectivity of 87% ee (Scheme 36). This catalyst worked even in water: it afforded products in up to 93% yield, an anti/syn ratio of 94:6 and enantioselectivity of 88% ee in toluene/water (7:3). In contrast, reacting aliphatic aldehydes with nitroethane gave an anti/syn ratio of only 1.6 and enantioselectivity of only 68% ee.
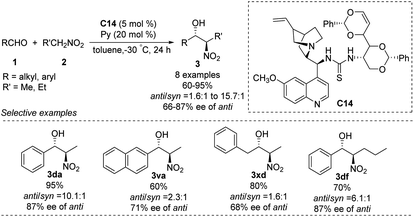 |
| | Scheme 36 Cinchona alkalloid catalyst C14 catalyzed Henry reactions. | |
6. Conclusions
Although lagging behind the extensive literature on asymmetric Henry reactions to form chiral β-nitro alcohols, the development of one-pot catalytic diastereoselective nitromethane-free Henry reactions to generate chiral β-nitro alcohol scaffolds with four adjacent stereogenic centers has been impressive. Here we have reviewed several reactions using metal- or organo-catalytic systems to react unfunctionalized higher nitroalkanes such as nitroethane or nitropropane with carbonyl compounds in a highly enantio- and diasteroselective manner. Despite these advances, at least three substantial barriers remain. First, direct catalytic asymmetric Henry reactions in which the less reactive ketone carbonyl can serve as electrophile remain rare. Second, stereoselectivity is often poor when the substrate is a substituted nitroalkane such as bromonitromethane, 1-bromo-2-nitroethane or 2-nitroethanol. Third, chiral ligands are usually expensive to purchase or difficult to synthesize. As more efficient catalytic systems are developed, we believe that these problems will be solved, and the range of applications for these Henry reactions will expand.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We are grateful for the financial support from the NSFC (21572138), Key Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, Sichuan Research Center for Drug Precision Industrial Technology, 111 Project B18035 and “the Fundamental Research Funds for the Central Universities”.
Notes and references
- For reviews on the metal-catalyzed asymmetric Henry reactions:
(a) F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS;
(b) C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442–5444 CrossRef CAS PubMed;
(c) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561–2574 CrossRef CAS;
(d) G. Murugavel, P. Sadhu and T. Punniyamurthy, Chem. Rec., 2016, 16, 1906–1917 CrossRef CAS PubMed;
(e) S. Saranya, N. A. Harry, S. M. Ujwaldev and G. Anilkumar, Asian J. Org. Chem., 2017, 6, 1349–1360 CrossRef CAS;
(f) S. Zhang, Y.-N. Li, Y.-G. Xu and Z.-Y. Wang, Chin. Chem. Lett., 2018, 29, 873–883 CrossRef CAS.
- For reviews on the organocatalytic asymmetric Henry reactions:
(a) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315–3326 CrossRef CAS;
(b) T. Marcelli, R. N. S. Haas, J. H. Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 929–931 CrossRef CAS PubMed;
(c) Y. A. Casao, E. M. Lopez and R. P. Herrera, Symmetry, 2011, 3, 220–245 CrossRef.
-
(a) G. Rosini, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and C. H. Heathcock, Pergamon, New York, 1991, Vol. 2, pp. 321–340 Search PubMed;
(b) M. Shibasaki and H. Groger, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, Vol III, pp. 1075–1090 Search PubMed;
(c) R. S. Varma, R. Dahiya and S. Kumar, Tetrahedron Lett., 1997, 38, 5131–5134 CrossRef CAS;
(d) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001, pp. 30–69 CrossRef;
(e) W. E. Noland, Chem. Rev., 1955, 55, 137–155 CrossRef CAS.
- H. Sasai, T. Suzuki, S. Arai and M. Shibasaki, J. Am. Chem. Soc., 1992, 114, 4418–4420 CrossRef CAS.
- S. E. Milner, T. S. Moody and A. R. Maguire, Eur. J. Org. Chem., 2012, 3059–3067 CrossRef CAS.
-
(a) J.-H. Liu, L.-M. Yang and E. Ganz, Energy Environ. Mater., 2019, 2, 193–200 CrossRef CAS;
(b) J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC;
(c) J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 11944–11952 RSC;
(d) J.-H. Liu, L.-M. Yang and E. Ganz, RSC Adv., 2019, 9, 27710–27719 RSC;
(e) L.-M. Yang, V. Bacic, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, J. Am. Chem. Soc., 2015, 137, 2757–2762 CrossRef CAS PubMed;
(f) L. Xu, L.-M. Yang and E. Ganz, Theor. Chem. Acc., 2018, 137, 98 Search PubMed;
(g) Y. Liu, L.-M. Yang and E. Ganz, Condens. Matter, 2019, 4, 65 Search PubMed;
(h) J.-H. Liu, L.-M. Yang and E. Ganz, ACS Sustainable Chem. Eng., 2018, 6, 15494–15502 Search PubMed;
(i) B. Song, Y. Zhou, H.-M. Yang, J.-H. Liao, L.-M. Yang, X.-B. Yang and E. Ganz, J. Am. Chem. Soc., 2019, 141, 3630–3640 CrossRef CAS PubMed.
- For reviews, see:
(a) H. C. Aspinall, Chem. Rev., 2002, 102, 1807–1850 CrossRef CAS PubMed;
(b) H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389–2404 CrossRef CAS PubMed;
(c) D. Parker, Chem. Soc. Rev., 2004, 33, 156–165 RSC.
- H. Sasai, T. Tokunaga, S. Watenabe, T. Suzuki, N. Itoh and M. Shibasaki, J. Org. Chem., 1995, 60, 7388–7389 CrossRef CAS.
- S. Handa, K. Nagawa, Y. Sohtome, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2008, 47, 3230–3233 CrossRef CAS PubMed.
-
(a) B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed;
(b) J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- T. Mashiko, K. Hara, D. Tanaka, Y. Fujiwara, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 11342–11343 CrossRef CAS PubMed.
- T. Nitabaru, N. Kumagai and M. Shibasaki, Tetrahedron Lett., 2008, 49, 272–276 CrossRef CAS.
- B. Lecea, A. Arrieta, I. Morao and F. P. Cossio, Chem.–Eur. J., 1997, 3, 20–28 CrossRef CAS.
- T. Nitabaru, A. Nojiri, M. Kobayashi, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 13860–13869 CrossRef CAS PubMed.
- T. Ogawa, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2013, 52, 6196–6201 CrossRef CAS PubMed.
- A. Nonoyama, K. Hashimoto, A. Saito, N. Kumagai and M. Shibasaki, Tetrahedron Lett., 2016, 57, 1815–1819 CrossRef CAS.
- T. Karasawa, R. Oriez, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2018, 140, 12290–12295 CrossRef CAS PubMed.
- T. Karasawa, N. Kumagai and M. Shibasaki, Org. Lett., 2018, 20, 308–311 CrossRef CAS PubMed.
- For reviews on copper catalyzed asymmetric reactions:
(a) A. Alexakis, J. E. Backvall, N. Krause, O. Pamies and M. Dieguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed;
(b) K. Yamada and K. Tomioka, Chem. Rev., 2008, 108, 2874–2886 CrossRef CAS PubMed;
(c) T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS PubMed;
(d) C. Deutsch and N. Krause, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- C. Christensen, K. Juhl and K. A. Jørgensen, Chem. Commun., 2001, 2222–2223 RSC.
- K. Ma and J.-S. You, Chem.–Eur. J., 2007, 13, 1863–1871 CrossRef CAS PubMed.
- L. Cheng, J. X. Dong, J.-S. You, G. Gao and J. B. Lan, Chem.–Eur. J., 2010, 16, 6761–6765 CrossRef CAS PubMed.
- Y. Wei, L. Yao, B.-L. Zhang, W. He and S.-Y. Zhang, Tetrahedron, 2011, 67, 8552–8558 CrossRef CAS.
- S. Shaw and J. D. White, Chem. Rev., 2019, 119, 9381–9426 CrossRef CAS PubMed.
-
(a) L. Borer, L. Thalken, C. Ceccarelli, M. Glick, J.-H. Zhang and W. M. Reiff, Inorg. Chem., 1983, 22, 1719–1724 CrossRef CAS;
(b) D. Chen, A. E. Martell and Y.-Z. Sun, Inorg. Chem., 1989, 28, 2647–2652 CrossRef CAS.
- J. D. White and S. Shaw, Org. Lett., 2012, 14, 6270–6273 CrossRef CAS PubMed.
- A. Das, R. I. Kureshy, K. J. Prathap, M. K. Choudhary, G. V. S. Rao, N. H. Khan, S. H. R. Abdi and H. C. Bajaj, Appl. Catal., A, 2013, 459, 97–105 CrossRef CAS.
- Z.-L. Guo, S. Zhong, Y.-B. Li and G. Lu, Tetrahedron: Asymmetry, 2011, 22, 238–245 CrossRef CAS.
- A. Chougnet, G.-Q. Zhang, K.-G. Liu, D. Haussinger, A. Kagi, T. Allmendinger and W. D. Woggon, Adv. Synth. Catal., 2011, 353, 1797–1806 CrossRef CAS.
- D.-D. Qin, W. Yu, J.-D. Zhou, Y.-C. Zhang, Y.-P. Ruan, Z.-H. Zhou and H.-B. Chen, Chem.–Eur. J., 2013, 19, 16541–16544 CrossRef CAS PubMed.
- M. A. Dickson, R. D. Carvajal, A. H. J. Merrill, M. Gonen, L. M. Cane and G. K. Schwartz, Clin. Cancer Res., 2011, 17, 2484–2492 CrossRef CAS PubMed.
- G.-Y. Lai, F.-F. Guo, Y.-Q. Zheng, Y. Fang, H.-G. Song, K. Xu, S.-J. Wang, Z.-G. Zha and Z.-Y. Wang, Chem.–Eur. J., 2011, 17, 1114–1117 CrossRef CAS PubMed.
- K. Xu, G.-Y. Lai, Z.-G. Zha, S.-S. Pan, H.-W. Chen and Z.-Y. Wang, Chem.–Eur. J., 2012, 18, 12357–12362 CrossRef CAS PubMed.
- S. Handa, K. Nagawa, Y. Sohtome, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2008, 47, 3230–3233 CrossRef CAS PubMed.
- Y. Li, P. Deng, Y.-M. Zeng, Y. Xiong and H. Zhou, Org. Lett., 2016, 18, 1578–1581 CrossRef CAS PubMed.
- T. Arai, M. Watanabe, A. Fujiwara, N. Yokoyama and A. Yanagisawa, Angew. Chem., Int. Ed., 2006, 45, 5978–5981 CrossRef CAS PubMed.
- T. Arai, M. Watanabe and A. Yanagisawa, Org. Lett., 2007, 9, 3595–3597 CrossRef CAS PubMed.
- A. Noole, K. Lippur, A. Metsala, M. Lopp and T. Kanger, J. Org. Chem., 2010, 75, 1313–1316 CrossRef CAS PubMed.
- Y.-R. Zhou, J.-F. Dong, F.-L. Zhang and Y.-F. Gong, J. Org. Chem., 2011, 76, 588–600 CrossRef CAS PubMed.
- W. Jin, X.-C. Li, Y.-B. Huang, F. Wu and B.-S. Wan, Chem.–Eur. J., 2010, 16, 8259–8261 CrossRef CAS PubMed.
- W. Jin, X.-C. Li and B. Wan, J. Org. Chem., 2011, 76, 484–491 CrossRef CAS PubMed.
- T. Arai, R. Takashita, Y. Endo, M. Watanabe and A. Yanagisawa, J. Org. Chem., 2008, 73, 4903–4906 CrossRef CAS PubMed.
- F. Liu, S.-H. Gou and L. Li, Appl. Organomet. Chem., 2014, 28, 186–193 CrossRef CAS.
- D. Scharnagel, A. Mìller, F. Prause, M. Eck, J. Goller, W. Milius and M. Breuning, Chem.–Eur. J., 2015, 21, 12488–12500 CrossRef CAS PubMed.
- For reviews on chiral N-oxides in asymmetric catalysis, see:
(a) A. V. Malkov and P. Kocovsky, Curr. Org. Chem., 2003, 7, 1737–1757 CrossRef CAS;
(b) G. Chelucci, G. Murineddu and G. A. Pinna, Tetrahedron: Asymmetry, 2004, 15, 1373–1389 CrossRef CAS;
(c) X.-H. Liu, L.-L. Lin and X.-M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed;
(d) K. Shen, X.-H. Liu, L. L. Lin and X.-M. Feng, Chem. Sci., 2012, 3, 327–334 RSC;
(e) X.-H. Liu, L.-L. Lin and X.-M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC.
- B. Qin, X. Xiao, X.-H. Liu, J.-L. Huang, Y.-H. Wen and X.-M. Feng, J. Org. Chem., 2007, 72, 9323–9328 CrossRef CAS PubMed.
- H.-J. Mei, X. Xiao, X.-H. Zhao, B. Fang, X.-H. Liu, L.-L. Lin and X.-M. Feng, J. Org. Chem., 2015, 80, 2272–2280 CrossRef CAS PubMed.
- G. Blay, L. R. Domingo, V. H. Olmos and J. R. Pedro, Chem.–Eur. J., 2008, 14, 4725–4730 CrossRef CAS PubMed.
- G. Blay, V. H. Olmos and J. R. Pedro, Chem. Commun., 2008, 4840–4842 RSC.
- S. Kezuka, T. Mita, N. Ohtsuki, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2001, 74, 1333–1342 CrossRef CAS.
- S. Kezuka, Y. Kogami, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2003, 76, 49–58 CrossRef CAS.
- J. Park, K. Lang, K. A. Abboud and S. Hong, J. Am. Chem. Soc., 2008, 130, 16484–16485 CrossRef CAS PubMed.
- J. Park, K. Lang, K. A. Abboud and S. Hong, Chem.–Eur. J., 2011, 17, 2236–2245 CrossRef CAS PubMed.
- K. Lang, J. Park and S. Hong, Angew. Chem., Int. Ed., 2012, 51, 1620–1624 CrossRef CAS PubMed.
-
(a) Y. Sohtome, Y. Hashimoto and K. Nawasaga, Eur. J. Org. Chem., 2006, 2006, 2894–2897 CrossRef;
(b) Y. Sohtome, N. Takemura, K. Takada, R. Takagi, I. Toshitsugu and K. Nawasaga, Chem.–Asian J., 2007, 2, 1150–1160 CrossRef CAS PubMed.
- K. Takada, N. Takemura, K. Cho, Y. Sohtome and K. Nagasawa, Tetrahedron Lett., 2008, 49, 1623–1626 CrossRef CAS.
-
(a) L. M. Ramos, M. O. Rodrigues and B. A. D. Neto, Org. Biomol. Chem., 2019, 17, 7260–7269 RSC;
(b) R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed;
(c) J. M. Crawford and M. S. Sigman, Synthesis, 2019, 1021–1036 CAS.
- H. Ube and M. Terada, Bioorg. Med. Chem. Lett., 2009, 19, 3895–3898 CrossRef CAS PubMed.
- J. V. Alegre-Requena, E. Marques-Lopez and R. P. Herrera, Adv. Synth. Catal., 2016, 358, 1801–1809 CrossRef CAS.
-
(a) H. Z. Ni, W. L. Chan and Y. X. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed;
(b) H. C. Guo, Y. C. Fan, Z. H. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- D. Uraguchi, S. Sakaki and T. Ooi, J. Am. Chem. Soc., 2007, 129, 12392–12393 CrossRef CAS PubMed.
- D. Uraguchi, S. Nakamura and T. Ooi, Angew. Chem., Int. Ed., 2010, 49, 7562–7565 CrossRef CAS PubMed.
- K. Kanagaraj, P. Suresh and K. Pitchumani, Org. Lett., 2010, 12, 4070–4073 CrossRef CAS PubMed.
- M.-X. Liu, N. Ji, L. Wang, P. Liu and W. He, Tetrahedron Lett., 2018, 59, 999–1004 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a and
Fen-Er Chen
*ab
a and
Fen-Er Chen
*ab