
Nano-immunoimaging
Ping
Wang†
 ab,
Taeho
Kim†
ac,
Masako
Harada
ac,
Christopher
Contag
abcd,
Xuefei
Huang
ace and
Bryan Ronain
Smith
*acf
ab,
Taeho
Kim†
ac,
Masako
Harada
ac,
Christopher
Contag
abcd,
Xuefei
Huang
ace and
Bryan Ronain
Smith
*acf
aInstitute for Quantitative Health Science and Engineering, Michigan State University, 775 Woodlot Drive, Room #1118, East Lansing, MI 488824, USA. E-mail: smit2901@msu.edu
bPrecision Health Program, Michigan State University, East Lansing, MI 488824, USA
cDepartment of Biomedical Engineering, Michigan State University, East Lansing, MI 488824, USA
dDepartment of Microbiology & Molecular Genetics, Michigan State University, East Lansing, MI 488824, USA
eDepartment of Chemistry, Michigan State University, East Lansing, MI 488824, USA
fDepartment of Radiology, Stanford University, Stanford, CA 94306, USA
First published on 11th December 2019
Abstract
Immunoimaging is a rapidly growing field stoked in large part by the intriguing triumphs of immunotherapy. On the heels of immunotherapy's successes, there exists a growing need to evaluate tumor response to therapy particularly immunotherapy, stratify patients into responders vs. non-responders, identify inflammation, and better understand the fundamental roles of immune system components to improve both immunoimaging and immunotherapy. Innovative nanomaterials have begun to provide novel opportunities for immunoimaging, in part due to their sensitivity, modularity, capacity for many potentially varied ligands (high avidity), and potential for multifunctionality/multimodality imaging. This review strives to comprehensively summarize the integration of nanotechnology and immunoimaging, and the field's potential for clinical applications.
 Ping Wang | Peter (Ping) Wang received his bachelor's degree in Clinical Medicine from Shanxi Medical University, Taiyuan, China, followed by residency training in the First Affiliated Hospital of Shanxi Medical University. Then he pursued doctoral research in Imaging and Nuclear Medicine at the Fudan University, Shanghai, China. Following his doctoral work and clinical fellow training, Dr Wang moved to the United States to pursue postdoctoral research at the Molecular Imaging Laboratory in the Athinoula A Martinos Center for Biomedical Imaging, Massachusetts General Hospital, Harvard Medical School. He is now an Assistant Professor in the Precision Health Program and member of the Institute for Quantitative Health Science and Engineering at Michigan State University. |
 Taeho Kim | Taeho Kim is an Assistant Professor in the Department of Biomedical Engineering and a member of the Institute for Quantitative Health Science and Engineering at Michigan State University. His research team at MSU focuses on developing nanoscale inorganic materials as novel contrast agents and therapeutics for medical applications. Dr Kim completed his PhD in chemical and biological engineering at Seoul National University, including two years as a visiting researcher at Johns Hopkins Radiology. He did his postdoctoral training in molecular imaging and nanotechnology at the University of California, San Diego. |
 Masako Harada | Masako Harada is an assistant professor in the Department of Biomedical Engineering at Michigan State University who was recruited from Stanford to join the Institute for Quantitative Health Science and Engineering. The ultimate goal of her research is to translate basic scientific findings into clinical applications. Dr Harada received her undergraduate degree in molecular genetics from King's College London. She earned her PhD in experimental oncology from Karolinska Institute in Sweden and conducted her postdoctoral training in cancer chemotherapy at Stanford University. |
 Christopher Contag | Christopher Contag, the inaugural James and Kathleen Cornelius Endowed Chair, was recruited to Michigan State University from Stanford to lead the new Institute for Quantitative Health Science and Engineering. A pioneer in molecular imaging, Dr Contag also serves as the Chair of the Department of Biomedical Engineering. Prior to joining MSU, he held many roles at Stanford, including associate chief of Neonatal and Developmental Medicine, director of Stanford's Center for Innovation in In vivo Imaging, and co-director of the Molecular Imaging Program. Dr Contag is a fellow and past president of the World Molecular Imaging Society. He is a founding member and past president of the Society for Molecular Imaging (SMI), and recipient of the Achievement Award from the SMI and the Britton Chance Award from SPIE for his fundamental contributions to optics. Dr Contag graduated from the University of Minnesota with a BS in biology and a PhD in microbiology. |
 Xuefei Huang | Xuefei Huang is a professor in the Departments of Biomedical Engineering and Chemistry, member of the Institute for Quantitative Health Science and Engineering, and MSU Foundation Professor. The multi-disciplinary approach of his group interfaces biology, chemistry, and engineering to develop next-generation tools for diagnosis and treatment of diseases. Prior to joining MSU in 2008, Dr Huang held appointments at the University of Toledo and Columbia University. He is a graduate of the University of Science and Technology of China and earned his PhD from Columbia University. |
 Bryan Ronain Smith | Bryan R. Smith is an Associate Professor of Biomedical Engineering and a member of the Institute for Quantitative Health Science and Engineering at Michigan State University. His lab blends engineering, nanochemistry, biology, physics, and medicine to develop new imaging and therapeutic approaches. He is developing novel nanotechnology-based strategies to harness the power of the immune system, creating novel diagnostic imaging and therapeutic agents for translational applications to combat diseases including cancer, atherosclerosis, and neurodegeneration. Dr Smith received his BS in Physics, Mathematics, and Biomedical Engineering from Tufts University, PhD from The Ohio State University, and did his post-doctoral work at Stanford University. Dr Smith was recently recruited to MSU from Stanford, where he has been a Visiting Associate Professor. |
1. Introduction
A variety of imaging modalities and approaches have been developed as tools to track disease initiation and progression, and to inform clinical practice with diagnostic, prognostic, staging and response-to-therapy data. Because the immune system is intimately involved in the progression of many diseases, a burgeoning sub-discipline within imaging is to monitor immune cell trafficking patterns and cell–cell and cell–tissue interactions as well as assess immune cell mediators, such as secreted cytokines and immune cell-derived extracellular vesicles. This field—immunoimaging—can yield insights into disease states based on the body's native immune responses,1–3 and in some cases can be engineered to also manipulate immune functions.4,5 Indeed, the recent successes of immunotherapy have helped drive the surge in interest in the in vivo interactions of the immune system with normal processes and disease targets, which has led to rapidly emerging immunoimaging technology and development. These strategies will advance our understanding of the clinical utility of the immune response; although still in its infancy, immunoimaging promises significant predictive capabilities for assessing disease progression and response to therapies, particularly in the development and use of immunotherapies. Yet many challenges remain in immunoimaging, including: (i) identifying suitable immune cell types and/or related molecules as imaging biomarkers; (ii) efficient and durable immune cell labeling for informative measures; (iii) developing approaches that facilitate detection without perturbing cell functions or phenotype; (iv) designing strategies that persist though possible dramatic expansion and maturation of immune function in response to an infectious or malignant insult; and (v) establishing procedures and methods for each imaging modality that provides the appropriate parameters, including sufficient quantitation, localization, resolution, penetration depth, and contrast to be informative for the investigator or clinician.Early advances in immunoimaging using fluorescent tags to image at the microscale6,7 have led to tremendous immunological insights in pre-clinical studies, and the early studies with nuclear medicine tools such as positron emission tomography (PET)8,9 planar imaging with gamma cameras, and single photon emission tomography (SPECT) have provided useful information on immune cell distribution for decades in the clinic. In contrast to these approaches that use labeled molecules, imaging with nanomaterials offers some advantages that include mitigating the need for radioactivity, affording intelligent control over delivery, and increasing diagnostic sensitivity and specificity that would not be otherwise possible. Nanomaterials are particularly valuable in immunoimaging based on their strong signal, enabling detection sensitivity down to single cell visualization in some cases (e.g., with optical and magnetic modalities),10–12 high specific immune cell selectivity/avidity,10 and the potential for multifunctionality including targeting, imaging diagnostics, and therapeutics (e.g., theranostics).13,14 The first immunoimaging nanomaterials were primarily iron oxide particles used to label immune cells, particularly macrophages, to track cells using modalities such as magnetic resonance imaging (MRI) and SPECT.15
In this review, we strive to provide a foundation for asking immunological questions through imaging followed by a comprehensive overview of nano-immunoimaging, focused on the nexus between imaging, nanotechnology, and immunology. We define nanoparticles as particles that are specifically engineered between 1 and 100 nm in at least one dimension critical to its application.16,17 Furthermore, we will: (i) focus on the use of nanomaterials for imaging the immune system, particularly its many cellular subsets; (ii) review only nanomaterials that have been used in vivo for imaging (i.e., they have been injected into living subjects); and (iii) concentrate on insights from the last 20 years (roughly the start of immunoimaging). The sections are divided without regard to specific diseases, and are intended to provide an introduction to the key areas that comprise nano-immunoimaging.
2. Short primers
i. Imaging
Imaging technologies used for nano-immunoimaging span the suite of available modalities (Table 1), which are categorized into magnetic, optical, acoustic, nuclear and multimodality imaging.16 There have been a number of recent reviews of these modalities, nonetheless, we briefly review these imaging tools below as a foundation for our discussion of nano-immunoimaging. The multimodality instruments and approaches, such as photoacoustic imaging (optical and acoustic), comprise two or more modalities from the four categories of imaging approaches (magnetic, optical, acoustic, and nuclear), and thus our description of the four categories provides a convenient framework for nanomaterial imaging in both single and multimodality approaches. We will acknowledge when multimodality approaches offer imaging benefits as we describe the applications and questions in nano-immunoimaging.| Magnetic imaging (MRI & MPI) | Nuclear imaging (PET & SPECT) | Computed tomography | Optical imaging | Ultrasound | Photoacoustics | |
|---|---|---|---|---|---|---|
| Mechanism of contrast | Difference in nuclear magnetic moment | Gamma emitting radiotracer | Absorption of X-rays by tissue | Fluorescence or bioluminescence | Difference in tissue density | Difference in light absorption |
| Tissue depth | Unlimited | Unlimited | Unlimited | <1 cm | Several cm | Several cm |
| Temporal res. | Min–hours | Minutes | Minutes | Seconds–minutes | Seconds | Seconds–minutes |
| Spatial res. | Sub-mm | 2 mm | 0.5 mm | 2–3 mm | mm | mm |
| Use in clinic | Routine | Routine | Routine | Limited | Routine | Limited |
| Cost | $$$$ | $$$ | $$ | $ | $ | $$ |
| Advantages | Soft tissue contrast; depth | Established; sensitivity; multiplexing (SPECT) | High resolution; accessibility | Many reporters; multiplexing | Rapid; inexpensive; accessibility | Minimize optical scatter; depth (vs. optical); multiplexing |
Computed tomography (CT) is a related imaging technique, also capable of whole body imaging, with high spatial resolution and rapid image acquisition. In CT imaging, the X-ray tube and detectors rotate 360° during which the absorption of X-rays is measured from many different angles. Subsequently, three-dimensional, tomographic images of the internal structures are reconstructed from these X-ray intensity profiles by computational algorithms.40 CT has been widely used in the clinic as an anatomical imaging tool, particularly for visualizing bone structures because of the apparent contrast difference between electron-dense bones and surrounding electron-poor soft tissues. However, due to the inherent low sensitivity of CT, contrast agents are required to visualize the changes that occur in soft tissues.41 These agents must display fairly high electron densities to produce robust CT signal, and nanomaterials are among the optimal types of agents to do so based on the ability to fabricate particles of high-electron density entities with hundreds to millions of atoms, e.g., gold,42 bismuth,43 tantalum, and silver. If a CT scanner is equipped with dual-energy or filters, differential absorption of X-rays by nanoparticles of different material composition can result in multispectral CT, allowing multiplexing. Given the anatomic information that CT provides, it is sometimes used as the anatomical overlay for functional imaging modalities such as PET and MPI that do not intrinsically provide anatomic structural contrast.
ii. Nanoparticles
Nanoparticles (NPs) enable imaging and tracking targeted cells and molecules deep within tissues of the living body using a variety of imaging modalities. These include magnetic, nuclear, optical, acoustic, and ‘adaptable’ nanotechnologies, and combinations thereof.16 NPs display a variety of advantageous properties, including multifunctionality and intelligent control. NPs attain imaging capabilities based on their unique functional properties (e.g., magnetic, optical, X-ray attenuation) originating from their component materials and nanoscale dimensions.44 For example, in MRI, superparamagnetic iron oxide NPs (SPIONs; with size below 20 nm) have been extensively used as T2* MRI contrast agents due to the fact that these particles induce a local perturbation of magnetic fields in the MRI scanner, resulting in the shortening of T2 relaxation and generating hypo-intense signals (dark contrast) on T2*-weighted MR imaging.45 SPIONs are considered safe and allow ultrasensitive detection, and some iron oxide particles can be visualized even down to the single cell/particle level.12 Materials with a high atomic number can absorb X-ray photons more effectively,46 making NPs comprising high-Z elements excellent CT contrast agents. Gold NPs tend to be the most attractive owing to their excellent biocompatibility, facile synthesis, and ease of bioconjugation.41,47,48 However, high masses of materials are often required due to the relatively low sensitivity of the approach.16NPs for immune cell imaging can be made fluorescent with linked fluorochromes. However, the photo-stability, high quantum yield of quantum dots (QDs) and their size-tunable emission, made QDs robust fluorescent tags of widespread utility for optical immune imaging.49,50 Recently, to overcome the shallow tissue penetration depth of UV excitation, lanthanide-based upconversion NPs (UCNPs), which fluoresce by a reverse-Stokes process upon near-infrared light activation (typically 600–900 nm), have emerged as new fluorescent NPs for immune imaging with low backgrounds.51,52 Nuclear nanomaterials are often formulated via the surface decoration of NPs with radioisotopes (e.g., 64Cu, 124I, 89Zr) through chelation chemistry, or by incorporation of the radioisotope within the NP.53,54 Upon accumulation at a target site, radioisotopes in nuclear nanomaterials produce radioactive decays for detection by a nuclear imaging modality such as PET or SPECT.
Many nanomaterials display superior flexibility and modularity in chemical design over small-molecule or protein-based imaging agents.55 Multimodal imaging NPs detectable by two (or more) imaging modalities that can interact synergistically may significantly improve diagnostics capabilities.56,57 For example, MRI and PET are complementary modalities, that can, for example, provide structural (anatomic) and functional information, with the high sensitivity of PET balanced by the high spatial resolution of MRI.58 Various methods to prepare composite nanomaterials and endow them with multimodal imaging properties have been developed (e.g., doping of radioisotopes into magnetic NPs, radiolabeled gold NPs, nanoscale assembly of magnetic NPs with quantum dots (QDs)).16,59,60 Upon loading therapeutic payloads into the nanoscale matrix, imaging NPs can also achieve multifunctional utility, e.g., theranostics (combination of diagnostic imaging and drug delivery). Examples of adaptable/multifunctional nanomaterials include liposomes, micelles, polymeric NPs, inorganic silicate, and natural nanomaterials (e.g., exosomes, lipoproteins).16,61–63
iii. Immunology
The immune system is a highly-regulated, complex, dynamic and interactive collection of cells and molecules that mediate responses to insults such as infection and malignancy. The dynamic nature of the immune system is required to combat a wide variety of insults that are endogenous, i.e., derived from self (e.g. cancer) with very subtle differences from normal cells, or exogenous, such as bacteria and viruses. Viruses and some bacteria can live inside the host cells making them difficult targets for the immune system, and there are commensal viruses and bacteria that live in the body without causing disease, necessitating the need for the immune system to assess the level of threat for each entity it encounters. To accomplish this very complex set of tasks, the immune system can both simply react without prior exposure, and it can “learn” from previous encounters with the presumed threat; these two fundamental functions are referred to as the innate and the adaptive immune systems, respectively. These two aspects of the immune system comprise immune cells and effector molecules that are distinct, yet overlapping.The key cell types of the innate and adaptive immune system are organized by their primary functions, e.g., phagocytic innate cells such as macrophages, monocytes, and neutrophils that engulf pathogens and foreign bodies. There are also antigen-presenting cells such as dendritic cells, macrophages, and monocytes that collect the markers (antigens) of the attacking entity and present these markers to other (adaptive) cells to determine if they comprise a threat or not. This is called antigen presentation. Once presented, there are a variety of responses that the organism can take based on this initial assessment. The immune system can launch a cellular attack on the invader that possesses the particular marker, and this can be a cell-to-cell attack by adaptive cytotoxic T lymphocytes (CTL) or a molecular response with B lymphocytes making antibodies that bind to the threatening agent. There are also helper cells that support this attack that are also antigen-specific, called helper T cells. If the marker that is presented is nonthreatening, then antigen presentation can lead to an immunosuppressive response in which regulatory T lymphocytes (Treg) and other cells diminish the immune response.
The innate immune response has been hardwired into the system and responds to entities that appear foreign by comparing these markers to self or by an inherited knowledge of what is foreign using cell surface molecules such as those in the Toll-like family of receptors.64,65 Cells of the innate immune system include macrophages, foreign body-engulfing neutrophils, and natural killer (NK) and natural killer T cells (NK-T) that effectively assess the molecular surface of the attacking entity and compare it to the appearance of self and kill those cells appearing to be non-self with perforating molecules (perforin) and toxins (e.g. granulysin). This is all tightly regulated by markers on the cell surface such as CD47 that tell phagocytic cells not to engulf them or markers that suppress the immune response—the so-called check point inhibitors. Both CD47 and checkpoint inhibitors are being targeted in the emerging cancer immunotherapeutic space.66 There are circumstances when self-nonself recognition fails and this leads to autoimmune diseases such as type 1 diabetes, multiple sclerosis, and rheumatoid arthritis. The fact that immune cells can “learn” and change, circulate around the body looking for foreign invaders, have a wide variety and varying levels of surface markers used for their identification and in vivo targeting, and typically divide rapidly upon encountering invaders, presents unique challenges—and opportunities—to the development of effective imaging tools for assessing immune functions and tracking responses over time.
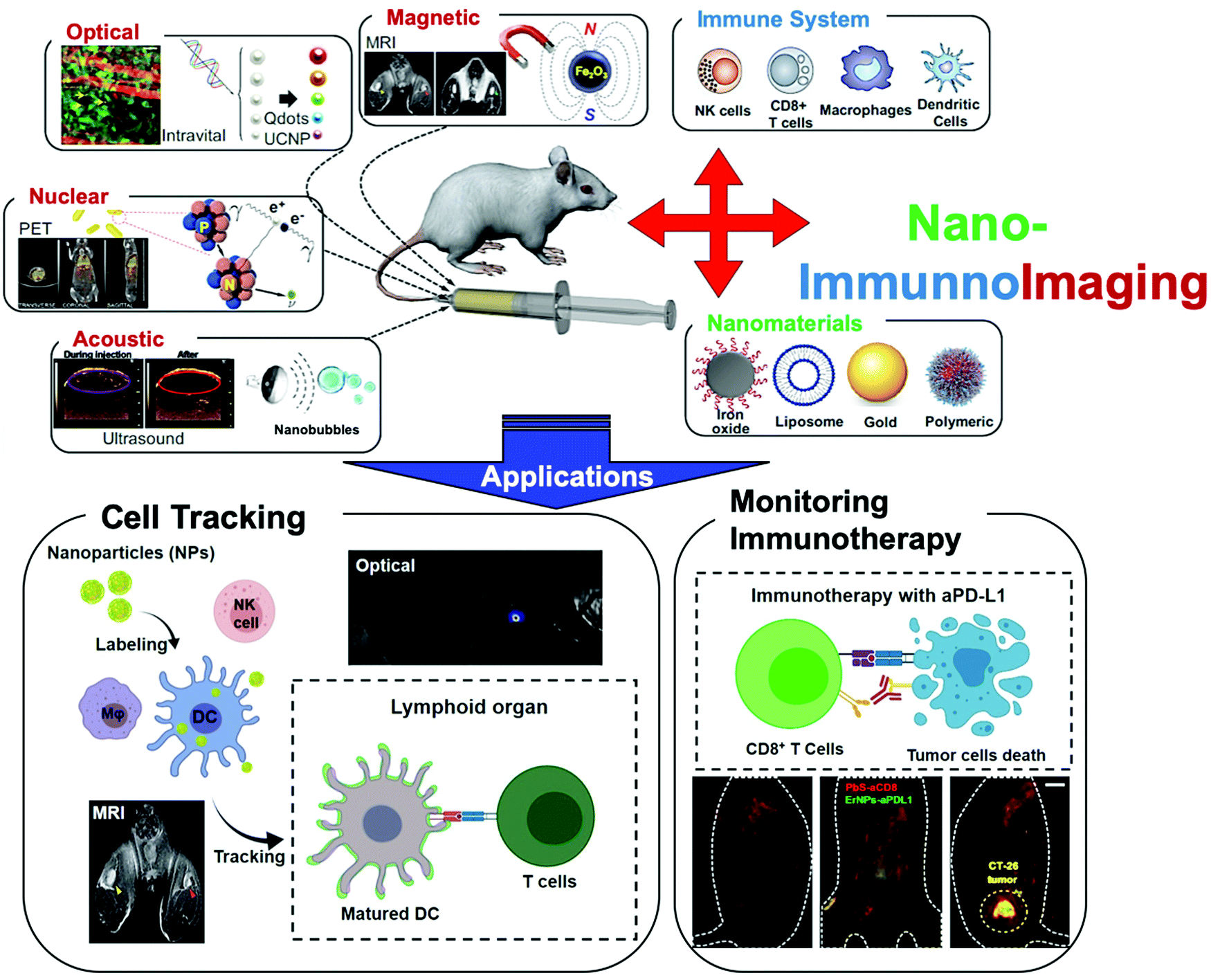 | ||
| Fig. 1 Nano-immunoimaging. This review systematically summarizes the integration of nanotechnology and immunoimaging toward both a fundamental in vivo understanding of the roles of the immune system and advancement of clinical diagnostics and therapeutics. | ||
3. Nano-immunoimaging applications
i. Learning basic biology via immunoimaging
One of the great challenges, and opportunities, in immunoimaging is due to the changing and migratory nature of components of the immune system. Immune cells may originate in the bone marrow or spleen then migrate via the blood stream to sites of infection, malignant growth, or immune stimulation, where they may collect an antigen or pathogen and then migrate through the lymphatics to draining lymph nodes or to the spleen. Throughout this process the immune cells will often mature, change their phenotypes, and divide to increase their numbers. This necessitates imaging over a range of scales, from macro- to micro-scopic imaging and incorporating imaging markers that persist through changing phenotypes and, in the future, can compensate for signal dilution due to cell division. The imaging strategies that provide a macroscale view can examine populations of immune cells in relatively large numbers as they accumulate in tissue targets, but can not, usually, be used to examine the phenotype or cellular activity of the labeled cells; this normally requires microscale imaging. Nearly all imaging strategies have been directed at these problems of determining cellular localization, concentration, and activity for cells of the immune system and many of them offer opportunities to understand immunology and guide the development and clinical application of immunotherapies. In each case, these imaging modalities can benefit from the use of NPs as contrast and in many cases NPs have been developed as multimodality agents that serve as an ideal link from macro- to micro-scopic imaging and in some cases can be used to reveal functional changes and migratory patterns.16,67Nanomaterials have provided immunological insights across the spectrum of microscale imaging modalities, including IVM, OCT, photoacoustic microscopy, and ultrasound.11,29,71,77–79 IVM has shown the trafficking of inflammatory monocytes and macrophages in brain tumors with subcellular detail,29,67 while OCT has shown tumor-associated macrophage migration patterns in brain cancer with high penetration depths.71
Imaging of cells of the immune system began initially as a means of seeking sites of inflammation with application to clinical infection80,81 and malignancy,82 and transitioned to imaging of immune activation83 as a potential tool for guiding therapies including immunotherapies in the clinic. These studies were performed using molecular contrast that had been developed for cancer imaging, particularly radiolabeled glucose (2-[18F]fluoro-2-deoxy-D-glucose, FDG) and imaged with PET and have been further developed to use molecular contrast that is specifically directed at immune cell function.8 MRI, despite its tremendous soft tissue contrast, has considerable limitations as a molecular or NP imaging modality with regard to sensitivity and the loss of signal used to localize magnetic NPs. To address the poor sensitivity of MRI, magnetic NP complexes have been used to increase the contrast.12 The iron oxide NPs used for MRI which lead to negative contrast, or ‘signal dropout’ under standard sequences, are not ideal for radiologists reading the images. MRI, and other such modalities, PET, SPECT, CT, and MPI, are used for whole body, macroscopic imaging and have utility for clinical imaging.
ii. Immune cell trafficking
NP probes, targeted to immune cells, can enable the detection and tracking of immune cells for monitoring disease progression and immune responses. The delivery of NPs to immune cells in living subjects can be broadly divided into two categories: (1) ex vivo labeling, outside the body, often with the aid of transfection agents; (2) in vivo targeting, which can be divided into passive targeting and active targeting. Passive targeting typically takes the form of immune cells which take up NPs non-specifically through phagocytosis. In active targeting, the NPs are typically functionalized with targeting ligands, which can selectively bind with proteins or other receptors on the cell surface to increase the accumulation of NPs into particular immune cell types (described in detail in Section 3(v)a).Beside macrophages, other types of innate immune cells including natural killer (NK) cells and dendritic cells (DC) have been labeled in vitro with SPIOs to support immunoimaging. A novel cell line of cytotoxic NK cells, KHYG-1, was developed for immunotherapy against prostate cancer.89 Incubation of KHYG-1 with SPIOs labeled with fluorophore rhodamine B loaded the NK cells with the NPs.90 Administration of the NPs to tumor bearing mice subcutaneously, but not intravenously or intraperitoneally, led to MR signal changes at the edge of tumor tissues detected by MRI in vivo (Fig. 2a and b). Incorporation of rhodamine B allowed matched ex vivo fluorescence imaging that showed extensive fluorescence labeling inside tumor (Fig. 2c), suggesting—as one might expect—higher sensitivity for ex vivo fluorescence detection of NK cell penetration to tumor tissues compared to MRI and highlighting why multimodal imaging can be critical to more fully understand immunological processes.
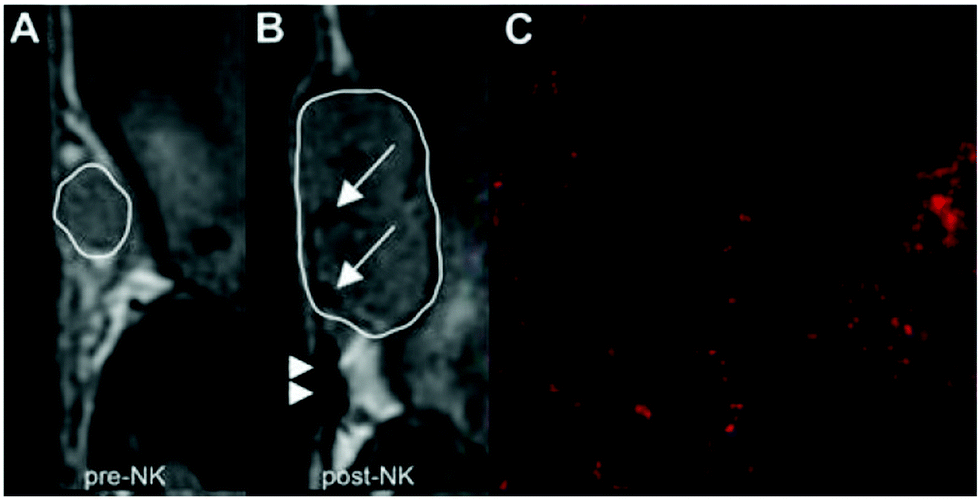 | ||
| Fig. 2 Signal loss in MRI after subcutaneous injection of rhodamine B, SPIO labeled KHYG-1 cells were observed at the edge of mouse tumor 4 days after np administration. (A) MRI image from the day before KHYG-1 administration. (B) MRI image from 4 days after KHYG-1 administration. Signal losses around the margin of tumor that received subcutaneous KHYG-1 were marked by the white arrows. The large signal loss areas marked with the arrowheads were the site of injection. (C) Fluorescence imaging showed extensive rhodamine signals from the center of the tumors 5 days after KHYG-1 administration ex vivo. | ||
Revealing DC trafficking patterns is important because of DCs’ role in detecting, processing, and presenting antigenic materials, often at different anatomic sites. To label DCs, SPIOs coated with a negatively charged block copolymer, poly(aspartic acid)-b-poly(ε-caprolactone) were incubated with the cells.91 Labeled DCs were unaffected in their viability, proliferation, and differentiation capacity, and exhibited excellent MRI sensitivity in vitro. The cells were detected by MRI in vivo in the draining lymph nodes of mice upon injections into the foot pad, demonstrating a proof-of-principle of whole-body DC tracking in live animals.
NK cells play important roles in innate immunity, and adoptive transfer of NK cells has attracted significant recent interest as a novel anti-cancer immunotherapy.92 Thus, the ability to image and track NK cells following administration in vivo can greatly aid in the monitoring of NK-based therapy.93 NK cells tend to display inherently low phagocytic activity. As a result, electroporation or a transfection agent such as protamine or lipofectin (lipofectin gave higher transfection efficiency than protamine)94,95 is often utilized to introduce SPIOs and other NPs into them. These approaches can disrupt normal cellular function, with increasing SPIO concentrations leading to lower cell viability.94 Nevertheless, SPIO-loaded NK cells were injected into liver cancer rat models. NK cells were observed in vivo by T2*-weighted MRI for up to 24 hours. Histological measurements confirmed increased NK cell delivery to tumor, which correlated well with a decrease in T2* relaxation time in livers of tumor bearing mice compared to those in normal mice.
Another method to assist NK cell transfection is to use a strong magnetic field.96 Incubation of organic dye (Cy5.5) labeled SPIOs with NK-92MI cells placed under an external magnetic field loaded the NPs into NK cells. Real-time fluorescence imaging of mice following NK administration allowed visualization of NK cell accumulation at the tumor site. Interestingly, infiltration of NK cells into tumor could be enhanced 17-fold by placing an external magnet by the tumor.
While optical imaging provides outstanding spatiotemporal resolution to visualize physiological and pathological processes, its wide application in living subjects is hampered by the limited penetration of photons through mammalian tissue. UCNPs designed to permit deeper tissue penetration with strong upconversion luminescence were synthesized and coated with positively charged polyethylenimine (PEI) or oligo-arginine.52,97 The greater tissue penetration at these longer wavelengths is due in large part to the reduced absorption of light by hemoglobin in the 800–1100 nm region of the spectrum, as well as decreased optical scattering. Upon incubation with DCs, UCNPs were engulfed. When hypodermically injected into mouse footpads, UCNP-labeled DCs homed to draining lymph nodes, as detected by luminescence imaging (Fig. 3).52 Furthermore, a model antigen (ovalbumin) was adsorbed onto the UCNPs, inducing DC maturation and cytokine release, thereby providing a platform for combined (i) imaging of lymph node drainage and (ii) immune activation leading to enhanced T cell proliferation, interferon gamma (IFN-γ) production, and CTL-mediated responses. A second example of imaging deeply via optical strategies is the use of OCT to study immune cell movement and function in the tumor microenvironment. To enable visualization of tumor associated macrophages (TAM) by OCT, TAM were externally labeled with large gold nanorods as optical scatter-based contrast.71 This approach can potentially allow deeper TAM tracking than would otherwise be possible using any standard IVM technique, enabling tracking potentially deep into biologically-complex tumor hypoxic or necrotic regions.
 | ||
| Fig. 3 UCL-labeled DC migration from footpad to draining lymph node as detected by in vivo UCL imaging. Labeled DCs were injected into the right footpad of a mouse. 48 h after injection, strong UCL signals from the draining lymph node were observed with in vivo UCL imaging (UCL panel). Overlay of the bright field image with UCL image (merge panel) localized the position of the NPs within the draining lymph node. | ||
In addition to innate immune cells, adaptive immune cells have also been labeled ex vivo. SPIOs were functionalized with a positively-charged cell-penetrating peptide ‘TAT’ to enhance their cellular uptake98 into CD8+ cytotoxic T lymphocytes via incubation. This strategy allowed detection of cells via MRI with a detection threshold of 2 cells/voxel in vitro and ∼3 cells/voxel in live mice. This method enabled high resolution, clinically-relevant non-invasive imaging and spatiotemporal localization of T-cell recruitment to tumors in vivo.
Traditional MR imaging monitors changes in relaxation time of water molecules, which has relatively low sensitivity due to the high background from water. 19F-MRI is a promising approach for in vivo imaging since natural levels of 19F in the body are very low, resulting in essentially no background in 19F-MRI, while conventional proton-based MRI can provide anatomical context. In an instance of immunotherapeutic monitoring, T cells labeled with a nano-emulsion containing perfluorinated polyether were injected into mice intraperitoneally in an inflammation model.99 Longitudinal 19F-MRI over 21 days revealed dynamic accumulation and clearance of T cells in the draining lymph nodes, with T cell numbers quantifiable via19F MRI data (Fig. 4). Similar NP strategies have been applied to label T cells for tracking diabetogenic T cells to the pancreas in a diabetic mouse model,100 to track Chimeric Antigen Receptor (CAR) T-cells in patients,101 to track macrophages in vivo with 19F for detection of acute allograft rejection via19F-MRI,102 and recently to perform multimodal 19F-MRI and ultrasound for visualizing the localization of therapeutic DCs.103
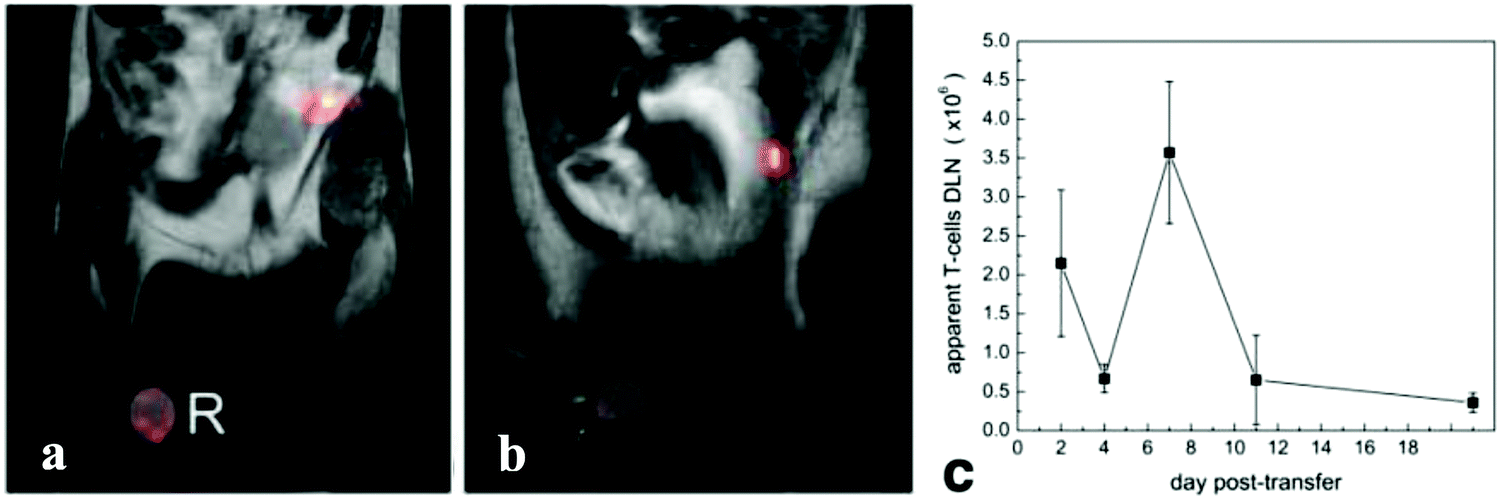 | ||
| Fig. 4 In vivo MRI of 19F-labeled T cells in a mouse inflammation model. (a) 4 days, (b) 21 days after administration of labeled T cells. 19F is shown in pseudocolor with 1 h shown in grayscale. T cells were detected primarily in the draining lymph nodes. The external reference tube used for quantification was labeled R. (c) Apparent T-cell quantification in the draining lymph node from in vivo19F-MRI data. | ||
Nuclear imaging, with its ultra-high contrast, has also been tested to monitor CAR-T cell therapy. Electro-transferred gold NPs (GNPs) functionalized with 64Cu2+ were used to track clinical-grade CAR-T cells. MicroPET/CT revealed the in vivo biodistribution of CAR-T cells, ensuring these cells were at the intended site, but not elsewhere potentially producing side effects, thereby highlighting the utility of radiolabeled GNPs for PET-based CAR-T cell imaging.104
Endogenous SPIO-labeling of macrophages can also be used to track the innate immune response to stem cell transplants. The host immune system may often reject allogeneic cell transplants, but the time-scale and mechanism is not well-understood. By combining MRI and IVM in a form of multimodality imaging that bridges the micro- to the macroscales, stem cell rejections were detected, yielding insights neither alone could have provided.111 In this study, rhodamine-conjugated ferumoxytol was pre-injected i.v. to C57BL/6-Tg (Csf1r-EGFP-NGFR/FKBP1A/TNFRSF6) 2Bck/J mice, which yields green fluorescent protein (GFP) expression under the control of the Csf1r promoter in macrophages (which also tended to take up ferumoxytol). 48 hours later, calvarial defects were created, and immune-matched or mismatched stem cells in an alginate scaffold were implanted into these defects. Immune-mismatched cell implants demonstrated stronger MR signals than immune-matched cells due to increased stem cell death and subsequent recruitment of SPIO-laden macrophages to engraftments. MR signal changes correlated well with IVM images, due to increasing quantities of GFP-expressing and rhodamine-positive macrophages in the mismatched transplants.
NP-based tools can thus help overcome the challenging immunogenicity (or allogenicity) that may occur in cell therapy due to consequent inflammation or immune-related conditions. Smart NPs were recently designed to reveal early immune rejection-induced apoptosis of MSCs. These nanoprobes are composed of an SPIO backbone and peptides with activatable fluorophore “light-up” features in the presence of activated caspase-3,112 an apoptosis marker. NPs were exogenously labeled to either immune-matched (mouse MSCs) or mismatched stem cells (pig MSCs), and the cells were transplanted into cartilage defects in mice. T2*-weighted MRI signals from SPIOs successfully localized in cell engraftments throughout the body. Once localized, the intense “turn-on” fluorescence signals were observed via IVM imaging in apoptotic pig MSCs with a decrease in the overall cell numbers. While such strategies display the promise of NP-based probes, they will likely require a mechanism to control potential unexpected proliferation or differentiation of MSCs for successful clinical translation.
Micrometer-sized iron oxide magnetic particles (MPIOs) have higher relaxivities per particle compared to SPIOs so that a single MPIO particle—and thus single cell—may be visualized by MRI.12,113 Imparting high sensitivity, MPIOs were administered intravenously to rats receiving a heart transplantation. Macrophages readily took up the particles in vivo and infiltrated the heart as observed by MRI in live animals, providing a macrophage-based proxy to sensitively detect acute cardiac rejection after solid organ transplantation.
In an example of an immunoimaging approach that is made possible by nanotechnology, magneto-motive ultrasound imaging (MM-US) was employed using intravenously-injected SPIOs. MM-US, like MPI, requires magnetic NPs as contrast in order to generate images, with signal existing where NPs localize and nowhere else; this results in extremely high contrast-to-noise ratios akin to nuclear imaging modalities such as PET and SPECT. MM-US creates signal by focusing strong magnetic fields to induce motion in tissue, which in this case was measured using ultrasound,114 but can also be measured using OCT or PAI.16 After injection, the pattern and dynamics of macrophage SPIO uptake in the liver was readily quantified. Imaging techniques in which NPs are required such as MM-US and MPI are gaining visibility, and particularly have strong potential for sensitive, non-radioactive whole-body immune cell tracking applications.
Nuclear imaging has likewise proven valuable to visualize macrophages. Tumor-associated macrophages (TAMs) are cancer progression indicators, and TAM levels likely influence therapeutic responses to immunotherapy. Therefore, a 64Cu-labeled polyglucose NP was developed for TAM quantification. Intriguingly, PET data showed that TAM-rich tumors accumulated >700% higher amounts of a model therapeutic NP compared to TAM-deficient tumors, suggesting that the radiolabeled TAM-targeted polyglucose NP strategy could be eventually used for patient stratification in immunotherapy trials.115
iii. Monitor inflammation/cellular activation
Commonly-used, clinically viable non-invasive imaging approaches with PET using tracer FDG, or with MRI using gadolinium as the contrast agent, can be used to provide the metabolic and anatomic information (respectively) that is often associated with the pathophysiology of inflammation.116,117 FDG–PET has tremendous sensitivity with spatial resolutions limited to about 0.5 cm in humans, whereas MRI provides higher-resolution images but with much lower sensitivity to molecular contrast. FDG–PET signal is dictated by glucose utilization in vivo, and since activated immune cells increase their energy needs, they can be preferentially visualized. However, the heart, brain, and malignancies are tissues with high glucose utilization, so their inflammation can not easily be imaged with FDG. Other PET tracers have been developed for T cell activation to provide insights into inflammation and immune cell therapies, but these are not yet widely used clinically.8,118 The multi-functional capability of NP-based probes can provide several types of contrast, a major strength that has propelled their use as multimodality imaging agents.13 The purpose of this approach is to synergistically combine the advantages of each modality and overcome their limitations. As opposed to a single contrast agent or molecular tracer, NPs are customizable for multifunctionality based on their large surface area and structural diversity, offering significant opportunities for imaging given their long circulation time in blood. These aspects of nanomaterials make them an attractive option to existing, and future, small molecule agents for in vivo molecular imaging.119Multifunctional imaging NPs based on a magneto-fluorescent NP structure were used for cardiovascular MRI to detect myocarditis. These NPs effectively discriminated between grades of inflammation in experimental autoimmune myocarditis (EAM) rat models as they were taken up into various inflammatory cells within the inflamed myocardium. The NPs thereby enabled assessment of the progression of myocardial inflammation in the same animal over time.120 In another example, the chemokine receptor 5 (CCR5), was targeted at a late stage of atherosclerosis using 64Cu-DOTA-DAPTA-comb NPs to examine receptor specific uptake in PET/CT imaging.121 These NPs accumulated in the lesions to a greater extent than in normal tissues based on targeting to various immune cells including macrophages, DCs, and T cells which express CCR5 for native homing purposes.
iv. Monitoring immunotherapy as a theranostic
The ability to monitor the efficacy of an immune treatment in real time is often critical for better therapeutic outcomes for patients.122 Clinical readouts of efficacy generally rely on indirect measurements, which often do not fully reflect actual effects. All major in vivo imaging modalities have already been applied for monitoring immunotherapy in vivo, including using various NPs. While various applications of immunotherapy monitoring were discussed in Section 3(ii)a, here we focus our discussion on the theranostic applications of nano-immunoimaging. Theranostics is an emerging biomedical branch nourished by development in the diverse fields of materials science, synthetic chemistry, and molecular imaging. ‘Theranostic’ is defined as a combination of therapy and diagnostic imaging,123 merging these conventionally separate approaches into a single delivery ‘package’ which has the potential to overcome undesirable differences in biodistribution and selectivity that currently exist between distinct imaging and therapeutic agents.124,125 Immunotheranostic NPs can be highly beneficial both for drug delivery and monitoring immunotherapy in translational medicine.124One recent example of ICB imaging adopted optical imaging in the near-infrared window to afford high-resolution imaging at sub-centimeter tissue depths. Multiplexed molecular imaging can be configured to enable simultaneous optical monitoring of several different molecular targets, including PD-L1 expression and immune cell status to enhance prediction of immunotherapeutic response. Zn-Doped α-phase (cubic-phase) ErNPs (α-NaYbF4:Er,Ce,Zn@NaYF4) exhibit strong downconversion luminescence in the near-infrared-IIb (NIR-IIb) (1500–1700 nm) window (Fig. 5a). Using anti-PD-L1 mAb-labeled ErNPs, PD-L1 expression was measured in vivo in CT-26 colon tumors (which over-express PD-L1), with a tumor-to-normal tissue signal ratio of ∼40 (Fig. 5b).128 These NPs have millisecond-scale lifetimes (∼4.3 ms), compared to PbS QDs with much shorter microsecond lifetimes (∼4.6 μs), in the same ∼1600 nm window. Time-resolved imaging was thus possible, enabling 2-plex in vivo molecular imaging with anti-PD-L1 mAb-labeled ErNPs (targeting PD-L1) and anti-CD8α mAb-labeled PbS QDs (targeting CD8+ T cells). This novel nano-immunoimaging strategy successfully uncovered spatially heterogeneous bio-distributions of PD-L1 and CD8+ cytotoxic T lymphocyte (CTLs) in tumors and altered CD8 signals in tumor and spleen due to immune activation (Fig. 5c and d). Given that PD-L1 is a checkpoint related to CD8+ T cell ability to destroy tumor cells, simultaneous quantification of the localization of PD-L1 and cytotoxic CD8+ tumor cells could help predict whether anti-PD-L1 therapy is likely to be successful in individual patients.
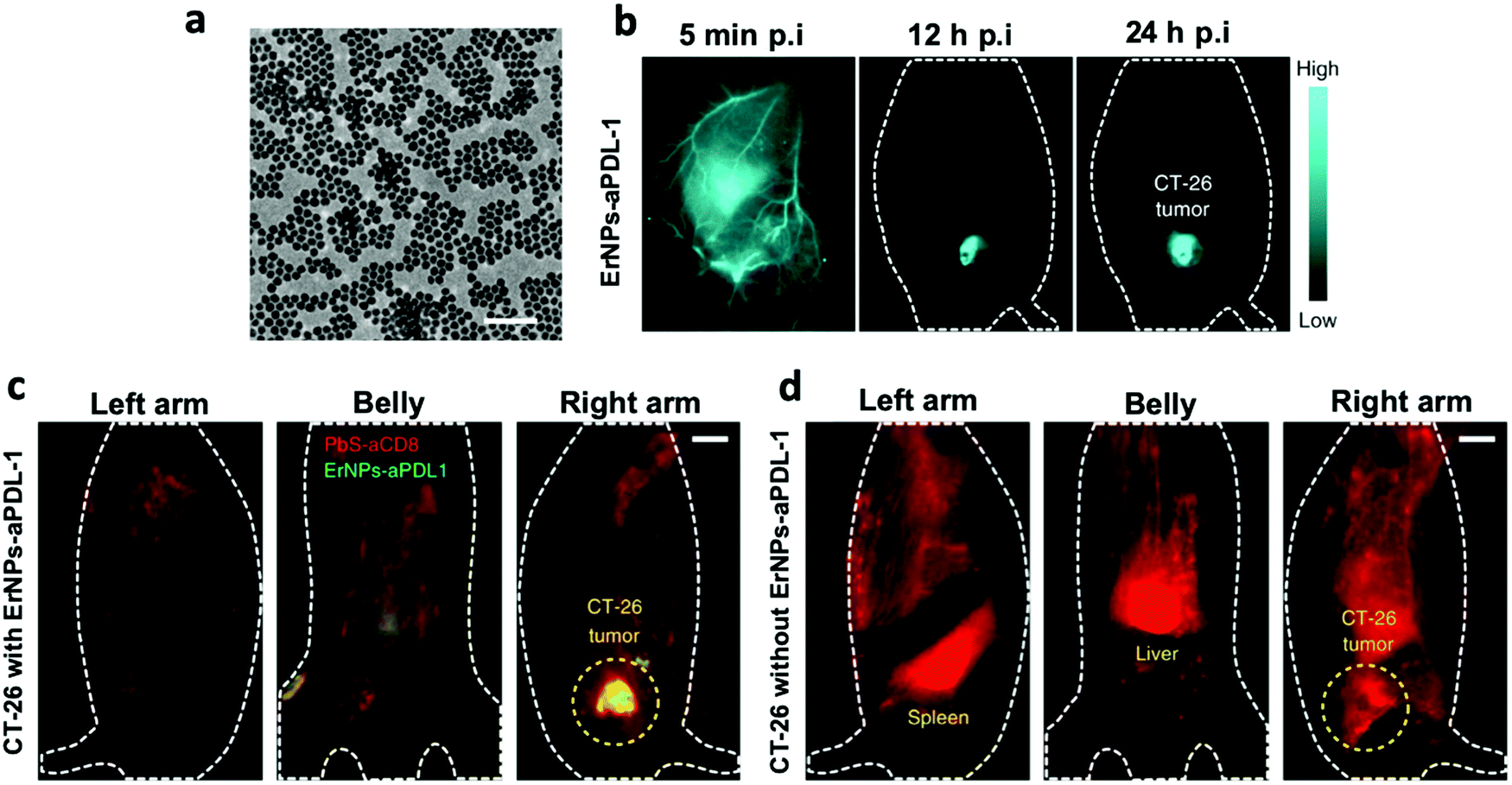 | ||
| Fig. 5 (a) Transmission electron microscopy image of ultra-bright NIR-IIb luminescence emitting ErNPs (α-NaYbF4:Er,Ce,Zn@NaYF4). Scale bar = 100 nm. (b) The wide-field fluorescence images of CT-26 tumor mice (n = 5) treated with ErNPs–aPDL1 at different time points p.i. (5 min, 12 h and 24 h). The ErNPs–aPDL1 nanoparticles primarily accumulated in tumor and the tumor-to-normal tissue (t/nt) signal ratio peaked at ∼42.2 ± 3.7 at 24 h p.i. (c and d) Wide-field fluorescence images of mice from different directions (left arm, belly and right arm) reveal the in vivo bio-distributions of ErNPs–aPDL1 and PbS–aCD8 in a CT-26 tumor mouse intravenously injected with mixed ErNPs–aPDL1 and PbS–aCD8 (c), with only PbS–aCD8 (d). Green color, ErNPs–aPDL1; red color, PbS–aCD8. Scale bars = 5 mm. The tumor accumulation of PbS–aCD8 in the immune-activated group (with ErNPs–aPDL1 treatment) was high, which suggested the T cells in spleen were infiltrating the tumor bed to exert the killing the cancer cells. By contrast, highly prominent PbS–aCD8 signals in the liver and spleen, with reduced tumor signals, were observed in tumor-bearing mice injected with PbS–aCD8 alone without PD-L1 antibody treatment. | ||
Human cytokine-induced killer (CIK) cells loaded with gold nanorods (AuNRs) were used in a nano-immunotheranostic approach which paired photoacoustic imaging with photothermal therapy to diagnose/treat gastric cancer.130 Human CIKs were labeled with silica-modified AuNRs by incubation. Labeled CIK cells inhibited the growth of gastric cancer MGC803 cells in vitro by inducing cell apoptosis and killed MGC803 cells under low power density near-infrared (NIR) laser treatment. The labeled CIK cells actively targeted and allowed imaging of subcutaneous gastric cancer in vivo via photoacoustic imaging at 4 h post-injection. These cells enhanced immunotherapy efficacy by up-regulating cytokines, including IL-1, IL-2, IL-4, IL-12, IL-17, and IFN-γ, and successfully killed gastric cancer tissues via photothermal irradiation using AuNRs.
Novel carbon nanodots (CDs) were incorporated in an ordered framework of mesoporous silica (CD@MSNs) as a different type of cancer nano-immunotheranostic strategy.131 These NPs display biodegradable features via swelling induction and gathering dispersive CDs for elevated targeting accumulation/retention in the tumor with subsequent photothermal imaging-guided Photo Thermal Therapy (PTT). In a novel advance, biodegraded debris could selectively enter immune organs with accompanying tumor-associated antigens (TAAs) from the malignant tumor cells. Therefore, CD@MSNs-mediated PTT synergistically achieved immune-mediated inhibition of distant tumor metastasis by stimulating the proliferation and activation of NK cells and macrophages, with simultaneous cytokine upregulation (IFN-γ and Granzyme B). Such work provides innovative insights into biodegradable NP-associated anticancer immunity. Nevertheless, the potential for clinical translation in these studies may be limited for several reasons: (i) optical imaging and photothermal imaging suffer from limited signal penetration due to tissue attenuation, which renders them relevant only to very specific superficial human applications; and (ii) length of degradation time, tumor tissue selectivity, and potential NP toxicity-induced side effects.
v. Molecular nano-immunoimaging
I. Carbohydrate targeting. Carbohydrates are ubiquitous in nature, and play many important roles in biological recognition including immune cell interactions with their targets.132 NPs coated with carbohydrates on the surface can potentially mimic immune recognition processes, allowing NP interaction with immune cells. For example, CD44 is a cell adhesion molecule over-expressed on the surface of immune cells, including macrophages.133 The major endogenous ligand for CD44 is hyaluronan (HA), a polysaccharide consisted of repeating glucuronic acid disaccharides linked with N-acetyl glucosamine;134 HA can also bind ICAM-1, LYVE-1, RHAMM, and TLR-4 expressed on macrophages. To target macrophages, SPIOs were coated with HA on the external surface through amide bonds (HA-SPIOs).135,136 These NPs were biocompatible with cells and colloidally stable in the presence of serum proteins. The HA immobilized on SPIOs retained its biological recognition of CD44. Cellular studies demonstrated HA-SPIO uptake by activated macrophages was HA- and CD44-dependent, and the level of HA-SPIO uptake was 4 times higher than that of SPIOs without HA coating.
HA-SPIOs can be used to non-invasively detect inflammatory atherosclerotic plaques.135 In a rabbit model of atherosclerosis, upon administration of the HA-SPIOs, significant contrast changes in aorta walls were observed in T2*-weighted MR images, allowing detection of atherosclerotic plaques. With the advantage that HA-SPIOs strongly bind to CD44-expressing macrophages, the dosage of HA-SPIO (0.21 mg Fe per kg of body weight) needed was much lower than that typically used for SPIOs without HA coating (56 mg Fe per kg of body weight), highlighting one benefit of NP molecular targeting.
One potential drawback for SPIOs is their inherent inflammatory activities, including induction of oxidative stress and toxicity in cells.137 While HA coating reduced inflammatory responses, upon incubation with macrophages in vitro or intravenous injection into mice, HA-SPIOs can elicit significant inflammatory cytokine production, such as TNF-α. Interestingly, nanomaterial morphology significantly impacted their inflammatory activity. Worm-like iron oxide nanoworms (NWs) were synthesized and functionalized with HA,138 and were shown to induce much lower inflammatory responses in macrophages in vitro and in mice than the corresponding spherically-shaped HA-SPIOs at the same dose, suggesting key NP shape effects.75,139 Cellular uptake of HA-NWs was also significantly higher than that of spherical HA-SPIOs. Thus, HA-NWs were able to detect atherosclerotic plaque in a mouse model of atherosclerosis (Fig. 6) based on their superior immune cell-targeting, safety, and in vivo performance properties.
 | ||
| Fig. 6 T 2*-Weighted MR images of ApoE knockout mouse aorta (a) before injection of HA-NWs and different time intervals after injection of HA-NWs (b–g). Areas of the aortic walls which have undergone contrast changes are highlighted by white arrows. | ||
Besides imaging, HA-NPs can have therapeutic effects. A type of HA-NP was prepared by reacting amine-functionalized oligomeric hyaluronan with cholanic ester and radiolabeled. Atherosclerotic rabbits received macrophage-targeted 89Zr-HA-NPs and were imaged by PET/MRI. PET/MRI of rabbits demonstrated 6-fold higher standardized uptake values compared to the control tissue. Therapeutic effects of HA-NPs were studied on atherosclerosis in ApoE−/− mice, which received weekly injections of HA-NPs for 12 weeks. The results showed the HA-NP-treated mouse lesions were significantly smaller than the control group, and contained approximately 30% fewer macrophages. Histological examination further revealed that both local and systemic HA-NP-immune cell interactions significantly decreased throughout disease progression.140
Carbohydrate mimetics as targeting agents. Selectins are a class of cell adhesion molecules involved in lymphocyte homing to inflammatory sites, as well as cancer metastasis, with their major ligand a tetrasaccharide sialyl LewisX (sLeX).141 E-selectin is expressed on activated endothelium, which mediates cell tethering and rolling interactions through the recognition of sLeX expressed on circulating leukocytes. During inflammation, the levels of E-selectins are upregulated, which can lead to enhanced binding of sLeX-coated NPs, and thus serve as a proxy for enhanced leukocyte trafficking. Due to the synthetic challenges in accessing sLeX, sLeX mimetics such as 1 have been designed to interact with selectin; these mimetics have been immobilized on NPs, polymers, or lipids (Fig. 7).142–144 When injected intravenously into a murine hepatitis model, sLeX-SPIOs induced a significantly lower attenuation of MR liver signal intensity compared to healthy mice or uncoated SPIOs in hepatitis mice, suggesting that sLeX-SPIOs are suitable for MRI diagnosis of inflammation.
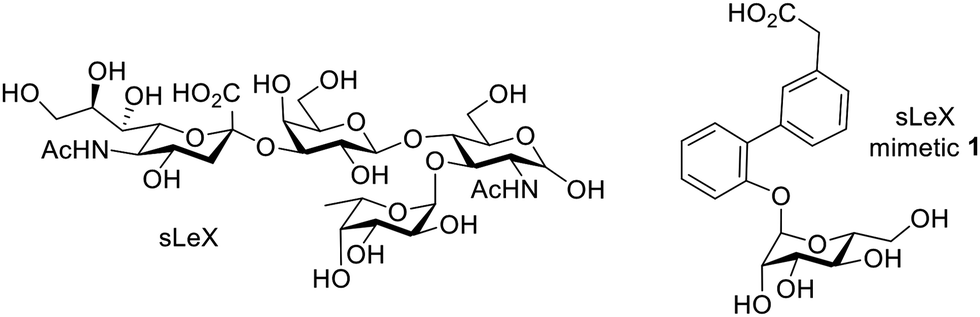 | ||
| Fig. 7 Structures of sLeX and sLeX mimetic 1 used as a targeting ligand for NP imaging. | ||
Sialoadhesin (Siglec-1 or CD169) is a member of the sialic acid binding Ig-like lectin (siglec) family expressed on macrophages.145 Its macrophage-specific expression makes it an attractive target for antigen delivery through Siglec-1 mediated uptake.146 Siglec-1 can bind sialyl-LacNAc trisaccharide 2. Structure-based modification based on molecule 2 led to the analog 3 (Fig. 8), which has significantly enhanced selectivity toward siglec-1 over other members of the siglec family. Fluorescently-labeled liposomes formulated with analog 3 were internalized much more readily by siglec-1-expressing cells than naked liposomes.146 Furthermore, over 10-fold improved selectivities were observed in Siglec-1 cells compared to cells expressing other siglec receptors. The Siglec-1-targeted liposomes dramatically enhanced the delivery of antigens to macrophages for presentation to, and enhanced proliferation of, antigen-specific T cells in mice.
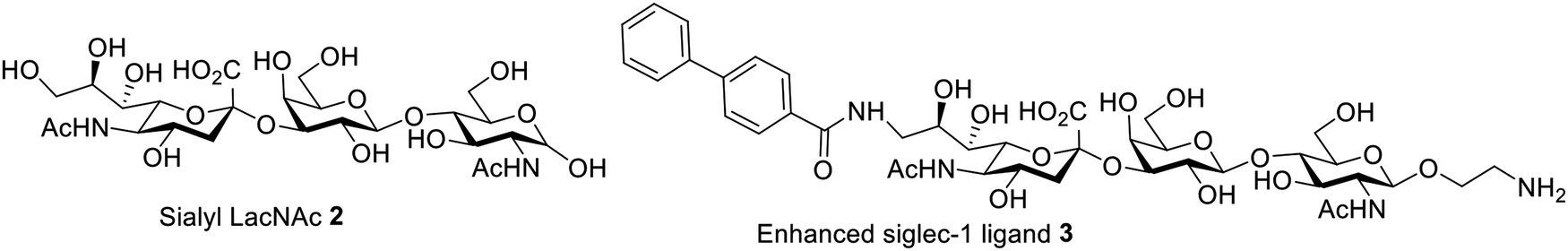 | ||
| Fig. 8 Structures of sialyl LacNAc 2 and the enhanced Siglec-1 ligand 3. | ||
II. Peptide and protein targeting. Beside carbohydrates and their mimics, peptides have been utilized as targeting agents. For example, peptide sequence CDSDSDITWDQLWDLMK, identified via phage display, binds tightly to E-selectin with Kd in the nM range. The peptide was immobilized on fluorescently-labeled SPIOs through the N-terminus cysteine,147 which was important to preserve the interactions with E-selectin. The selectin binding peptide-SPIOs were rapidly internalized by activated human umbilical vein endothelial cells (HUVECs) mediated by E-selectin, which was readily detected by optical fluorescence imaging versus SPIOs with a scrambled control peptide. The high selectivity of the peptide-SPIOs toward E-selectin was also confirmed by fluorescence imaging of the activated endothelium in a mouse tumor model. Thus, peptide-based selectin targeting complements choices of carbohydrate ligands as a useful approach for NP-directed E-selectin imaging in vitro and in vivo.
Vascular cell adhesion molecule-1 (VCAM-1) is a cell adhesion molecule that regulates inflammation-associated vascular adhesion and the transendothelial migration of leukocytes, such as macrophages and T cells (see also Sections 3.3 and 3.4c). It is associated with the progression of diseases such as atherosclerosis, rheumatoid arthritis, and cancer.148 To detect inflammatory cell activation in atherosclerosis, VCAM-1 targeting peptide VHPKQHR was conjugated to imaging contrast NPs such as SPIOs and Cy7-labeled polymer micelles.149 When evaluated using ApoE−/− mice, these VCAM-1 targeting nanoprobes not only helped identify inflammation in early atherosclerotic development, but also enabled real-time detection of VCAM-1 expression in vivo for monitoring pharmacotherapy-induced reductions of inflammation reflected in VCAM-1 expression levels.
To target dying cells in vivo, the protein Annexin V was bound to SPIOs and injected into a rabbit model of atherosclerosis, the Watanabe Heritable Hyperlipidemic-Myocardial Infarction rabbit.150 Since Annexin V can bind to phosphatidylserine presented on the outer membrane leaflets of apoptotic cells, injected SPIOs tended to localize in dying/dead macrophages and debris within the atherosclerotic plaque, providing an MRI-based assessment of plaque progression using T2*-weighted MRI. The use of this molecular targeting strategy not to a cell type, but to a particular cell state (apoptosis) primarily experienced by macrophages in plaques, permitted ∼200-fold less SPIOs to be injected for MR detection of inflamed plaque versus untargeted SPIOs.
A multifunctional core–shell NP system was developed for DC-based cancer immunotherapy.61 These NPs are composed of an SPIO core coated with a photonic zinc oxide (ZnO) shell with diameters of 16 nm. Novel peptides were developed to bind the ZnO shell, providing a new approach to anchor antigens on the NP surface and deliver those antigens to DCs. Interestingly, the Fe3O4–ZnO NPs were taken up more rapidly than Fe3O4 NPs without the ZnO shell, although the final levels of uptake were similar at 24 hours. Injection of DCs loaded with NPs into mice successfully boosted anti-cancer immunity, enhancing tumor antigen-specific T-cell responses, inhibiting tumor growth, and prolonging survival in immunized mice. MRI was used to track the NP-labeled DCs in draining lymph nodes after ipsilateral footpad injection in the animal models, ensuring that treatment occurred at the appropriate site and allowing longitudinal measurements (Fig. 9). This type of core–shell NP design could be a platform for DC-based immunotherapies that track DCs in vivo for consistent clinical efficacy.
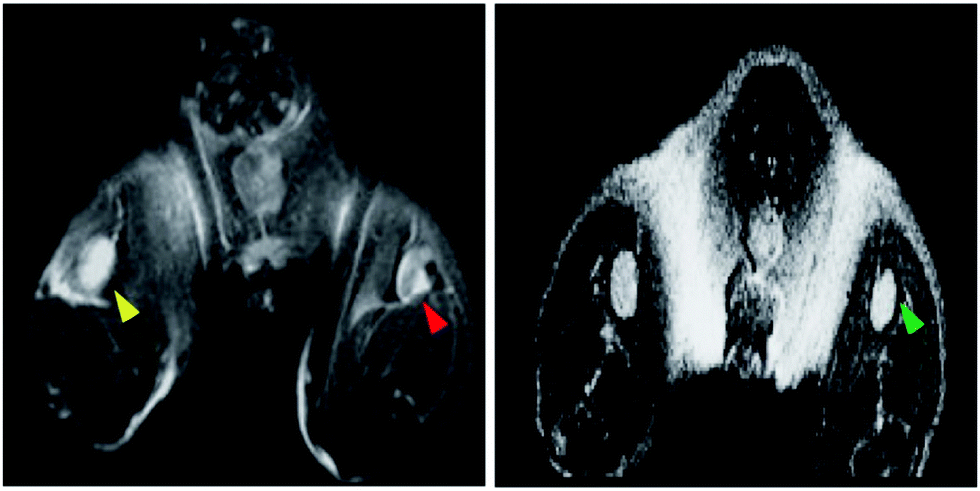 | ||
| Fig. 9 (left) In vivo T2 weighted MRI images of draining lymph nodes of a mouse 48 hours after injection with DCs labelled with Fe3O4–ZnO (red arrow) or ZnO NPs (yellow arrow) into the ipsilateral footpads. Localized hypointense regions within the lymph node marked with the red arrow were identified, indicating the presence of the NP-labelled cells. (right panel) A draining lymph node (green arrow) of cell-free Fe3O4–ZnO NP-injected mouse showed no contrast changes, indicating it is critical that Fe3O4–ZnO were encapsulated in DCs to lead to MRI contrast changes. | ||
Immunosuppression bestowed by Tregs in tumor microenvironments is a major mechanism of tumor immune escape and a significant hurdle to successful cancer immunotherapy.151 Neuropilin-1 (Nrp1) is a cell surface transmembrane glycoprotein that can serve as a Treg marker152 (Treg Nrp1 expression correlated with Foxp3 expression, Treg survival, and suppressive function). The tLyp1 peptide is a high affinity ligand for the Nrp1 receptor expressed on intratumoral Treg cells. tLyp1 peptide was functionalized with a phospholipid chain, and incorporated onto fluorescently labeled poly(D,L-lactide-co-glycolide) (PLGA) NPs.153 The inclusion of tLyp1 peptide increased cellular uptake of the NPs 3-fold compared to bare NPs. Upon administration of tLyp1-NPs into tumor bearing mice, fluorescence imaging exhibited significant signal from Tregs residing in tumor tissues. In contrast, little fluorescence was found in mice receiving NPs bearing the control peptide, demonstrating that tLyp1 effectively targets intratumoral Tregs. Understanding where and how much immunosuppression occurs in the context of disease is an important and rapidly burgeoning field, and diagnostic NPs that can spatiotemporally and quantitatively provide such information can lead the way. For clinical applications, it is likely that instead of fluorophores the NP will bear nuclear or magnetic contrast for PET, SPECT, or MR imaging of Tregs.
III. Antibody targeting. Antibodies can be attractive alternatives to carbohydrates and peptides for immune-targeting due to their potentially high specificities, as well as the widespread availability of monoclonal antibodies (mAbs) against many biologically-important targets. Disadvantages may include their relatively large size, particularly with respect to peptides, carbohydrates, aptamers, and small molecules. mAb-based inflammation detection is a rapidly growing sub-field. For example, dual-targeted MPIOs were prepared containing mAbs against P-selectin and VCAM-1 on their surface.154 The antibodies retained their binding specificities upon immobilization. When administered into ApoE knockout mice bearing atherosclerotic plaques, the probes bound aortic root endothelium overlaying atherosclerotic plaques, and were quantifiable by ex vivo MRI. Higher levels (5- to 7-fold) of the dual-targeting probes were found at the plaque sites compared to those with a single mAb against either P-selectin or VCAM-1, indicating the advantage of dual targeting. However, interestingly, detection of atherosclerotic plaques in vivo by MRI using mAb-MPIOs has yet to be demonstrated.
CD56 and CD16 are standard phenotypic markers of NK cells, suggesting that NK-92MI cells could be labeled with NPs bearing anti-CD56 or anti-CD16 mAbs.155 QDs are suitable for fluorescence imaging due to their high quantum yields and excellent photostability, which can improve detection and imaging sensitivities in vitro and in vivo.156 Near infrared-emitting QDs were conjugated to anti-CD56 mAb through thio-ether linkages. NK cells incubated with anti-CD56-QD retained their viability, as well as the ability to produce IFN-γ and lyse target cells. This approach, which combines ex vivo labeling and re-injection with molecular targeting, can be more advantageous than electroporation or using positively-charged transfection agents due to the mildness of the labeling method. After intratumoral injection into mouse tumor xenografts, NK cells tagged with QDs could be tracked up to 12 days through QD signals in human malignant melanoma xenografts in mice.155
CD8+ T cells are important effector cells of the immune system which can directly kill antigen-matched tumor cells. To target endogenous CD8+ T cells, anti-CD8 mAbs were reduced and cleaved.157 The resulting antibody binding fragments (Fab) were immobilized onto fluorophore DiD-labeled PLGA NPs bearing sulfhydryl-reactive maleimides. One hour after intravenous injection of the anti-CD8 NPs, over 90% of the CD8+ T cells in the blood, spleen and tumor tissues were bound by the NPs as detected by DiD fluorescence. The NPs persisted on CD8+ T cells for more than 48 h. Importantly, because only Fab fragments were used, cells labeled by the NPs remained viable without being recognized and killed by Fc receptor-bearing NK cells. In addition, immune activating agents such as TLR7/8 agonists and inhibitors of TGFβ signaling could be encapsulated inside the NPs for CD8+ T cell-targeted delivery to enhance the effects of immunotherapy. This type of theranostic NP can co-deliver various therapeutic agents and imaging agents to target cells and tissues (see Section 3(iv)). Therefore, suitably-designed theranostic NPs that can monitor immune cells or immune responses provide intriguing opportunities for rapid evaluation of therapeutic effects as well as optimization of treatment processes.
Immune cell NP targeting by mAb is modular. Judicious choice of the mAb on NPs could enable targeting of somewhat defined subsets of endogenous immune cells (although many cells require multiple surface markers to identify them, leading to binding of unintended cells ‘off-target’ in this strategy, careful choice in targeting ligand can still lead to efficacious results). Future in vivo imaging strategies could apply such NPs, such as, for example, human caveoline-1 NPs (termed caveospheres) functionalized with anti-CD4, anti-CD20, and anti-CCR5 mAbs to target CD4+ T cells, B cells, and memory CD4+ T cells, respectively. These fluorescently-labeled NPs158 were able to selectively recognize the appropriate target cells within physiological mixtures of primary human blood immune cells. We anticipate that such efficient and flexible immune cell-targeted NP systems will be increasingly employed for the development of advanced nano-immunoimaging approaches, immunotherapeutics, and vaccines.
IV. Intrinsic selectivity. Some NPs display an apparently intrinsic cellular selectivity. While many NPs are preferentially taken up by cells like macrophages due to phagocytic prowess, NPs which are themselves intrinsically selective can provide many advantages due to the ability to: (i) target cell subsets other than macrophages, and (ii) inject the NPs in vivo without requiring cell removal via blood draw, isolation, and reinjection. The cost, time, and risk of the cell phenotype changing during the reinjection process makes intrinsic in vivo labeling an extremely attractive alternative to ex vivo labeling. Typically, the biggest challenge is to target a particular cellular sub-population selectively enough, given that most cell types are identified by a set of multiple cell surface receptors (e.g., by flow cytometric analyses). Nevertheless, while delivery strategies generally do not require 100% accuracy in targeting to be effective in imaging (and therapy), there must be relatively high specificity with little off-target uptake. Intriguingly, extremely high selectivity of single-walled carbon nanotubes (SWNTs) to inflammatory monocytes, a subset of monocytes, has been observed.10 Upon injection, nearly 100% of circulating Ly-6Chi murine monocytes in blood took up SWNTs, and less than 3% of any other circulating cell type, including other monocyte subsets, took them up. IVM visualization of these processes revealed extremely rapid uptake of SWNTs into inflammatory monocytes and subsequent ‘Trojan Horse’-like deposition into the tumor as observed by IVM10 (Fig. 10) as well as PAI.13 The challenge to increase the number of different immune cell subsets (e.g., high-value targets such as CD8+ T cells, NK, and NKT cells) that NPs can target with ultra-high selectivity is a difficult, yet high-reward objective in nano-immunoimaging, one that is critical especially to minimize off-target uptake and potentially confounding image signal.
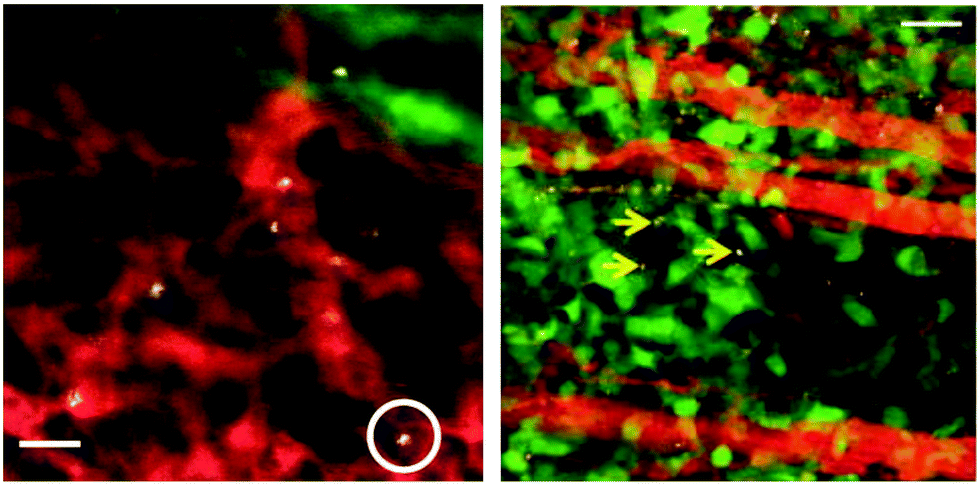 | ||
| Fig. 10 SWNT uptake into circulating cells and deposition into tumour in a mouse model of glioblastoma multiforme (u87-mg cells). (left) Representative intravital fluorescence image of single-walled carbon nanotube (SWNT)-laden circulating cells in tumour vasculature (one example cell is circled). Grayscale – SWNT-Cy5.5; red – long-circulating dye highlighting tumour blood vessels; green – EGFP (enhanced green fluorescent protein)-transfected tumour cells. Scale-bar: 40 μm. (right) A representative intravital micrograph of a tumour region (tumour cells, green; blood vessels, red; SWNTs, grayscale). Yellow arrows point to several SWNT-laden monocytes within the tumour interstitium. Scale bar: 50 μm.10 Reproduced by Permission of Nature. | ||
The first report on EV-mediated immune response showed that exosomes bearing MHC-II released from B lymphocytes can present peptide–MHC-II complexes to activate T-cell responses.177 Numerous exogenous and endogenous methods have been reported for labeling EV membranes or cargo including fluorescent or bioluminescent reporter molecules,178,179 ultrasmall superparamagnetic iron oxide (USPIO),180 and clinical grade radioisotopes such as 111In-oxine181 and Technetium-99m (99mTc)182 for SPECT imaging both in living cells and animals. In vivo use of these tools is providing useful information on cell–cell communication during pathogenic processes in addition to potentially uncovering novel immune cell responses in the form of released vesicles. For example, EVs can be indirectly labeled with bioluminescent molecules such as gaussia luciferase by fusing the gene to the EV binding domain lactadherin, a membrane-associated EV protein.183 SPIO- and 99mTc-labeled EVs can allow visualization of their native communications with lymph nodes when they are derived from tumor cells, as they traffic to a specific microanatomical location.184,185 EVs derived from red blood cells have been imaged communicating with tissue-resident hepatic macrophages.186,187 Limitations related to EV-mediated nano-immunoimaging imaging include difficulties in co-isolation of unwanted proteins during EV purification; moreover, since EVs are derived from cells and not fully engineered de novo, they may display unanticipated in vivo behaviors.188 While EV imaging remains in its infancy, we project that it will continue to grow eventually into a clinical proxy of disease pathogenesis, dissemination, communication, and outcome.
Key issues in nano-immunoimaging. Although there is agreement on the potential held by the field of nano-immunoimaging, critical issues must be addressed before clinical translation of related nanomaterials: (1) a key challenge confronting both in vitro and in vivo loading of NP-based imaging agents to targeted cells is that some NPs (e.g., those incorporating radioisotopes189) affect the viability and functions of labeled cells. (2) Precise quantification of cell numbers in vivo: cells take up NPs unevenly,190 and imaging accuracy and sensitivity based on NP labels in cells decreases over time due to cell division. (3) Evading off-target uptake of NPs is an ubiquitous problem in the field of in vivo NP labeling of cells. For instance, injected NPs are invariably taken up by the mononuclear phagocyte system, in which NPs are rapidly shuttled out of the circulation to non-target organs such as the spleen and liver, which can lead to serious adverse side effects and simultaneously decrease the amount of NPs available to home to the target tissue. For this reason, maximizing selective interactions of NPs with their immune targets, typically via NP surface engineering, is critical. (4) In nano-immunotheranostics, a hybrid between nano-immunoimaging and nano-immunotherapy, a common issue is the difference in the required mass administered and/or circulation time between a therapeutic and NP imaging contrast probes. In vivo imaging strategies aside from nuclear imaging approaches often require higher doses (compared to therapy) to readily discern signal from the area of interest compared to the surrounding tissue/background (which is the reason why most imaging tracer/contrast probes are designed to be quickly cleared from the blood). Conversely, therapeutics usually are designed for the opposing effect: to have longer circulation times in order to ensure adequate uptake and sufficient therapeutic effects in the target cells/tissues. Balancing the diagnostic and therapeutic parameters requires forethought, thorough understanding of the underlying biological mechanisms, and sometimes good fortune.
4. Potential for clinical translation
While nano-focused immunoimaging has a key role to play in pre-clinical imaging (such as elucidating the roles of immune cells and guiding optimal pre-clinical therapeutic approaches), it must succeed in the clinic to fulfill its compelling promise. Currently, the great potential of nano-immunoimaging lies in its ability to quantify if adequate numbers of immune cells traffic to the site of interest, whether and when they are activated, and how long they persist.8 These efforts could make possible a diverse array of revolutionary strategies: (1) ideal patient stratification for a particular therapy, especially immunotherapy, to bolster patient therapeutic outcome; (2) rapid assessment of patient therapeutic response; (3) companion monitoring tools for patient management to inform when to switch drugs, and which new drugs to select; (4) assessment of delivery localization for locally or systemically injected therapies; and (5) potentially as a surrogate for disease diagnosis and particularly prognosis.i. Early examples
Several pioneering human nano-immunoimaging trials pave the route toward future applications in cancer, atherosclerosis, and host of other inflammatory and immune-related conditions.13,67,104 Most trials involve iron oxide nanomaterials, an in vivo labeling agent which tends to tag highly phagocytic cells such as macrophages (but also potentially including monocytes and immature dendritic cells67). Because of the lack of selectivity, these agents have informed on the approximate biodistribution and dynamics, but thus far have not provided a true representation about macrophage location or activity. On the other hand, Figdor, Bulte, and colleagues magnetically labeled autologous dendritic cells with iron oxides and re-injected them into melanoma patients’ lymph nodes.191 Their MRI strategy ensured accurate delivery of the cells within the lymph nodes and tracked inter- and intranodal migration patterns—key data points that could not have been obtained otherwise, such as with scintigraphy. A potential limitation of this strategy is that MRI signals may not accurately reflect the labeled autologous dendritic cells. If/when labeled cells die, NPs might be released and could be free within the interstitial space or taken up by other cells; thus, under some conditions, some MRI signal could be false positive. Nevertheless, these groundbreaking examples of clinical nano-immunoimaging help direct pre-clinical focus to fulfill the promise of the field.ii. Vision for the field and clinical roadmap
The last approximately half a decade has seen explosive growth in pre-clinical nano-immunoimaging, driven by the successes of immunotherapy in the clinic. Nevertheless, there remains outstanding opportunity in the as-yet-unexplored areas of the field. Innovations in nanomaterials chemistry will inevitably drive enhanced nano-based multifunctionality, theranostics, as well as newer imaging modalities that require NPs to function such as MPI and MM-US,13 but key to the growth of the field and its clinical applications is to match nanomaterials, immune cells, and disease models with the appropriate application. Too often, nanomaterials are employed with the easiest available model to provide proof-of-principle immunoimaging, rather than developing the collaborations with physicians and related scientists that are necessary to pinpoint the most relevant or appropriate application. Likewise, improvement in animal models of disease, particularly those with complete, unadulterated immune systems, is a related field that will strongly impact the clinical translatability of nano-immunoimaging.Another major pre-clinical field that is likely to increasingly impact nano-immunoimaging involves living nano-contrast agents (e.g., contrast agent-producing bacteria) that were recently introduced and that may be capable of dividing intracellularly and thus be used to address the problem of probe dilution during cell division (recently reviewed).192,193 These ‘living NPs’ loaded into the cytoplasm of living mammalian cells have been used to image labeled cancer cells in animal models of human disease194,195 but the opportunities offered by living contrast agents to counteract dilution due to cell division have yet not been realized, and the agents have not yet been used in immune cells.
The initial nano-immunoimaging patient applications most likely to be successful will minimize disruption to clinical workflow. Thus, given the needs of its sister discipline immunotherapy, immunotheranostics and basic cell tracking are likely to be the first clinical applications. For instance, CAR-T cells are generally already engineered outside the body, therefore ex vivo labeling using iron oxide or gold NPs for MRI, PET, and/or CT (or some combination thereof) is likely to be relatively inexpensive and straightforward in order to track their localization, persistence, and dynamics in patients. Transplanted pancreatic islet cells loaded with contrast NPs may likewise serve as a strong early clinical application to track persistence and survival of pancreatic islet grafts as a proxy for immune rejection.
5. Conclusions
Nano-immunoimaging is a rapidly burgeoning field. It is limited by many of the same challenges that face the fields that comprise it: in nano, there are materials, size, and surface coating-based toxicity barriers to overcome in order to attain regulatory approvals and common clinical usage; in imaging, one faces the limitations inherent in each imaging modality (e.g., MRI is expensive, uses large magnetic fields, has fairly low sensitivity, and takes fairly long times; PET and SPECT employ radioactivity and have relatively poor spatial resolution, etc.); in immunology, one is limited by both the basic biology of the immune system, such as the molecular markers and functions that differentiate one cell type from another, and by our knowledge of it. Aside from the specter of improving basic immunobiology, all the other challenges are currently being investigated feverishly in the respective fields. We therefore anticipate that in the near-term, initial clinical applications in cell tracking will showcase the utility of nano-immunoimaging, while pre-clinical studies will drive the field toward its full potential. In the medium- to longer-term, at their best, nano-immunoimaging agents will integrate seamlessly into the clinical workflow, helping physicians stratify their patients for treatment planning, monitor their patients’ therapeutic responses to rapidly change treatment plans as necessary, and ultimately to treat and transmit diagnostic and prognostic information simultaneously as nano-immunotheranostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate support from the American Association of Cancer Research (AACR) – Translational Breast Cancer Research Award (17-20-26-SMIT) and the American Heart Association (18TPA34230113) (BRS). We gratefully acknowledge Dr LeAnn Matta for helping to identify articles of interest from the literature.Notes and references
- K. E. de Visser, A. Eichten and L. M. Coussens, Paradoxical Roles of the Immune System during Cancer Development, Nat. Rev. Cancer, 2006, 6(1), 24–37 CrossRef CAS PubMed.
- J. L. Markman and S. L. Shiao, Impact of the Immune System and Immunotherapy in Colorectal Cancer, J. Gastrointest. Oncol., 2015, 6(2), 208–223 Search PubMed.
- E. Porta-Pardo and A. Godzik, Mutation Drivers of Immunological Responses to Cancer, Cancer Immunol. Res., 2016, 4(9), 789–798 CrossRef CAS PubMed.
- W. Park, Y.-J. Heo and D. K. Han, New Opportunities for Nanoparticles in Cancer Immunotherapy, Biomater. Res., 2018, 22, 24 CrossRef PubMed.
- C. B. Rodell, S. P. Arlauckas, M. F. Cuccarese, C. S. Garris, R. Li, M. S. Ahmed, R. H. Kohler, M. J. Pittet and R. Weissleder, TLR7/8-Agonist-Loaded Nanoparticles Promote the Polarization of Tumour-Associated Macrophages to Enhance Cancer Immunotherapy, Nat. Biomed. Eng., 2018, 2, 578–588 CrossRef CAS PubMed.
- P. Bousso and H. D. Moreau, Functional Immunoimaging: The Revolution Continues, Nat. Rev. Immunol., 2012, 12(12), 858–864 CrossRef CAS PubMed.
- M. J. Miller, S. H. Wei, I. Parker and M. D. Cahalan, Two-Photon Imaging of Lymphocyte Motility and Antigen Response in Intact Lymph Node, Science, 2002, 296(5574), 1869–1873 CrossRef CAS PubMed.
- A. T. Mayer and S. S. Gambhir, The Immunoimaging Toolbox, J. Nucl. Med., 2018, 59(8), 1174–1182 CrossRef CAS PubMed.
- J. Tang, N. van Panhuys, W. Kastenmüller and R. N. Germain, The Future of Immunoimaging--Deeper, Bigger, More Precise, and Definitively More Colorful, Eur. J. Immunol., 2013, 43(6), 1407–1412 CrossRef CAS PubMed.
- B. R. Smith, E. E. B. Ghosn, H. Rallapalli, J. A. Prescher, T. Larson, L. A. Herzenberg and S. S. Gambhir, Selective Uptake of Single-Walled Carbon Nanotubes by Circulating Monocytes for Enhanced Tumour Delivery, Nat. Nanotechnol., 2014, 9(6), 481–487 CrossRef CAS PubMed.
- B. R. Smith, C. Zavaleta, J. Rosenberg, R. Tong, J. Ramunas, Z. Liu, H. Dai and S. S. Gambhir, High-Resolution, Serial Intravital Microscopic Imaging of Nanoparticle Delivery and Targeting in a Small Animal Tumor Model, Nano Today, 2013, 8(2), 126–137 CrossRef CAS PubMed.
- E. M. Shapiro, S. Skrtic, K. Sharer, J. M. Hill, C. E. Dunbar and A. P. Koretsky, MRI Detection of Single Particles for Cellular Imaging, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(30), 10901–10906 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Nanomaterials for In Vivo Imaging, Chem. Rev., 2017, 117, 3 Search PubMed.
- J. H. Ryu, S. Lee, S. Son, S. H. Kim, J. F. Leary, K. Choi and I. C. Kwon, Theranostic Nanoparticles for Future Personalized Medicine, J. Controlled Release, 2014, 190, 477–484 CrossRef CAS PubMed.
- T.-C. Yeh, W. Zhang, S. T. Ildstad and C. Ho, Intracellular Labeling of T-Cells with Superparamagnetic Contrast Agents, Magn. Reson. Med., 1993, 30(5), 617–625 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Nanomaterials for In Vivo Imaging, Chem. Rev., 2017, 117(3), 901–986 CrossRef CAS PubMed.
- B. R. Smith, M. Ruegsegger, P. A. Barnes, M. Ferrari and S. C. Lee, Nanodevices in Biomedical Applications, in BioMEMS and Biomedical Nanotechnology, ed. M. Ferrari, A. P. Lee and L. J. Lee, Springer US, Boston, MA, 2006, pp. 363–398 Search PubMed.
- N. Lee and T. Hyeon, Designed Synthesis of Uniformly Sized Iron Oxide Nanoparticles for Efficient Magnetic Resonance Imaging Contrast Agents, Chem. Soc. Rev., 2012, 41(7), 2575–2589 RSC.
- M. F. Kircher and J. K. Willmann, Molecular Body Imaging: MR Imaging, CT, and US. Part I. Principles, Radiology, 2012, 263(3), 633–643 CrossRef PubMed.
- E. T. Ahrens and J. W. M. Bulte, Tracking Immune Cells In Vivo Using Magnetic Resonance Imaging, Nat. Rev. Immunol., 2013, 13(10), 755–763 CrossRef CAS PubMed.
- B. Zheng, M. P. von See, E. Yu, B. Gunel, K. Lu, T. Vazin, D. V. Schaffer, P. W. Goodwill and S. M. Conolly, Quantitative Magnetic Particle Imaging Monitors the Transplantation, Biodistribution, and Clearance of Stem Cells In Vivo, Theranostics, 2016, 6(3), 291–301 CrossRef CAS PubMed.
- X. Zhu, J. Li, P. Peng, N. Hosseini-Nassab and B. R. Smith, Quantitative Drug Release Monitoring in Tumors of Living Subjects by Magnetic Particle Imaging Nanocomposite, Nano Lett., 2019, 19(10), 6725–6733 CrossRef CAS PubMed.
- B. J. Vakoc, D. Fukumura, R. K. Jain and B. E. Bouma, Cancer Imaging by Optical Coherence Tomography: Preclinical Progress and Clinical Potential, Nat. Rev. Cancer, 2012, 12(5), 363–368 CrossRef CAS PubMed.
- M. Ahmad, G. Pratx, M. Bazalova and L. Xing, X-Ray Luminescence and X-Ray Fluorescence Computed Tomography: New Molecular Imaging Modalities, IEEE Access, 2014, 2, 1051–1061 Search PubMed.
- C. H. Contag and M. H. Bachmann, Advances In Vivo Bioluminescence Imaging of Gene Expression, Annu. Rev. Biomed. Eng., 2002, 4, 235–260 CrossRef CAS PubMed.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Semiconductor Nanocrystals as Fluorescent Biological Labels, Science, 1998, 281(5385), 2013–2016 CrossRef CAS PubMed.
- E. J. Sutton, T. D. Henning, B. J. Pichler, C. Bremer and H. E. Daldrup-Link, Cell Tracking with Optical Imaging, Eur. Radiol., 2008, 18(10), 2021–2032 CrossRef PubMed.
- X. H. Gao, Y. Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, In Vivo Cancer Targeting and Imaging with Semiconductor Quantum Dots, Nat. Biotechnol., 2004, 22(8), 969–976 CrossRef CAS PubMed.
- B. R. Smith, E. E. B. Ghosn, H. Rallapalli, J. A. Prescher, T. Larson, L. A. Herzenberg and S. S. Gambhir, Selective Uptake of Single-Walled Carbon Nanotubes by Circulating Monocytes for Enhanced Tumour Delivery, Nat. Nanotechnol., 2014, 9(6), 481–487 CrossRef CAS PubMed.
- B. R. Smith, P. Kempen, D. Bouley, A. Xu, Z. Liu, N. Melosh, H. Dai, R. Sinclair and S. S. Gambhir, Shape Matters: Intravital Microscopy Reveals Surprising Geometrical Dependence for Nanoparticles in Tumor Models of Extravasation, Nano Lett., 2012, 12(7), 3369–3377 CrossRef CAS PubMed.
- B. R. Smith, Z. Cheng, A. De, A. L. Koh, R. Sinclair and S. S. Gambhir, Real-Time Intravital Imaging of RGD-Quantum Dot Binding to Luminal Endothelium in Mouse Tumor Neovasculature, Nano Lett., 2008, 8(9), 2599–2606 CrossRef CAS PubMed.
- V. Ntziachristos, C. Bremer and R. Weissleder, Fluorescence Imaging with Near-Infrared Light: New Technological Advances That Enable In Vivo Molecular Imaging, Eur. Radiol., 2003, 13(1), 195–208 CrossRef PubMed.
- L. Abou-Elkacem, S. V. Bachawal and J. K. Willmann, Ultrasound Molecular Imaging: Moving toward Clinical Translation, Eur. J. Radiol., 2015, 84(9), 1685–1693 CrossRef PubMed.
- K. E. Wilson, T. Y. Wang and J. K. Willmann, Acoustic and Photoacoustic Molecular Imaging of Cancer, J. Nucl. Med., 2013, 54(11), 1851–1854 CrossRef PubMed.
- S. R. Wilson and P. N. Burns, Microbubble-Enhanced US in Body Imaging: What Role?, Radiology, 2010, 257(1), 24–39 CrossRef PubMed.
- C. Kim, T. N. Erpelding, L. Jankovic, M. D. Pashley and L. H. V. Wang, Deeply Penetrating In Vivo Photoacoustic Imaging Using a Clinical Ultrasound Array System, Biomed. Opt. Express, 2010, 1(1), 278–284 CrossRef PubMed.
- J. Weber, P. C. Beard and S. E. Bohndiek, Contrast Agents for Molecular Photoacoustic Imaging, Nat. Methods, 2016, 13(8), 639–650 CrossRef CAS PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Molecular Imaging with PET, Chem. Rev., 2008, 108(5), 1501–1516 CrossRef CAS PubMed.
- W. B. Cai, K. Chen, K. A. Mohamedali, Q. Z. Cao, S. S. Gambhir, M. G. Rosenblum and X. Y. Chen, PET of Vascular Endothelial Growth Factor Receptor Expression, J. Nucl. Med., 2006, 47(12), 2048–2056 CAS.
- L. W. Goldman, Principles of CT and CT Technology, J. Nucl. Med. Technol., 2007, 35(3), 115–128 CrossRef PubMed.
- T. Kim, N. Lee, D. R. Arifin, I. Shats, M. Janowski, P. Walczak, T. Hyeon and J. W. M. Bulte, In Vivo Micro-CT Imaging of Human Mesenchymal Stem Cells Labeled with Gold-Poly-l-Lysine Nanocomplexes, Adv. Funct. Mater., 2017, 27(3), 1604213 CrossRef PubMed.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Gold Nanoparticles: A New X-Ray Contrast Agent, Br. J. Radiol., 2006, 79(939), 248–253 CrossRef CAS PubMed.
- O. Rabin, J. M. Perez, J. Grimm, G. Wojtkiewicz and R. Weissleder, An X-Ray Computed Tomography Imaging Agent Based on Long-Circulating Bismuth Sulphide Nanoparticles, Nat. Mater., 2006, 5(2), 118–122 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Gold Nanoparticles for Biology and Medicine, Angew. Chem., Int. Ed., 2010, 49(19), 3280–3294 CrossRef CAS PubMed.
- J. W. M. Bulte and D. L. Kraitchman, Iron Oxide MR Contrast Agents for Molecular and Cellular Imaging, NMR Biomed., 2004, 17(7), 484–499 CrossRef CAS PubMed.
- S. B. Yu and A. D. Watson, Metal-Based X-Ray Contrast Media, Chem. Rev., 1999, 99(9), 2353–2377 CrossRef CAS PubMed.
- R. Meir, K. Shamalov, O. Betzer, M. Motiei, M. Horovitz-Fried, R. Yehuda, A. Popovtzer, R. Popovtzer and C. J. Cohen, Nanomedicine for Cancer Immunotherapy: Tracking Cancer-Specific T-Cells In Vivo with Gold Nanoparticles and CT Imaging, ACS Nano, 2015, 9(6), 6363–6372 CrossRef CAS PubMed.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, Antibiofouling Polymer-Coated Gold Nanoparticles as a Contrast Agent for In Vivo x-Ray Computed Tomography Imaging, J. Am. Chem. Soc., 2007, 129(24), 7661–7665 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Quantum Dot Bioconjugates for Imaging, Labelling and Sensing, Nat. Mater., 2005, 4(6), 435–446 CrossRef CAS PubMed.
- E. B. Voura, J. K. Jaiswal, H. Mattoussi and S. M. Simon, Tracking Metastatic Tumor Cell Extravasation with Quantum Dot Nanocrystals and Fluorescence Emission-Scanning Microscopy, Nat. Med., 2004, 10(9), 993–998 CrossRef CAS PubMed.
- Y. I. Park, K. T. Lee, Y. D. Suh and T. Hyeon, Upconverting Nanoparticles: A Versatile Platform for Wide-Field Two-Photon Microscopy and Multi-Modal In Vivo Imaging, Chem. Soc. Rev., 2015, 44(6), 1302–1317 RSC.
- J. Xiang, L. G. Xu, H. Gong, W. W. Zhu, C. Wang, J. Xu, L. Z. Feng, L. Cheng, R. Peng and Z. Liu, Antigen-Loaded Upconversion Nanoparticles for Dendritic Cell Stimulation, Tracking, and Vaccination in Dendritic Cell-Based Immunotherapy, ACS Nano, 2015, 9(6), 6401–6411 CrossRef CAS PubMed.
- X. Sun, W. Cai and X. Chen, Positron Emission Tomography Imaging Using Radiolabeled Inorganic Nanomaterials, Acc. Chem. Res., 2015, 48(2), 286–294 CrossRef CAS PubMed.
- H. Hong, Y. Zhang, J. Sun and W. Cai, Molecular Imaging and Therapy of Cancer with Radiolabeled Nanoparticles, Nano Today, 2009, 4(5), 399–413 CrossRef CAS PubMed.
- H. Kobayashi, M. R. Longmire, M. Ogawa and P. L. Choyke, Rational Chemical Design of the next Generation of Molecular Imaging Probes Based on Physics and Biology: Mixing Modalities, Colors and Signals, Chem. Soc. Rev., 2011, 40(9), 4626–4648 RSC.
- J. Cheon and J. H. Lee, Synergistically Integrated Nanoparticles as Multimodal Probes for Nanobiotechnology, Acc. Chem. Res., 2008, 41(12), 1630–1640 CrossRef CAS PubMed.
- L. Marti-Bonmati, R. Sopena, P. Bartumeus and P. Sopena, Multimodality Imaging Techniques, Contrast Media Mol. Imaging, 2010, 5(4), 180–189 CrossRef CAS PubMed.
- B. J. Pichler, M. S. Judenhofer and H. F. Wehrl, PET/MRI Hybrid Imaging: Devices and Initial Results, Eur. Radiol., 2008, 18(6), 1077–1086 CrossRef PubMed.
- R. Chakravarty, H. F. Valdovinos, F. Chen, C. M. Lewis, P. A. Ellison, H. Luo, M. E. Meyerand, R. J. Nickles and W. Cai, Intrinsically Germanium-69-Labeled Iron Oxide Nanoparticles: Synthesis and In Vivo Dual-Modality PET/MR Imaging, Adv. Mater., 2014, 26(30), 5119–5123 CrossRef CAS PubMed.
- S. B. Lee, S. W. Lee, S. Y. Jeong, G. Yoon, S. J. Cho, S. K. Kim, I. K. Lee, B. C. Ahn, J. Lee and Y. H. Jeon, Engineering of Radioiodine-Labeled Gold Core Shell Nanoparticles As Efficient Nuclear Medicine Imaging Agents for Trafficking of Dendritic Cells, ACS Appl. Mater. Interfaces, 2017, 9(10), 8480–8489 CrossRef CAS PubMed.
- N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, A Multifunctional Core-Shell Nanoparticle for Dendritic Cell-Based Cancer Immunotherapy, Nat. Nanotechnol., 2011, 6(10), 675–682 CrossRef CAS PubMed.
- M. Lameijer, T. Binderup, M. M. T. van Leent, M. L. Senders, F. Fay, J. Malkus, B. L. Sanchez-Gaytan, A. J. P. Teunissen, N. Karakatsanis and P. Robson, et al., Efficacy and Safety Assessment of a TRAF6-Targeted Nanoimmunotherapy in Atherosclerotic Mice and Non-Human Primates, Nat. Biomed. Eng., 2018, 2(5), 279–292 CrossRef CAS PubMed.
- J. E. Lee, N. Lee, T. Kim, J. Kim and T. Hyeon, Multifunctional Mesoporous Silica Nanocomposite Nanoparticles for Theranostic Applications, Acc. Chem. Res., 2011, 44(10), 893–902 CrossRef CAS PubMed.
- T. Taguchi and K. Mukai, Innate Immunity Signalling and Membrane Trafficking, Curr. Opin. Cell Biol., 2019, 59, 1–7 CrossRef CAS PubMed.
- G. Natoli and R. Ostuni, Adaptation and Memory in Immune Responses, Nat. Immunol., 2019, 20(7), 783–792 CrossRef CAS PubMed.
- X. Liu, H. Kwon, Z. Li and Y.-X. Fu, Is CD47 an Innate Immune Checkpoint for Tumor Evasion?, J. Hematol. Oncol., 2017, 10(1), 12 CrossRef PubMed.
- R. Weissleder, M. Nahrendorf and M. J. Pittet, Imaging Macrophages with Nanoparticles, Nat. Mater., 2014, 13(2), 125–138 CrossRef CAS PubMed.
- M. J. Pittet and R. Weissleder, Intravital Imaging, Cell, 2011, 147(5), 983–991 CrossRef CAS PubMed.
- M. Owyong, N. Hosseini-Nassab, G. Efe, A. Honkala, R. J. E. van den Bijgaart, V. Plaks and B. R. Smith, Cancer Immunotherapy Getting Brainy: Visualizing the Distinctive CNS Metastatic Niche to Illuminate Therapeutic Resistance, Drug Resist. Updates, 2017, 33–35 Search PubMed.
- B. J. Vakoc, D. Fukumura, R. K. Jain and B. E. Bouma, Cancer Imaging by Optical Coherence Tomography: Preclinical Progress and Clinical Potential, Nat. Rev. Cancer, 2012, 12(5), 363–368 CrossRef CAS PubMed.
- E. D. SoRelle, D. Yecies, O. Liba, F. C. Bennett, C. M. Graef, R. Dutta, S. S. Mitra, L.-M. Joubert, S. H. Cheshier and G. A. Grant, et al., Wide-Field Dynamic Monitoring of Immune Cell Trafficking in Murine Models of Glioblastoma, bioRxiv, 2017, 220954 Search PubMed.
- L. V. Wang, S. Hu, J. P. Culver, V. Ntziachristos, M. J. Holboke, A. G. Yodh, L. V. Wang, L. V. Wang, Z. Guo and L. Li, et al., Photoacoustic Tomography: In Vivo Imaging from Organelles to Organs, Science, 2012, 335(6075), 1458–1462 CrossRef CAS PubMed.
- R. N. Germain, E. a Robey and M. D. Cahalan, A Decade of Imaging Cellular Motility and Interaction Dynamics in the Immune System, Science, 2012, 336(6089), 1676–1681 CrossRef CAS PubMed.
- T. R. Mempel, M. L. Scimone, J. R. Mora and U. H. von Andrian, In Vivo Imaging of Leukocyte Trafficking in Blood Vessels and Tissues, Curr. Opin. Immunol., 2004, 16(4), 406–417 CrossRef CAS PubMed.
- X. Zhu, C. Vo, M. Taylor and B. R. Smith, Non-Spherical Micro- and Nanoparticles in Nanomedicine, Mater. Horiz., 2019, 6, 1094–1121 RSC.
- V. R. Taqueti and F. A. Jaffer, High-Resolution Molecular Imaging via Intravital Microscopy: Illuminating Vascular Biology In Vivo, Integr. Biol., 2013, 5(2), 278–290 CrossRef CAS PubMed.
- B. Wang, P. Joshi, V. Sapozhnikova, J. Amirian, S. H. Litovsky, R. Smalling, K. Sokolov and S. Emelianov, in Intravascular Photoacoustic Imaging of Macrophages Using Molecularly Targeted Gold Nanoparticles, ed. A. A. Oraevsky and L. V. Wang, 2010, p. 75640A Search PubMed.
- K. T. Warzecha, M. Bartneck, D. Möckel, L. Appold, C. Ergen, W. Al Rawashdeh, F. Gremse, P. M. Niemietz, W. Jahnen-Dechent and C. Trautwein, et al., Targeting and Modulation of Liver Myeloid Immune Cells by Hard-Shell Microbubbles, Adv. Biosyst., 2018, 2(5), 1800002 CrossRef PubMed.
- J. R. Cook, W. Frey and S. Emelianov, Quantitative Photoacoustic Imaging of Nanoparticles in Cells and Tissues, ACS Nano, 2013, 7(2), 1272–1280 CrossRef CAS PubMed.
- F. De Winter, D. Vogelaers, F. Gemmel and R. Dierckx, Promising Role of 18-F-Fluoro-D-Deoxyglucose Positron Emission Tomography in Clinical Infectious Diseases, Eur. J. Clin. Microbiol. Infect. Dis., 2002, 21(4), 247–257 CrossRef CAS PubMed.
- S. M. Bakheet, J. Powe, A. Kandil, A. Ezzat, A. Rostom and J. Amartey, F-18 FDG Uptake in Breast Infection and Inflammation, Clin. Nucl. Med., 2000, 25(2), 100–103 CrossRef CAS PubMed.
- R. Hustinx, R. J. Smith, F. Benard, D. I. Rosenthal, M. Machtay, L. A. Farber and A. Alavi, Dual Time Point Fluorine-18 Fluorodeoxyglucose Positron Emission Tomography: A Potential Method to Differentiate Malignancy from Inflammation and Normal Tissue in the Head and Neck, Eur. J. Nucl. Med., 1999, 26(10), 1345 CrossRef CAS PubMed.
- I. S. Alam, A. T. Mayer, I. Sagiv-Barfi, K. Wang, O. Vermesh, D. K. Czerwinski, E. M. Johnson, M. L. James, R. Levy and S. S. Gambhir, Imaging Activated T Cells Predicts Response to Cancer Vaccines, J. Clin. Invest., 2018, 128(6), 2569–2580 CrossRef PubMed.
- A. S. Arbab, G. T. Yocum, H. Kalish, E. K. Jordan, S. A. Anderson, A. Y. Khakoo, E. J. Read and J. A. Frank, Efficient Magnetic Cell Labeling with Protamine Sulfate Complexed to Ferumoxides for Cellular MRI, Blood, 2004, 104, 1217–1223 CrossRef CAS PubMed.
- A. S. Arbab, L. A. Bashaw, B. R. Miller, E. K. Jordan, B. K. Lewis, H. Kalish and J. A. Frank, Characterization of Biophysical and Metabolic Properties of Cells Labeled with Superparamagnetic Iron Oxide Nanoparticles and Transfection Agent for Cellular MR Imaging, Radiology, 2003, 229, 838–846 CrossRef PubMed.
- A. S. Arbab, L. A. Bashaw, B. R. Miller, E. K. Jordan, J. W. Bulte and J. A. Frank, Intracytoplasmic Tagging of Cells with Ferumoxides and Transfection Agent for Cellular Magnetic Resonance Imaging after Cell Transplantation: Methods and Techniques, Transplantation, 2003, 76, 1123–1130 CrossRef CAS PubMed.
- I. Raynal, P. Prigent, S. Peyramaure, A. Najid, C. Rebuzzi and C. Corot, Macrophage Endocytosis of Superparamagnetic Iron Oxide Nanoparticles: Mechanisms and Comparison of Ferumoxides and Ferumoxtran-10, Invest. Radiol., 2004, 39, 56–63 CrossRef CAS PubMed.
- M. Selt, A. Tennstaedt, A. Beyrau, M. Nelles, G. Schneider, C. Löwik and M. Hoehn, In Vivo Non-Invasive Tracking of Macrophage Recruitment to Experimental Stroke, PLoS One, 2016, 11, e0156626 CrossRef PubMed.
- M. Yagita, C. Huang, H. Umehara, Y. Matsuo, R. Tabata, M. Miyake, Y. Konaka and K. Takatsuki, A Novel Natural Killer Cell Line (KHYG-1) from a Patient with Aggressive Natural Killer Cell Leukemia Carrying a P53 Point Mutation, Leukemia, 2000, 14, 922–930 CrossRef CAS PubMed.
- C. L. Mallett, C. Mcfadden, Y. Chen and P. J. Foster, Migration of Iron-Labeled KHYG-1 Natural Killer Cells to Subcutaneous Tumors in Nude Mice, as Detected by Magnetic Resonance Imaging, Cytotherapy, 2012, 14, 743–751 CrossRef CAS PubMed.
- C. Wu, Y. Ye Xu, L. Yang, J. Wu, W. Zhu, D. Li, Z. Cheng, C. Xia, Y. Guo and Q. Gong, et al., Negatively Charged Magnetite Nanoparticle Clusters as Efficient MRI Probes for Dendritic Cell Labeling and In Vivo Tracking, Adv. Funct. Mater., 2015, 25, 3581–3591 CrossRef CAS.
- C. Guillerey, N. D. Huntington and M. J. Smyth, Targeting Natural Killer Cells in Cancer Immunotherapy, Nat. Immunol., 2016, 17(9), 1025–1036 CrossRef CAS PubMed.
- N. S. Sta Maria, S. R. Barnes and R. E. Jacobs, In Vivo Monitoring of Natural Killer Cell Trafficking During Tumor Immunotherapy, Magn. Reson. Insights, 2014, 7, 15–21 Search PubMed.
- A. Sheu, Z. Zhang, R. Omary and A. Larson, MRI-Monitored Transcatheter Intra-Arterial Delivery of SPIO-Labeled Natural Killer Cells to Hepatocellular Carcinoma: Preclinical Studies in a Rodent Model, Invest. Radiol., 2013, 48, 492–499 CrossRef PubMed.
- R. Meier, D. Golovko, S. Tavri, T. D. Henning, C. Knopp, G. Piontek, M. Rudelius, P. Heinrich, W. S. Wels and H. Daldrup-Link, Depicting Adoptive Immunotherapy for Prostate Cancer in an Animal Model with Magnetic Resonance Imaging, Magn. Reson. Med., 2011, 65, 756–763 CrossRef CAS PubMed.
- E. S. Jang, J. H. Shin, G. Ren, M. J. Park, K. Cheng, X. Chen, J. C. Wu, J. B. Sunwoo and Z. Cheng, The Manipulation of Natural Killer Cells to Target Tumor Sites Using Magnetic Nanoparticles, Biomaterials, 2012, 33, 5584–5592 CrossRef CAS PubMed.
- C. Wang, L. Cheng, H. Xu and Z. Liu, Towards Whole-Body Imaging at the Single Cell Level Using Ultra-Sensitive Stem Cell Labeling with Oligo-Arginine Modified Upconversion Nanoparticles, Biomaterials, 2012, 33, 4872–4881 CrossRef CAS PubMed.
- M. F. Kircher, J. R. Allport, E. E. Graves, V. Love, L. Josephson, A. H. Lichtman and R. Weissleder, In Vivo High Resolution Three-Dimensional Imaging of Antigen-Specific Cytotoxic T-Lymphocyte Trafficking to Tumors, Cancer Res., 2003, 63, 6836–6846 Search PubMed.
- M. Srinivas, M. S. Turner, J. M. Janjic, P. A. Morel, D. H. Laidlaw and E. T. Ahrens, In Vivo Cytometry of Antigen-Specific t Cells Using 19F MRI, Magn. Reson. Med., 2009, 62, 747–753 CrossRef CAS PubMed.
- M. Srinivas, P. A. Morel, L. A. Ernst, D. H. Laidlaw and E. T. Ahrens, Fluorine-19 MRI for Visualization and Quantification of Cell Migration in a Diabetes Model, Magn. Reson. Med., 2007, 58(4), 725–734 CrossRef CAS PubMed.
- C. F. O’Hanlon, T. Fedczyna, S. Eaker, W. D. Shingleton and B. M. Helfer, Integrating a 19F MRI Tracer Agent into the Clinical Scale Manufacturing of a T-Cell Immunotherapy, Contrast Media Mol. Imaging, 2017, 2017, 9548478 Search PubMed.
- T. K. Hitchens, Q. Ye, D. F. Eytan, J. M. Janjic, E. T. Ahrens and C. Ho, 19F MRI Detection of Acute Allograft Rejection with In Vivo Perfluorocarbon Labeling of Immune Cells, Magn. Reson. Med., 2011, 65, 1144–1153 CrossRef PubMed.
- O. Koshkina, G. Lajoinie, F. Baldelli Bombelli, E. Swider, L. J. Cruz, P. B. White, R. Schweins, Y. Dolen, E. A. W. van Dinther and N. K. van Riessen, et al., Multicore Liquid Perfluorocarbon-Loaded Multimodal Nanoparticles for Stable Ultrasound and 19 F MRI Applied to In Vivo Cell Tracking, Adv. Funct. Mater., 2019, 29(19), 1806485 CrossRef.
- P. Bhatnagar, Z. Li, Y. Choi, J. Guo, F. Li, D. Y. Lee, M. Figliola, H. Huls, D. A. Lee and T. Zal, et al., Imaging of Genetically Engineered T Cells by PET Using Gold Nanoparticles Complexed to Copper-64, Integr. Biol., 2013, 5(1), 231–238 CrossRef CAS PubMed.
- J. B. Morris, A. R. Olzinski, R. E. Bernard, K. Aravindhan, R. C. Mirabile, R. Boyce, R. N. Willette and B. M. Jucker, P38 MAPK Inhibition Reduces Aortic Ultrasmall Superparamagnetic Iron Oxide Uptake in a Mouse Model of Atherosclerosis. MRI Assessment, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 265–271 CrossRef CAS PubMed.
- M. Sigovan, L. Boussel, A. Sulaiman, D. Sappey-Marinier, H. Alsaid, C. Desbleds-Mansard, D. Ibarrola, D. Gamondès, C. Corot and E. Lancelot, et al., Rapid-Clearance Iron Nanoparticles for Inflammation Imaging of Atherosclerotic Plaque: Initial Experience in Animal Model, Radiology, 2009, 252, 401–409 CrossRef PubMed.
- S. G. Ruehm, C. Corot, P. Vogt, S. Kolb and J. F. Debatin, Magnetic Resonance Imaging of Atherosclerotic Plaque With Ultrasmall Superparamagnetic Particles of Iron Oxide in Hyperlipidemic Rabbits, Circulation, 2001, 103, 415–422 CrossRef CAS PubMed.
- K. Morishige, D. F. Kacher, P. Libby, L. Josephson, P. Ganz, R. Weissleder and M. Aikawa, High-Resolution Magnetic Resonance Imaging Enhanced With Superparamagnetic Nanoparticles Measures Macrophage Burden in Atherosclerosis, Circulation, 2010, 122, 1707–1715 CrossRef CAS PubMed.
- M. A. Miller, S. Arlauckas and R. Weissleder, Prediction of Anti-Cancer Nanotherapy Efficacy by Imaging, Nanotheranostics, 2017, 1(3), 296–312 CrossRef PubMed.
- A. Fu, R. J. Wilson, B. R. Smith, J. Mullenix, C. Earhart, D. Akin, S. Guccione, S. X. Wang and S. S. Gambhir, Fluorescent Magnetic Nanoparticles for Magnetically Enhanced Cancer Imaging and Targeting in Living Subjects, ACS Nano, 2012, 6(8), 6862–6869 CrossRef CAS PubMed.
- H. E. Daldrup-Link, C. Chan, O. Lenkov, S. Taghavigarmestani, T. Nazekati, H. Nejadnik, F. Chapelin, A. Khurana, X. Tong and F. Yang, et al., Detection of Stem Cell Transplant Rejection with Ferumoxytol MR Imaging: Correlation of MR Imaging Findings with Those at Intravital Microscopy, Radiology, 2017, 284(2), 495–507 CrossRef PubMed.
- K. Li, C. T. Chan, H. Nejadnik, O. D. Lenkov, C. Wolterman, R. Paulmurugan, H. Yang, S. S. Gambhir and H. E. Daldrup-Link, Ferumoxytol-Based Dual-Modality Imaging Probe for Detection of Stem Cell Transplant Rejection, Nanotheranostics, 2018, 2(4), 306–319 CrossRef PubMed.
- K. A. Hinds, J. M. Hill, E. M. Shapiro, M. O. Laukkanen, A. C. Silva, C. A. Combs, T. R. Varney, R. S. Balaban, A. P. Koretsky and C. E. Dunbar, Highly Efficient Endosomal Labeling of Progenitor and Stem Cells with Large Magnetic Particles Allows Magnetic Resonance Imaging of Single Cells, Blood, 2003, 102, 867–872 CrossRef CAS.
- J. Oh, M. D. Feldman, J. Kim, C. Condit, S. Emelianov and T. E. Milner, Detection of Magnetic Nanoparticles in Tissue Using Magneto-Motive Ultrasound, Nanotechnology, 2006, 17(16), 4183–4190 CrossRef CAS.
- H. Y. Kim, R. Li, T. S. C. Ng, G. Courties, C. B. Rodell, M. Prytyskach, R. H. Kohler, M. J. Pittet, M. Nahrendorf and R. Weissleder, et al., Quantitative Imaging of Tumor-Associated Macrophages and Their Response to Therapy Using (64)Cu-Labeled Macrin, ACS Nano, 2018, 12(12), 12015–12029 CrossRef CAS PubMed.
- M. Pacilio, C. Lauri, D. Prosperi, A. Petitti and A. Signore, New SPECT and PET Radiopharmaceuticals for Imaging Inflammatory Diseases: A Narrative Review, Semin. Nucl. Med., 2018, 48(3), 261–276 CrossRef PubMed.
- C. Rischpler, N. Langwieser, M. Souvatzoglou, A. Batrice, S. van Marwick, J. Snajberk, T. Ibrahim, K.-L. Laugwitz, S. G. Nekolla and M. Schwaiger, PET/MRI Early after Myocardial Infarction: Evaluation of Viability with Late Gadolinium Enhancement Transmurality vs. 18F-FDG Uptake, Eur. Heart J. Cardiovasc. Imaging, 2015, 16(6), 661–669 Search PubMed.
- S. S. Yaghoubi, M. C. Jensen, N. Satyamurthy, S. Budhiraja, D. Paik, J. Czernin and S. S. Gambhir, Noninvasive Detection of Therapeutic Cytolytic T Cells with 18F–FHBG PET in a Patient with Glioma, Nat. Clin. Pract. Oncol., 2009, 6(1), 53–58 CrossRef CAS.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Multifunctional Nanoparticles for Multimodal Imaging and Theragnosis, Chem. Soc. Rev., 2012, 41(7), 2656–2672 RSC.
- H. Moon, H. E. Park, J. Kang, H. Lee, C. Cheong, Y. T. Lim, S.-H. Ihm, K.-B. Seung, F. A. Jaffer and J. Narula, et al., Noninvasive Assessment of Myocardial Inflammation by Cardiovascular Magnetic Resonance in a Rat Model of Experimental Autoimmune Myocarditis, Circulation, 2012, 125(21), 2603–2612 CrossRef.
- H. P. Luehmann, E. D. Pressly, L. Detering, C. Wang, R. Pierce, P. K. Woodard, R. J. Gropler, C. J. Hawker and Y. Liu, PET/CT Imaging of Chemokine Receptor CCR5 in Vascular Injury Model Using Targeted Nanoparticle, J. Nucl. Med., 2014, 55(4), 629–634 CrossRef CAS PubMed.
- E. D. Wieder, Real-Time Monitoring of Immune Responses, Cytotherapy, 2002, 4(4), 347–352 CrossRef CAS PubMed.
- S. S. Kelkar and T. M. Reineke, Theranostics: Combining Imaging and Therapy, Bioconjugate Chem., 2011, 22(10), 1879–1903 CrossRef CAS.
- X. Ma, Y. Zhao and X. J. Liang, Theranostic Nanoparticles Engineered for Clinic and Pharmaceutics, Acc. Chem. Res., 2011, 44(10), 1114–1122 CrossRef CAS PubMed.
- P. Wang, M. V. Yigit, C. Ran, A. Ross, L. Wei, G. Dai, Z. Medarova and A. Moore, A Theranostic Small Interfering RNA Nanoprobe Protects Pancreatic Islet Grafts from Adoptively Transferred Immune Rejection, Diabetes, 2012, 61(12), 3247–3254 CrossRef CAS PubMed.
- H. Borghaei, L. Paz-Ares, L. Horn, D. R. Spigel, M. Steins, N. E. Ready, L. Q. Chow, E. E. Vokes, E. Felip and E. Holgado, et al., Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer, N. Engl. J. Med., 2015, 373(17), 1627–1639 CrossRef CAS PubMed.
- R. Meir, K. Shamalov, T. Sadan, M. Motiei, G. Yaari, C. J. Cohen and R. Popovtzer, Fast Image-Guided Stratification Using Anti-Programmed Death Ligand 1 Gold Nanoparticles for Cancer Immunotherapy, ACS Nano, 2017, 11(11), 11127–11134 CrossRef CAS.
- Y. Zhong, Z. Ma, F. Wang, X. Wang, Y. Yang, Y. Liu, X. Zhao, J. Li, H. Du and M. Zhang, et al., In Vivo Molecular Imaging for Immunotherapy Using Ultra-Bright near-Infrared-IIb Rare-Earth Nanoparticles, Nat. Biotechnol., 2019, 37(11), 1322–1331 CrossRef CAS PubMed.
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, In Situ Programming of Leukaemia-Specific T Cells Using Synthetic DNA Nanocarriers, Nat. Nanotechnol., 2017, 12(8), 813–820 CrossRef CAS PubMed.
- Y. Yang, J. Zhang, F. Xia, C. Zhang, Q. Qian, X. Zhi, C. Yue, R. Sun, S. Cheng and S. Fang, et al., Human CIK Cells Loaded with Au Nanorods as a Theranostic Platform for Targeted Photoacoustic Imaging and Enhanced Immunotherapy and Photothermal Therapy, Nanoscale Res. Lett., 2016, 11(1), 285 CrossRef PubMed.
- M. Qian, L. Chen, Y. Du, H. Jiang, T. Huo, Y. Yang, W. Guo, Y. Wang and R. Huang, Biodegradable Mesoporous Silica Achieved via Carbon Nanodots-Incorporated Framework Swelling for Debris-Mediated Photothermal Synergistic Immunotherapy, Nano Lett., 2019, 19(12), 8409–8417 CrossRef CAS PubMed.
- A. Varki, Biological Roles of Glycans, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- H. Ponta, L. Sherman and P. A. Herrlich, CD44: From Adhesion Molecules to Signalling Regulators, Nat. Rev. Mol. Cell Biol., 2003, 4, 33–45 CrossRef CAS PubMed.
- S. Banerji, A. J. Wright, M. Noble, D. J. Mahoney, I. D. Campbell, A. J. Day and D. G. Jackson, Structures of the CD44–Hyaluronan Complex Provide Insight into a Fundamental Carbohydrate Protein Interaction, Nat. Struct. Mol. Biol., 2007, 14, 234–239 CrossRef CAS PubMed.
- M. H. El-Dakdouki, K. El-boubbou, M. Kamat, R. Huang, G. S. Abela, M. Kiupel, D. C. Zhu and X. Huang, CD44 Targeting Magnetic Glyconanoparticles for Atherosclerotic Plaque Imaging, Pharm. Res., 2012, 31, 1426–1437 CrossRef PubMed.
- M. Kamat, K. El-boubbou, D. Zhu, T. Lansdell, X. Lu, W. Li and X. Huang, Hyaluronic Acid Immobilized Magnetic Nanoparticles for Active Targeting and Imaging of Macrophages, Bioconjugate Chem., 2010, 21, 2128–2135 CrossRef CAS PubMed.
- N. Singh, G. J. S. Jenkins, R. Asadi and S. H. Doak, Potential Toxicity of Superparamagnetic Iron Oxide Nanoparticles (SPION), Nano Rev., 2010, 1, 5358 CrossRef PubMed.
- S. Hossaini Nasr, A. Tonson, M. H. El-Dakdouki, D. C. Zhu, D. Agnew, R. Wiseman, C. Qian and X. Huang, Effects of Nanoprobe Morphology on Cellular Binding and Inflammatory Responses: Hyaluronan-Conjugated Magnetic Nanoworms for Magnetic Resonance Imaging of Atherosclerotic Plaques, ACS Appl. Mater. Interfaces, 2018, 10, 11495–11507 CrossRef CAS PubMed.
- B. R. Smith, P. Kempen, D. Bouley, A. Xu, Z. Liu, N. Melosh, H. Dai, R. Sinclair and S. S. Gambhir, Shape Matters: Intravital Microscopy Reveals Surprising Geometrical Dependence for Nanoparticles in Tumor Models of Extravasation, Nano Lett., 2012, 12(7), 3369–3377 CrossRef CAS PubMed.
- T. J. Beldman, M. L. Senders, A. Alaarg, C. Pérez-Medina, J. Tang, Y. Zhao, F. Fay, J. Deichmöller, B. Born and E. Desclos, et al., Hyaluronan Nanoparticles Selectively Target Plaque-Associated Macrophages and Improve Plaque Stability in Atherosclerosis, ACS Nano, 2017, 11(6), 5785–5799 CrossRef CAS PubMed.
- R. P. McEver, Selectins: Initiators of Leucocyte Adhesion and Signalling at the Vascular Wall, Cardiovasc. Res., 2015, 107, 331–339 CrossRef CAS.
- E. Jubeli, L. Moine, V. Nicolas and G. Barratt, Preparation of E-Selectin-Targeting Nanoparticles and Preliminary in Vitro Evaluation, Int. J. Pharm., 2012, 426, 291–301 CrossRef CAS PubMed.
- S. Laurent, D. Stanicki, S. Boutry, J. C. Roy, E. L. Vander and R. N. Muller, Development of a New Molecular Probe for the Detection of Inflammatory Process, J. Mol. Biol. Mol. Imaging, 2015, 2, id1013 Search PubMed.
- C. Chantarasrivong, A. Ueki, R. Ohyama, J. Unga, S. Nakamura, I. Nakanishi, Y. Higuchi, S. Kawakami, H. Ando and A. Imamura, et al., Synthesis and Functional Characterization of Novel Sialyl LewisX Mimic-Decorated Liposomes for E-Selectin-Mediated Targeting to Inflamed Endothelial Cells, Mol. Pharmaceutics, 2017, 14, 1528–1537 CrossRef CAS PubMed.
- N. R. Zaccai, K. Maenaka, T. Maenaka, P. R. Crocker, R. Brossmer, S. Kelm and E. Y. Jones, Structure-Guided Design of Sialic Acid-Based Siglec Inhibitors and Crystallographic Analysis in Complex with Sialoadhesin, Structure, 2003, 11, 557–567 CrossRef CAS PubMed.
- W. C. Chen, N. Kawasaki, C. M. Nycholat, S. Han, J. Pilotte, P. R. Crocker and J. C. Paulson, Antigen Delivery to Macrophages Using Liposomal Nanoparticles Targeting Sialoadhesin/CD169, PLoS One, 2012, 7(6), 1–9 Search PubMed.
- M. Funovics, X. Montet, F. Reynolds, R. Weissleder and L. Josephson, Nanoparticles for the Optical Imaging of Tumor E-Selectin, Neoplasia, 2005, 7, 904–911 CrossRef CAS PubMed.
- D. H. Kong, Y. K. Kim, M. R. Kim, J. H. Jang and S. Lee, Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer, Int. J. Mol. Sci., 2018, 19, E1057 CrossRef PubMed.
- K. Khodabandehlou, J. J. Masehi-Lano, C. Poon, J. Wang and E. J. Chung, Targeting Cell Adhesion Molecules with Nanoparticles Using In Vivo and Flow-Based in Vitro Models of Atherosclerosis, Exp. Biol. Med., 2017, 242, 799–812 CrossRef CAS PubMed.
- B. R. Smith, J. Heverhagen, M. Knopp, P. Schmalbrock, J. Shapiro, M. Shiomi, N. I. Moldovan, M. Ferrari and S. C. Lee, Localization to Atherosclerotic Plaque and Biodistribution of Biochemically Derivatized Superparamagnetic Iron Oxide Nanoparticles (SPIONs) Contrast Particles for Magnetic Resonance Imaging (MRI), Biomed. Microdevices, 2007, 9(5), 719–727 CrossRef PubMed.
- M. A. Curotto de Lafaille and J. J. Lafaille, Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor?, Immunity, 2009, 30, 626–635 CrossRef CAS PubMed.
- D. Bruder, M. Probst-Kepper, A. M. Westendorf, R. Geffers, S. Beissert, K. Loser, H. von Boehmer, J. Buer and W. Hansen, Neuropilin-1: A Surface Marker of Regulatory T Cells, Eur. J. Immunol., 2004, 34, 623–630 CrossRef CAS PubMed.
- W. Ou, R. K. Thapa, L. Jiang, Z. C. Soe, M. Gautam, J. H. Chang, J. H. Jeong, S. K. Ku, H. G. Choi and C. S. Yong, et al., Regulatory T Cell-Targeted Hybrid Nanoparticles Combined with Immuno-Checkpoint Blockage for Cancer Immunotherapy, J. Controlled Release, 2018, 281, 84–96 CrossRef CAS PubMed.
- M. A. McAteer, J. E. Schneider, Z. A. Ali, N. Warrick, C. A. Bursill, C. von zur Muhlen, D. R. Greaves, S. Neubauer, K. M. Channon and R. P. Choudhury, Magnetic Resonance Imaging of Endothelial Adhesion Molecules in Mouse Atherosclerosis Using Dual-Targeted Microparticles of Iron Oxide, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 77–83 CrossRef CAS.
- Y. T. Lim, M. Y. Cho, Y. W. Noh, J. W. Chung and B. H. Chung, Near-Infrared Emitting Fluorescent Nanocrystals-Labeled Natural Killer Cells as a Platform Technology for the Optical Imaging of Immunotherapeutic Cells-Based Cancer Therapy, Nanotechnology, 2009, 20, 475102 CrossRef PubMed.
- B. R. Smith, Z. Cheng, A. De, J. Rosenberg and S. S. Gambhir, Dynamic Visualization of RGD-Quantum Dot Binding to Tumor Neovasculature and Extravasation in Multiple Living Mouse Models Using Intravital Microscopy, Small, 2010, 6(20), 2222–2229 CrossRef CAS PubMed.
- D. Schmid, C. G. Park, C. A. Hartl, N. Subedi, A. N. Cartwright, R. B. Puerto, Y. Zheng, J. Maiarana, G. J. Freeman and K. W. Wucherpfennig, et al., T Cell-Targeting Nanoparticles Focus Delivery of Immunotherapy to Improve Antitumor Immunity, Nat. Commun., 2017, 8, 1747 CrossRef PubMed.
- J. J. Glass, D. Yuen, J. Rae, A. P. Johnston, R. G. Parton, S. J. Kent and R. De Rose, Human Immune Cell Targeting of Protein Nanoparticles--Caveospheres, Nanoscale, 2016, 8, 8255–8265 RSC.
- D. D. Lasic, F. J. Martin, A. Gabizon, S. K. Huang and D. Papahadjopoulos, Sterically Stabilized Liposomes: A Hypothesis on the Molecular Origin of the Extended Circulation Times, Biochim. Biophys. Acta, 1991, 1070, 187–192 CrossRef CAS.
- A. S. Abu Lila, H. Kiwada and T. Ishida, The Accelerated Blood Clearance (ABC) Phenomenon: Clinical Challenge and Approaches to Manage, J. Controlled Release, 2013, 172, 38–47 CrossRef CAS PubMed.
- A. V. Kroll, R. H. Fang and L. Zhang, Biointerfacing and Applications of Cell Membrane-Coated Nanoparticles, Bioconjugate Chem., 2017, 28, 23–32 CrossRef CAS PubMed.
- K. Jin, Z. Luo, B. Zhang and Z. Pang, Biomimetic Nanoparticles for Inflammation Targeting, Acta Pharm. Sin. B, 2018, 8(1), 23–33 CrossRef PubMed.
- C. M. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Erythrocyte Membrane-Camouflaged Polymeric Nanoparticles as a Biomimetic Delivery Platform, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- L. Rao, L.-L. Bu, Q. F. Meng, B. Cai, W.-W. Deng, A. Li, K. Li, S.-S. Guo, W.-F. Zhang and W. Liu, et al., Antitumor Platelet-Mimicking Magnetic Nanoparticles, Adv. Funct. Mater., 2017, 27, 1604774 CrossRef.
- C.-M. J. Hu, R. H. Fang, K. C. Wang, B. T. Luk, S. Thamphiwatana, D. Dehaini, P. Nguyen, P. Angsantikul, C. H. Wen and A. V. Kroll, et al., Nanoparticle Biointerfacing by Platelet Membrane Cloaking, Nature, 2015, 526, 118–121 CrossRef CAS PubMed.
- T. Kang, Q. Zhu, D. Wei, J. Feng, J. Yao, T. Jiang, Q. Song, X. Wei, H. Chen and X. Gao, et al., Nanoparticles Coated with Neutrophil Membranes Can Effectively Treat Cancer Metastasis, ACS Nano, 2017, 11, 1397–1411 CrossRef CAS PubMed.
- X. Cao, Y. Hu, S. Luo, Y. Wang, T. Gong, X. Sun, Y. Fu and Z. Zhang, Neutrophil-Mimicking Therapeutic Nanoparticles for Targeted Chemotherapy of Pancreatic Carcinoma, Acta Pharm. Sin. B, 2019, 9(3), 575–589 CrossRef PubMed.
- L. Rao, L.-L. Bu, B. Cai, J. H. Xu, A. Li, W.-F. Zhang, Z.-J. Sun, S. S. Guo, W. Liu and T. H. Wang, et al., Cancer Cell Membrane-Coated Upconversion Nanoprobes for Highly Specific Tumor Imaging, Adv. Mater., 2016, 28, 3460–3466 CrossRef CAS PubMed.
- R. H. Fang, C. M. Hu, B. T. Luk, W. Gao, J. A. Copp, Y. Tai, D. E. O’Connor and L. Zhang, Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery, Nano Lett., 2014, 14, 2181–2188 CrossRef CAS PubMed.
- M. Wu, W. Le, T. Mei, Y. Wang, B. Chen, Z. Liu and C. Xue, Cell Membrane Camouflaged Nanoparticles: A New Biomimetic Platform for Cancer Photothermal Therapy, Int. J. Nanomed., 2019, 14, 4431–4448 CrossRef CAS PubMed.
- G. van Niel, G. D’Angelo and G. Raposo, Shedding Light on the Cell Biology of Extracellular Vesicles, Nat. Rev. Mol. Cell Biol., 2018, 19(4), 213–228 CrossRef CAS PubMed.
- H. Kalra, G. Drummen and S. Mathivanan, Focus on Extracellular Vesicles: Introducing the Next Small Big Thing, Int. J. Mol. Sci., 2016, 17(2), 170 CrossRef PubMed.
- M. Tkach and C. Théry, Communication by Extracellular Vesicles: Where We Are and Where We Need to Go, Cell, 2016, 164(6), 1226–1232 CrossRef CAS PubMed.
- C. D’Souza-Schorey and J. W. Clancy, Tumor-Derived Microvesicles: Shedding Light on Novel Microenvironment Modulators and Prospective Cancer Biomarkers, Genes Dev., 2012, 26(12), 1287–1299 CrossRef PubMed.
- P. D. Robbins and A. E. Morelli, Regulation of Immune Responses by Extracellular Vesicles, Nat. Rev. Immunol., 2014, 14(3), 195–208 CrossRef CAS PubMed.
- Y. D. Dai and P. Dias, Exosomes or Microvesicles, a Secreted Subcellular Organelle Contributing to Inflammation and Diabetes, Diabetes, 2018, 67(11), 2154–2156 CrossRef CAS PubMed.
- G. Raposo, H. W. Nijman, W. Stoorvogel, R. Liejendekker, C. V. Harding, C. J. Melief and H. J. Geuze, B Lymphocytes Secrete Antigen-Presenting Vesicles, J. Exp. Med., 1996, 183(3), 1161–1172 CrossRef CAS PubMed.
- C. P. Lai, E. Y. Kim, C. E. Badr, R. Weissleder, T. R. Mempel, B. A. Tannous and X. O. Breakefield, Visualization and Tracking of Tumour Extracellular Vesicle Delivery and RNA Translation Using Multiplexed Reporters, Nat. Commun., 2015, 6(1), 7029 CrossRef CAS PubMed.
- C. P. Lai, O. Mardini, M. Ericsson, S. Prabhakar, C. A. Maguire, J. W. Chen, B. A. Tannous and X. O. Breakefield, Dynamic Biodistribution of Extracellular Vesicles In Vivo Using a Multimodal Imaging Reporter, ACS Nano, 2014, 8(1), 483–494 CrossRef CAS PubMed.
- A. Busato, R. Bonafede, P. Bontempi, I. Scambi, L. Schiaffino, D. Benati, M. Malatesta, A. Sbarbati and R. Mariotti, Magnetic Resonance Imaging of Ultrasmall Superparamagnetic Iron Oxide-Labeled Exosomes from Stem Cells: A New Method to Obtain Labeled Exosomes, Int. J. Nanomed., 2016, 2481 CAS.
- T. Smyth, M. Kullberg, N. Malik, P. Smith-Jones, M. W. Graner and T. J. Anchordoquy, Biodistribution and Delivery Efficiency of Unmodified Tumor-Derived Exosomes, J. Controlled Release, 2015, 199, 145–155 CrossRef CAS PubMed.
- D. W. Hwang, H. Choi, S. C. Jang, M. Y. Yoo, J. Y. Park, N. E. Choi, H. J. Oh, S. Ha, Y.-S. Lee and J. M. Jeong, et al., Noninvasive Imaging of Radiolabeled Exosome-Mimetic Nanovesicle Using (99m)Tc-HMPAO, Sci. Rep., 2015, 5, 15636 CrossRef CAS PubMed.
- Y. Takahashi, M. Nishikawa, H. Shinotsuka, Y. Matsui, S. Ohara, T. Imai and Y. Takakura, Visualization and In Vivo Tracking of the Exosomes of Murine Melanoma B16-BL6 Cells in Mice after Intravenous Injection, J. Biotechnol., 2013, 165(2), 77–84 CrossRef CAS PubMed.
- J. L. Hood, R. S. San and S. A. Wickline, Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis, Cancer Res., 2011, 71(11), 3792–3801 CrossRef CAS PubMed.
- L. Hu, S. A. Wickline and J. L. Hood, Magnetic Resonance Imaging of Melanoma Exosomes in Lymph Nodes, Magn. Reson. Med., 2015, 74(1), 266–271 CrossRef CAS PubMed.
- Z. Varga, I. Gyurkó, K. Pálóczi, E. I. Buzás, I. Horváth, N. Hegedűs, D. Máthé and K. Szigeti, Radiolabeling of Extracellular Vesicles with 99m Tc for Quantitative In Vivo Imaging Studies, Cancer Biother. Radiopharm., 2016, 31(5), 168–173 CrossRef CAS PubMed.
- P. Gangadaran, C. M. Hong, J. M. Oh, R. L. Rajendran, S. Kalimuthu, S. H. Son, A. Gopal, L. Zhu, S. H. Baek and S. Y. Jeong, et al., In Vivo Non-Invasive Imaging of Radio-Labeled Exosome-Mimetics Derived From Red Blood Cells in Mice, Front. Pharmacol., 2018, 9, 817 CrossRef PubMed.
- M. K. Islam, P. Syed, L. Lehtinen, J. Leivo, K. Gidwani, S. Wittfooth, K. Pettersson and U. Lamminmäki, A Nanoparticle-Based Approach for the Detection of Extracellular Vesicles, Sci. Rep., 2019, 9(1), 10038 CrossRef PubMed.
- F. J. Gildehaus, F. Haasters, I. Drosse, E. Wagner, C. Zach, W. Mutschler, P. Cumming, P. Bartenstein and M. Schieker, Impact of Indium-111 Oxine Labelling on Viability of Human Mesenchymal Stem Cells in Vitro, and 3D Cell-Tracking Using SPECT/CT In Vivo, Mol. Imaging Biol., 2011, 13(6), 1204–1214 CrossRef PubMed.
- S. Correia Carreira, J. P. K. Armstrong, A. M. Seddon, A. W. Perriman, R. Hartley-Davies and W. Schwarzacher, Ultra-Fast Stem Cell Labelling Using Cationised Magnetoferritin, Nanoscale, 2016, 8(14), 7474–7483 RSC.
- I. J. M. de Vries, W. J. Lesterhuis, J. O. Barentsz, P. Verdijk, J. H. van Krieken, O. C. Boerman, W. J. G. Oyen, J. J. Bonenkamp, J. B. Boezeman and G. J. Adema, et al., Magnetic Resonance Tracking of Dendritic Cells in Melanoma Patients for Monitoring of Cellular Therapy, Nat. Biotechnol., 2005, 23(11), 1407–1413 CrossRef CAS PubMed.
- Z. A. Nima, F. Watanabe, A. Jamshidi-Parsian, M. Sarimollaoglu, D. A. Nedosekin, M. Han, J. A. Watts, A. S. Biris, V. P. Zharov and E. I. Galanzha, Bioinspired Magnetic Nanoparticles as Multimodal Photoacoustic, Photothermal and Photomechanical Contrast Agents, Sci. Rep., 2019, 9, 887 CrossRef PubMed.
- G. Vargas, J. Cypriano, T. Correa, P. Leão, D. A. Bazylinski and F. Abreu, Applications of Magnetotactic Bacteria, Magnetosomes and Magnetosome Crystals in Biotechnology and Nanotechnology: Mini-Review, Molecules, 2018, 23(10), 2438 CrossRef PubMed.
- K. D. Brewer, R. Spitler, K. R. Lee, A. C. Chan, J. C. Barrozo, A. Wakeel, C. S. Foote, S. Machtaler, J. Rioux and J. K. Willmann, et al., Characterization of Magneto-Endosymbionts as MRI Cell Labeling and Tracking Agents, Mol. Imaging Biol., 2018, 20, 65–73 CrossRef CAS PubMed.
- K. R. Lee, A. Wakeel, P. Chakraborty, C. S. Foote, L. Kajiura, J. C. Barrozo, A. C. Chan, A. V. Bazarov, R. Spitler and P. M. Kutny, et al., Cell Labeling with Magneto-Endosymbionts and the Dissection of the Subcellular Location, Fate, and Host Cell Interactions, Mol. Imaging Biol., 2018, 20, 55–64 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |