
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origin of Sn(II) oxidation in tin halide perovskites†
Jorge
Pascual
 a,
Giuseppe
Nasti
b,
Mahmoud H.
Aldamasy
ac,
Joel A.
Smith
d,
Marion
Flatken
a,
Nga
Phung
a,
Diego
Di Girolamo
b,
Silver-Hamill
Turren-Cruz
a,
Meng
Li
ae,
André
Dallmann
*f,
Roberto
Avolio
*g and
Antonio
Abate
*ab
a,
Giuseppe
Nasti
b,
Mahmoud H.
Aldamasy
ac,
Joel A.
Smith
d,
Marion
Flatken
a,
Nga
Phung
a,
Diego
Di Girolamo
b,
Silver-Hamill
Turren-Cruz
a,
Meng
Li
ae,
André
Dallmann
*f,
Roberto
Avolio
*g and
Antonio
Abate
*ab
aHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Germany. E-mail: antonio.abate@helmholtz-berlin.de
bDepartment of Chemical Materials and Production Engineering, University of Naples Federico II, Piazzale Tecchio 80, 80125 Fuorigrotta, Naples, Italy
cEgyptian Petroleum Research Institute, Nasr City, P.O. 11727, Cairo, Egypt
dDepartment of Physics and Astronomy, University of Sheffield, UK
eInstitute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou 215123, China
fHumboldt Universität zu Berlin, Institut für Chemie, AG NMR, Germany
gNational Research Council of Italy, Institute for Polymers Composites and Biomaterials, Via Campi Flegrei 34, 80078 Pozzuoli (NA), Italy
First published on 9th July 2020
Abstract
Tin-halide perovskites have great potential as photovoltaic materials, but their performance is hampered by undesirable oxidation of Sn(II) to Sn(IV). In this work, we use nuclear magnetic resonance spectroscopy (NMR) to identify and describe the origins of Sn(IV) in Sn-based perovskites, mainly focusing on direct measurements of Sn oxidation states with 119Sn-NMR in solid-state and solution. We find that dimethylsulfoxide (DMSO), a typical solvent for Sn-based perovskites, oxidizes Sn(II) in acidic conditions under temperatures used for film annealing. We propose a redox reaction between DMSO and Sn(II), catalyzed by hydroiodic acid, with iododimethylsulfonium iodide intermediate. We find that lower temperatures and less acidic conditions abate this reaction, and we assess a range of compositions and solution components for this instability. These results suggest the need for strategies to prevent this reaction and shed light on other solution instabilities beyond Sn(IV) that must be mitigated to achieve high-performance lead-free perovskites.
Tin halide perovskite materials have recently gained the attention of the photovoltaic community due to their advantages over lead-based perovskites, such as their close-to ideal bandgap,1 lower environmental toxicity2 and potential to be the bottom cell in all-perovskite tandem solar cells.3 However, the efficiency of tin-based perovskite solar cells has only recently surpassed 10%,4 mainly due to the instability of the Sn(II) oxidation state, easily oxidised to Sn(IV).5 Although the presence of Sn(IV) in the films has been proven, it has not yet been fully explained how, and at which stage of the material and device processing, Sn(II) can be oxidised in the absence of any oxidant species, assuming that these materials are processed in inert atmospheres of shallow O2 content (i.e. <0.1 ppm).6
Several reports have attempted to quantify the Sn(IV) content in perovskite films utilising X-ray photoelectron spectroscopy (XPS) measurements. This technique is highly surface sensitive, probing depths usually below 10 nm. Therefore values obtained with XPS can be misleading and inaccurately represent the real Sn(IV) content in the film. Besides, the influence on Sn(IV) formation from ultra-high vacuum and X-rays, which are claimed to induce degradation in lead-based perovskite films,7 has not yet been studied in detail. While precisely measuring the proportion of these two oxidation states in perovskite thin films can be tricky, there are other facile and available techniques to investigate the origins of Sn(II) oxidation directly. Considering that the electronic environment of different oxidation states markedly differs, we should explore methods that are sensitive to changes in this property. In this regard, nuclear magnetic resonance (NMR) is an ideal technique with excellent capability to differentiate between chemical environments of each element. Mainly used in materials science for the characterisation of organic molecules, NMR has proven its utility for characterising organometallic systems.8 Although there are steadily increasing reports on its application for metal halide perovskites,9,10 its full potential remains underexplored.
In this work, we present the use of a range of NMR techniques as a straightforward and versatile tool for quantifying Sn(IV) content in Sn-based precursors and solutions. By making use of 119Sn-NMR, we identify and describe the origin of Sn(IV) formation from the reaction between DMSO, which is the most commonly used solvent to prepared devices, and Sn(II) at high temperature in acidic conditions, quantifying the amount of oxidised Sn(IV) being generated. This study aims to be a reproducible guideline for rapid and facile use of NMR for characterising the properties and chemical processes occurring in tin halide perovskite materials.
Intending to find when Sn(IV) is formed during the processing of perovskite, we identified three possible sources of oxidation: precursors, solutions and annealing (Fig. 1a). The versatility of NMR allowed for different experiments appropriate for each processing step. We first used solid-state 119Sn-NMR for characterising the content of the different oxidation states in SnI2, the precursor generally used for perovskite solution preparation. For the as-received material, Sn(II) signal appears at −529 ppm (Fig. 1b), but no Sn(IV) signal was detected. Consequently, the molar content of any Sn(IV) in the precursor was below the detection limit of the measurement, which we estimate to be ∼1.5 wt% from the signal-to-noise ratio. Exposure of the powder to air led to increasing Sn(IV) content with time, indicating that oxidised Sn(IV) from SnI2 shows up between −1740 and −1745 ppm. These results confirm that there is not a significant contribution from the precursor to the final Sn(IV) content into the perovskite.
 | ||
| Fig. 1 (a) Steps of tin-based perovskite material preparation studied in this work as possible sources of Sn(II) oxidation; (b) solid-state 119Sn-NMR spectra of SnI2 precursor with and without air exposure. | ||
Our next hypothesis involved the potential oxidation of Sn(II) through the preparation of the tin halide perovskite solution. In this step, we studied DMF and DMSO (both deuterated), the most common solvents in literature for these materials, as well as formamidinium iodide (FAI) as the organic salt for the preparation of FASnI3 perovskite. For this purpose, we measured freshly prepared solutions (no heating was applied unless stated otherwise) using liquid-state 119Sn-NMR (Fig. 2a) and found no Sn(IV) for either SnI2 or FASnI3 in both solvents. This finding agrees with the results from solid-state NMR. A 1.0 M sample of SnI4 in DMSO was measured to know the approximate expected shift, with Sn(IV) signal found at −2023 ppm (Fig. 2a). We found that the chemical shift of the SnI2 can vary significantly. Firstly, we found lower chemical shifts in DMSO than in DMF, which agrees with the more effective donation of electron density from DMSO to SnI2 and, to a lesser extent, between SnI2 and FASnI3 (Fig. 2a). In the latter case, the introduction of FAI can have an electron-withdrawing effect on SnI2 adducts, as NMR has already proven its complexing ability in solution.11 Interestingly, Fig. 2b shows that there is a strong dependence of the SnI2 chemical shift with its solution concentration in DMSO, which is less pronounced in DMF (Fig. S1 and S2, ESI†). The lower the concentration of SnI2, the more efficiently DMSO can solvate SnI2, shielding Sn(II) and shifting its NMR signal upfield.
 | ||
| Fig. 2 (a) 119Sn-NMR Sn(II) signals for SnI2 in DMF (green) and DMSO (red), FASnI3 in DMF (turquoise) and DMSO (yellow) and Sn(IV) signal for SnI4 in DMSO (purple); (b) dependence of the 119Sn-NMR Sn(II) signal chemical shift of SnI2 in DMSO and DMF with solution concentration; (c) FASnI3 (left) and FASnI3 in DMSO heated at 100 °C for 30 min (right); (d) 119Sn-NMR spectra in the Sn(IV) range of FASnI3 solutions in DMSO heated at 80 °C and 100 °C for 30 min. | ||
We also investigated the possible effect of concentration on the chemical shift of SnI4. However, no significant difference was found (Fig. S3, ESI†), suggesting that the configuration of SnI4 in solution prevents interaction between the solvent and the metallic centre. Therefore, solvation does not substantially affect its shift. Thus liquid-state NMR is not just useful for Sn(IV) detection but also provides information on the coordination chemistry in tin halide perovskite solutions.
After finding no significant formation of Sn(IV) from the previous steps, we decided to investigate the annealing process. We heated FASnI3 solutions at 100 °C for 30 min, to mimic the conditions used for annealing tin-based perovskite layers in the reported literature so far.12 Notably, we observed a change in the colour of the solution from orange to dark red (Fig. 2c), which is clear evidence of a chemical reaction occurring. This change in colour was recently reported also in the publication by Saidaminov and co-workers after heating the solution at 120 °C for 10 min.13 We characterised this solution by liquid-state 119Sn-NMR and found a peak corresponding to Sn(IV) (Fig. 2d).
To prove whether the reaction is spontaneous or depends on externally applied energy, we also heated a FASnI3 solution at 80 °C for 30 min, which did cause only a minor change in colour. As can be seen in Fig. 2d, no Sn(IV) formation is observed after this treatment, suggesting that performing annealing at lower temperatures could be a solution for preventing Sn(IV) formation. This result would agree with the fact that the current highest PCE reported for Sn-based perovskite solar cells was processed at 70 °C.4 We note that heating the solution is under excess-solvent conditions. Comparing it to the thin film preparation, solvent will remain present in the wet film at the start of annealing as well. Hence, it should be considered that similar effects may also occur during annealing to modify the Sn oxidation state ratio.
One of the most potent aspects of NMR characterisation is that the signal intensities from the different chemical environments in a sample are proportional to the number of nuclei, allowing the quantification or estimation of relative proportions of two different compounds. For our study, we prepared a solution of FASnI3 in DMSO deliberately substituting 1% SnI2 for SnI4, to confirm the detection limit for this method. With this sample, we proved that even this low Sn(IV) content could be easily identified by NMR (Fig. S4, ESI†). From this, we also determined the Sn(IV) content for FASnI3 and MASnI3 samples to be around 1%, considering that for the same signal-to-noise ratio and acquisition time, the signal intensities were approximately the same.
To have insights into the Sn(II) oxidation reaction, we also investigated the state of the organic parts (i.e. DMSO and FAI/MAI) by 1H- and 13C-NMR. As we can see from the 1H spectra in Fig. 3a, a signal is found in the 1.89–1.92 ppm range for both MA and FA containing solution. This trend fits the expected shift for dimethyl sulfide (DMS), that we determined by adding 1 equivalent of DMS to a FASnI3 sample (Fig. S5 and S6, ESI†). This was also confirmed with a 13C-NMR experiment, where we found the signal of the methyl groups from DMS at 17 ppm (Fig. S7, ESI†). In this respect, 13C-NMR is a more reliable method than 1H-NMR for monitoring DMS formation, considering that using DMSO-d6 leads to deuterated DMS. These results are in explicit agreement with the recent work on DMS formation, where the authors establish a relationship between the decomposition of DMSO and the emergence of the signal from DMS by 1H-NMR in Sn-based perovskite solutions, which they indirectly link to the oxidation of Sn(II).13 However, this correlation might present some drawbacks. At the same time, the authors also found traces of DMS for MAPbI3 solutions, as we did for unheated SnI2 or FASnI3 samples (Fig. S8, ESI†), which is reasonable considering that DMS is one of the main impurities found in DMSO.14 Also, DMS has such a low boiling point (37.3 °C) that it could potentially be lost during sample preparation. We see no longer an increase in the intensity of the DMS signal after heating at 100 °C for 2 or 4 days (Fig. S9, ESI†). We believe the direct identification of Sn(IV) by 119Sn-NMR that we present in this work is a more reliable approach to quantify the Sn(II) oxidation occurring in solution.
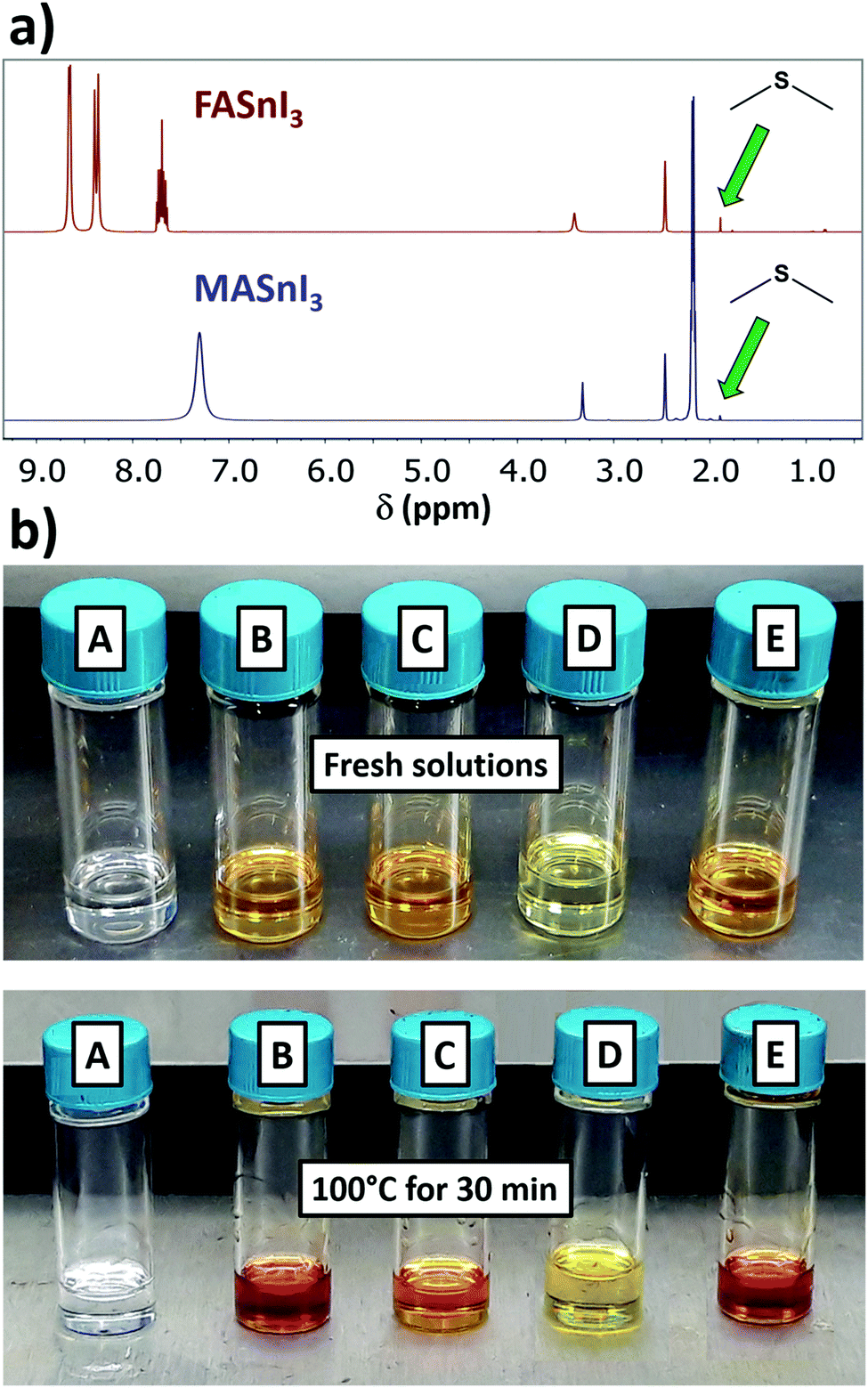 | ||
| Fig. 3 (a) 1H-NMR spectra of FASnI3 and MASnI3 in DMSO at 1 M after heating the solutions at 100 °C for 30 min. The green arrow highlights the peak of DMS originating from heating the solution; (b) vials containing different solutions before and after being heated at 100 °C for 30 min. A: FAI in DMSO; B: SnI2 in DMSO; C: FASnI3 in DMF; D: FASnI3 in DMSO with 10 mol% SnF2; E: CsSnI3 in DMSO. | ||
We performed 119Sn-NMR on heated FASnI3 solutions in DMF. The first observation was that the solution did not turn red during heating (Fig. 3b). Additionally, no Sn(IV) formation was detected by 119Sn-NMR, confirming that, among the conventional solvents, Sn(II) was exclusively being oxidised by DMSO through the following reaction, proposed in the recent viewpoint on DMS formation13 and previously described for tin chloride:15
| 2SnI2 + 2(CH3)2SO → SnO2 (s) + SnI4 + 2(CH3)S |
We were also interested in observing if the extensively used additive SnF2 could have any effect on this process. As we show in Fig. 3b, heating the solution containing 10 mol% SnF2 did not produce any significant change in colour, and showed no Sn(IV) signal (we investigated the complete spectral range in case new compounds were being formed). This result suggests the possibility of using certain additives to suppress or even prevent the redox reaction between DMSO and Sn(II). The different electronegativity of I− and F− might play an important role in the stabilization of Sn(II). In addition, a recent study has proposed the introduction of formic acid as an additive in the perovskite solution, owing to the higher stability of the Sn(II) oxidation state.17 While acidic conditions will stabilise Sn(II), we should be careful applying them when using DMSO as the solvent.
The results obtained so far point towards a redox reaction between DMSO and Sn(II) that is favoured in acidic conditions. The oxidation of many different types of organic compounds in DMSO in the presence of hydrogen halides has been widely studied.18–21 Through the in situ generation of halodimethylsulfonium halides19 it is possible to carry out oxidative halogenations on olefins20 or aromatic compounds,21 among others. In this sense, any I2 generated would also be able to oxidise SnI2 to SnI4. We confirmed this by adding I2 beads to a FASnI3 solution and found both Sn(II) and Sn(IV) presence (Fig. S11, ESI†).
Concerning this, the previously studied solution of FAI in DMSO did not change colour after heating at 100 °C for 30 min (Fig. 3b). If the DMSO reduction to DMS happened through iodide oxidation to I2, we should have seen this solution turn yellow, due to the formation of an I3− complex. This result would then suggest that the redox reaction might be directly between DMSO and Sn(II), and not through the formation of I2. However, the formation and stability of I2 might be influenced by whether a reducing agent like Sn(II) is present in solution to react with or not. If I2 has no compounds in solution to react with, the equilibrium might be shifted to the previous intermediates rather than to I2 formation. While I2 might be undetectable for FAI in DMSO, iododimethylsulfonium iodide might still be generated in situ in addition to DMS (Scheme 1), which could be related to there being two different signals in 1H-NMR in the range of DMS (1.89–2.05 ppm). The formation of this intermediate would explain the I2-mediated mechanism of Sn(II) oxidation in DMSO (Scheme 1). This mechanism involves the use of protons, which would agree with our previous results, in which we found that acidic conditions favour the reaction. The formation of SnO2 precipitates might also be postulated, originating from a secondary reaction between SnI4 and H2O, which would regenerate HI.
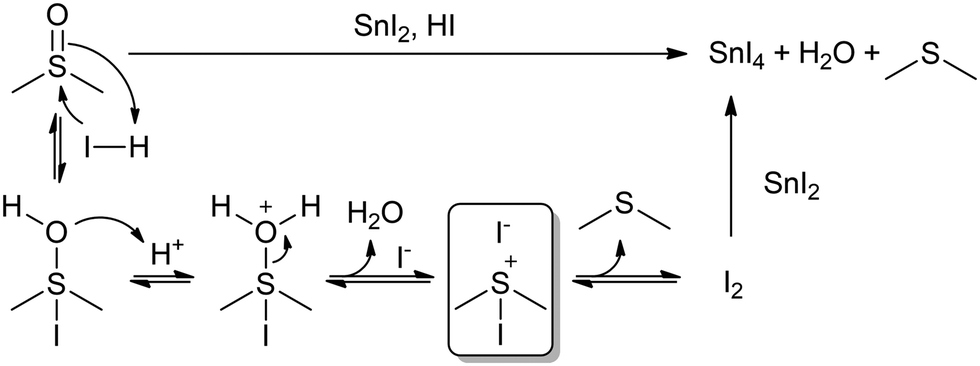 | ||
| Scheme 1 I2-Mediated mechanism of Sn(II) oxidation in DMSO. A possible intermediate species, iododimethylsulfonium iodide, is highlighted inside the square. | ||
Characterisation by NMR can also shed light on the decomposition of the organic components of the perovskite. As mentioned earlier, we heated FASnI3 solutions at 100 °C for 2 and 4 days. In contrast to the previous experiments, no signal was found for Sn(IV) in this case. Interestingly, we began to observe a precipitate, eliciting that extended heating can potentially lead to the precipitation of all Sn(IV) as SnO2. Additionally, we found a new group of signals in 1H spectra with shifts in the range of 6.80–7.20 ppm, which were increasing in intensity over time (Fig. S9, ESI†), suggesting that they belong to compounds forming during decomposition of the solution. At this point, we performed 2D-NMR experiments to understand the nature of these products. The 1H–1H correlation spectroscopy (COSY) (Fig. S12, ESI†) and rotating frame Overhauser effect spectroscopy (ROESY) results (Fig. S13, ESI†) confirmed that the signals are not coupled with each other. Still, they showed chemical exchange in between them and with the amine protons of FA (Fig. S13, ESI†), indicating that they have exchangeable protons. Therefore, these new signals might belong to decomposition products of FA that has only amine protons, meaning that these signals must belong to the ammonium ion. These signals have been previously assigned to NH4+ in the study on DMS generation,13 and are one of the decomposition products described by Clay Hamill et al. in perovskite solutions in DMSO.16 Other decomposition processes of DMSO such as its degradation to sulfate ion should always be considered, although, to the best of our knowledge, we believe our system does not provide the required conditions for this process to happen.
Currently FASnI3-based devices achieve Voc values between 0.4 and 0.6 V (with SnF2, Fig. S14, ESI†), while the material has potential for much higher values. This fact points out that there is a limiting factor, which would be strongly influenced by the so reviewed Sn4+ content.5 The oxidising effect of DMSO could be behind it, therefore alternative strategies that avoid the use of this solvent have to be discovered in order to improve the performance of these devices.
Conclusions
In conclusion, we have presented solid- and liquid-state 119Sn-NMR as a simple and straightforward technique for identifying Sn(II) oxidation and described its redox reaction with DMSO. Other methods traditionally used for oxidation state quantification in Sn-based perovskite materials show clear limitations. For instance, XPS not only requires extended measurement, ultra-high vacuum and X-rays, all of which may damage samples but also only gives information about the sample surface. At the same time, XANES depends on synchrotron facilities, making it a less available technique. These results also present NMR as a useful and accessible tool, with clear underexplored potential for understanding perovskite solutions, and point out the necessity of finding new solvents for Sn perovskites to substitute DMSO or otherwise suppressing its decomposition.Author contribution
A. A. supervised the project. R. A. and A. D. supervised the solid-state and liquid-state NMR measurements, respectively. J. P., M. H. A., J. A. S., M. F. and N. P. prepared the NMR samples. G. N. coordinated the solid-state NMR measurements. J. P., D. D. G. and A. A. designed the experimental plan. D. D. G., S.-H. T.-C. and M. L. helped with the coordination of the work. J. P. wrote the manuscript with the help of the rest of the authors. All authors discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. P. acknowledges Forschungszentrum Jülich GmbH (SolarWAVE project) for financial support. M. H. A. acknowledges DAAD and Egyptian missions for PhD scholarship. N. P. acknowledges the PhD program of University of Potsdam.Notes and references
- N. K. Noel, S. D. Stranks, A. Abate, C. Wehrenfennig, S. Guarnera, A.-A. Haghighirad, A. Sadhanala, G. E. Eperon, S. K. Pathak, M. B. Johnston, A. Petrozza, L. M. Herz and H. Snaith, Energy Environ. Sci., 2014, 7, 3061 RSC.
- J. Li, H.-L. Cao, W.-B. Jiao, Q. Wang, M. Wei, I. Cantone, J. Lü and A. Abate, Nat. Commun., 2020, 11, 310 CrossRef CAS PubMed.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu, M. Xiao, A. Li, J. Zhu, E. H. Sargent and H. Tan, Nat. Energy, 2019, 4, 864 CrossRef CAS.
- X. Jiang, F. Wang, Q. Wei, H. Li, Y. Shang, W. Zhou, C. Wang, P. Cheng, Q. Chen, L. Chen and Z. Ning, Nat. Commun., 2020, 11, 1245 CrossRef CAS PubMed.
- G. Nasti and A. Abate, Adv. Energy Mater., 2019, 1902467 Search PubMed.
- X. He, T. Wu, X. Liu, Y. Wang, X. Meng, J. Wu, T. Noda, X. Yang, Y. Moritomo, H. Segawa and L. Han, J. Mater. Chem. A, 2020, 8, 2760–2768 RSC.
- F. Zhang, S. H. Silver, N. K. Noel, F. Ullrich, B. P. Rand and A. Kahn, Adv. Energy Mater., 2020, 1903252 CrossRef CAS.
- B. E. G. Lucier, S. Chen and Y. Huang, Acc. Chem. Res., 2018, 51, 319–330 CrossRef CAS PubMed.
- W. M. J. Franssen and A. P. M. Kentgens, Solid State Nucl. Magn. Reson., 2019, 100, 36–44 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 14173–14180 CrossRef CAS PubMed.
- J. Pascual, S. Collavini, S. F. Völker, N. Phung, E. Palacios-Lidon, L. Irusta, H.-J. Grande, A. Abate, R. Tena-Zaera and J. L. Delgado, Sustainable Energy Fuels, 2019, 3, 2779–2787 RSC.
- X. Meng, J. Lin, X. Liu, X. He, Y. Wang, T. Noda, T. Wu, X. Yang and L. Han, Adv. Mater., 2019, 31, 1903721 CrossRef CAS PubMed.
- M. I. Saidaminov, I. Spanopoulos, J. Abed, W. Ke, J. Wicks, M. G. Kanatzidis and E. H. Sargent, ACS Energy Lett., 2020, 5, 1153 CrossRef CAS.
- S. P. Gamage, S. Garusinghe, M. Bessey, C. Boyd, M. Aghamoosa, B. G. Frederick, M. R. M. Bruce and A. E. Bruce, RSC Adv., 2015, 5, 40603 RSC.
- E. Glynn, Analyst, 1947, 72, 248–250 RSC.
- J. C. Hamill, J. C. Sorli, I. Pelczer, J. Schwartz and Y.-L. Loo, Chem. Mater., 2019, 31, 2114–2120 CrossRef CAS.
- X. Meng, T. Wu, X. Liu, X. He, T. Noda, Y. Wang, H. Segawa and L. Han, J. Phys. Chem. Lett., 2020, 11, 2965 CrossRef CAS PubMed.
- A. Monga, S. Bagchi and A. Sharma, New J. Chem., 2018, 42, 1551–1576 RSC.
- K. Mislow, T. Simmons, J. T. Melillo and A. L. Ternay, J. Am. Chem. Soc., 1964, 86, 1452–1453 CrossRef CAS.
- M. Karki and J. Magolan, J. Org. Chem., 2015, 80, 3701–3707 CrossRef CAS PubMed.
- G. Majetich, R. Hicks and S. Reister, J. Org. Chem., 1997, 62, 4321–4326 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on materials and methods and additional NMR spectra. See DOI: 10.1039/d0ma00245c |
| This journal is © The Royal Society of Chemistry 2020 |