
Beryllium coordination chemistry and its implications on the understanding of metal induced immune responses
Magnus R.
Buchner

Anorganische Chemie, Nachwuchsgruppe Hauptgruppenmetallchemie, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35032 Marburg, Germany. E-mail: magnus.buchner@chemie.uni-marburg.de; Fax: +49 (0)6421 2825669; Tel: +49 (0)6421 2825668
First published on 16th June 2020
Abstract
The inhalation of beryllium and its compounds can cause the development of chronic beryllium disease in susceptible individuals. This is caused by a distinct autoimmune process. Here, beryllium coordination compounds with biomimetic ligands are discussed, which are used to understand the coordination of Be2+ in the body and its effect on biomolecules. The advances in the development of precursors for the directed synthesis of these coordination complexes are presented as well as the potential use of non-aqueous solvents for these investigations.
 Magnus R. Buchner | Magnus R. Buchner studied chemistry at the Technische Universität München where he received his PhD in 2011 under the supervision of Klaus Ruhland. After postdoctoral stays in the groups of Florian Kraus (Munich), Robin Perutz (York) and Sjörd Harder (Erlangen) and a stint at the patent department of the Fraunhofer-Gesellschaft, he started his independent research at the Philipps-Universität Marburg in 2015, funded by the Deutsche Forschungs Gemeinschaft and since 2019 within the Emmy Noether program. His research interests lie in the coordination, organometallic and bioinorganic chemistry of hard (pseudo) main group metals, with a predilection for beryllium. |
1 Introduction
With the atomic number 4 beryllium is the second lightest and smallest metal. Metallic beryllium can withstand air and humidity even at elevated temperature, due to a passivating layer of beryllium oxide on its surface.1 When alloyed with other metals beryllium significantly increases the strength, fatigue resistance, ductility and elasticity in comparison to the pure metals. Additionally the weight of these alloys is drastically reduced, when a high beryllium content is used. This makes beryllium an indispensable element for the aeronautic and space industry but also for transportation and telecommunication applications. Furthermore, beryllium copper alloys have comparable mechanical properties to steel but are non-magnetic and non-sparking. Accordingly tools and components for magnetic resonance imaging, mining and oil and gas extraction consist of these alloys. Beryllium oxide has a high electric resistivity combined with a high thermal conductivity, which is only surpassed by diamond. But unlike the latter, beryllia ceramics are shock resistant and relatively break-proof. This renders them the material of choice for high frequency electronics.2 However, mining, extraction and production of beryllium metal is rather costly. Therefore, it is mainly used in high technology applications and the current annual production is around 300 t.3Additionally to these important material characteristics beryllium also exhibits unique chemical properties. It is the s-block element with by far the highest electronegativity, which is most closely matched by aluminium and zinc (Table 1).4 Hence, the covalency of beryllium element bonds is unusually high. The radius of the Be2+ ion is the smallest of all metal ions.5 Therefore, the charge to radius ratio of Be2+ is extremely high, which renders it one of the hardest known Lewis acids. Among the main group metals, this high charge density is only approached by Al3+. These chemical qualities would, in theory, make beryllium an ideal candidate for Lewis acid-induced conversions. However due to safety issues (vide infra), the chemistry of beryllium is extremely underdeveloped in comparison to all other non-radioactive elements.6 Nevertheless, the recent years have seen high interest in alkaline earth metal chemistry.7,8 Particularly dinuclear magnesium(I) compounds by Jones have proven as highly versatile and mild reducing agents.9 According to this, compounds with beryllium in the formal oxidation states zero10 and one11 as well as with unsupported Be–Al bonds have been realised recently.12 Despite these achievements, there is still a profound lack of general insight into beryllium chemistry and only in the last years a systematic reinvestigation of its coordination chemistry has begun.13
While the acute toxicity of beryllium salts is not higher than related cadmium, barium or arsenic derivatives,14 inhalation of air containing small amounts of beryllium compounds (2 μg m−3) can result in the development of chronic beryllium disease (CBD).15 The latency time for the onset of symptoms varies between one and over twenty years. The reason for this high variability is still unknown, but it was proposed that environmental changes might alter the beryllium species in vivo into a more soluble form. The higher bio-availability would then trigger the CBD.16 The development of CBD is well understood regarding the occupational exposure. In up to 16% of workers inhalation or skin exposure can result in beryllium sensitisation. This is evident from T-cells in their blood which proliferate in the presence of aqueous beryllium salt solutions. Only individuals with these lymphocytes can develop CBD upon inhalation of beryllium containing substances. Long term occupational health investigations suggest that a maximum of 11% of workers in the beryllium industry develop CBD. These have a granulomatous inflammation of the lung, which results in reduced oxygen uptake and sometimes death.17
The reason why such minute amounts of beryllium are sufficient to cause CBD is that the metal triggers an autoimmune mechanism.18 The exposure of cells in the lung or skin to beryllium or its compounds leads to their death and the release of cytokines, which activate dendritic cells. A beryllium species itself, which acts as a potent adjuvant, enhances this initial immune response.19 A soluble beryllium species is then bound by the dendritic cells together with a peptide on the major histocompatibility class II (MHCII) receptor and the dendritic cells migrate to the nearest lymph node.20 There, the peptide–beryllium complex is presented to naive T-cells and leads to the proliferation of beryllium sensitised T-cells.21 Beryllium-containing particles in the lung lead to the migration of these beryllium-specific T-cells to the particles, where an immune response is triggered. This results in macrophage accumulation around these particles, the inflammation of the surrounding tissue and the proliferation of more beryllium-sensitised T-cells in this area.20,21 In CBD patients the immune regulation is dysfunctional. Therefore, the inflammation is not dampened and leads to granulomata formation and progresses into fibrosis, which results in respiratory insufficiency.22 These three immunological steps are summarised in Scheme 1.
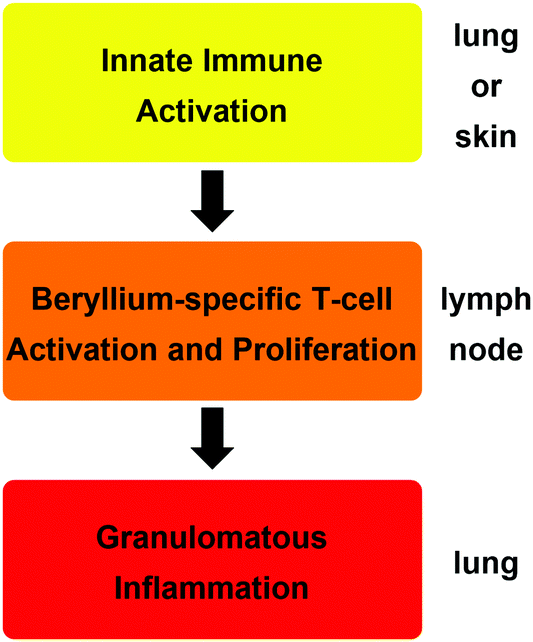 | ||
| Scheme 1 Consecutive immunologic effects and organ localisation, which lead to the development of chronic beryllium disease (CBD).14 | ||
Only in the last decades it was understood that all individuals with CBD have one distinct subtype of the MHCII. This receptor consists of four peptides: two α- and two β-chains. Only if a glutamic acid is at the 69th position of the β-chain beryllium exposure can result in CBD.23–25 It could also be shown that the beryllium species is bound within an acidic pocket formed by the MHCII/peptide complex. This pocket consists of three glutamic acid side chains in positions 26, 68 and 69 on the β-chain and an additional glutamic and aspartic acid side chain of the bound peptide. The presence of the beryllium species in this pocket reduces the surface electron density of the MHCII/peptide complex, which leads to immune recognition by the T-cells.26–30 However, due to the inherent low resolution in protein X-ray crystallography a direct localisation of the atoms inside the acidic pocket was not possible.30 Therefore, computational chemistry was used to evaluate the species bound inside. This resulted in two models, which either propose the coordination of a single Be2+ ion together with two Na+ ions31 or the presence of an oxygen centred [Be4O]6+ tetrahedron.32–34
However, due to the low electron number of Be2+ this cannot be verified with protein X-ray crystallography and the low solubility of beryllium species at biological pH values makes most solution analytics in water extremely difficult.35 Therefore, simple and low molecular model systems are necessary to understand how Be2+ ions interact with biomolecules and which beryllium-induced changes occur in their functional groups. Only with this a sufficient understanding on the interaction of Be2+ with the immune system will be possible. Due to the high selectivity of this interaction, these insights could also shed light on other metal induced immune responses like the action of Al3+ as an adjuvant for vaccinations.
2 Interaction of Be2+ with biomimetic ligands
To understand how Be2+ ions interact with biomolecules simple model systems have been used and a few of these are shown in Fig. 1. Considering the fact that biological processes occur in water all of these studies were conducted in aqueous media. However, due to the high excess of H2O molecules only chelating ligands are able to bind under these conditions.35 Reaction of catechol (1a) with Be(OH)2 under basic conditions results in the formation of a beryllate with the composition [Be(1a)2]2−.36 An aliphatic alcoholate forms a similar compound under these conditions.37 Electrospray ionisation mass spectrometry (ESI-MS) revealed that related 1,3-diketones were able to bind strongly to Be2+, while 1,2-diketones performed poorly.38 Salicylic acid (1b) and its derivatives as well as dicarboxylic acids like phtalic acid (1c) are able to form stable complexes with beryllium in acidic aqueous media.39,40 Reaction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of glycolic acid and sodium glycolate with Be(OH)2 in water yields solutions with a pH of 5.7, from which the hexanuclear complex anion [Be6(OCH2CO2)8]4− (2) was isolated (see also Fig. 8).41 However, at neutral pH only tridentate ligands like citric acid (1d), 2-hydroxyisophtalic acid (1e) and 2,3-dihydroxybenzoic acid (1f) are able to form soluble species with Be2+. This was attributed to the formation of dinuclear beryllium complexes, in which the hydroxy group is deprotonated and acts as a μ2-bridge between the two beryllium atoms.42,43 Nitrilotripropionic (1g) and related polyamino carboxylic acids were shown to bind strongly to beryllium ions.44 Subsequently ligands (e.g.1h) have been developed which are intended to selectively encapsulate Be2+.45–47
:1 mixtures of glycolic acid and sodium glycolate with Be(OH)2 in water yields solutions with a pH of 5.7, from which the hexanuclear complex anion [Be6(OCH2CO2)8]4− (2) was isolated (see also Fig. 8).41 However, at neutral pH only tridentate ligands like citric acid (1d), 2-hydroxyisophtalic acid (1e) and 2,3-dihydroxybenzoic acid (1f) are able to form soluble species with Be2+. This was attributed to the formation of dinuclear beryllium complexes, in which the hydroxy group is deprotonated and acts as a μ2-bridge between the two beryllium atoms.42,43 Nitrilotripropionic (1g) and related polyamino carboxylic acids were shown to bind strongly to beryllium ions.44 Subsequently ligands (e.g.1h) have been developed which are intended to selectively encapsulate Be2+.45–47
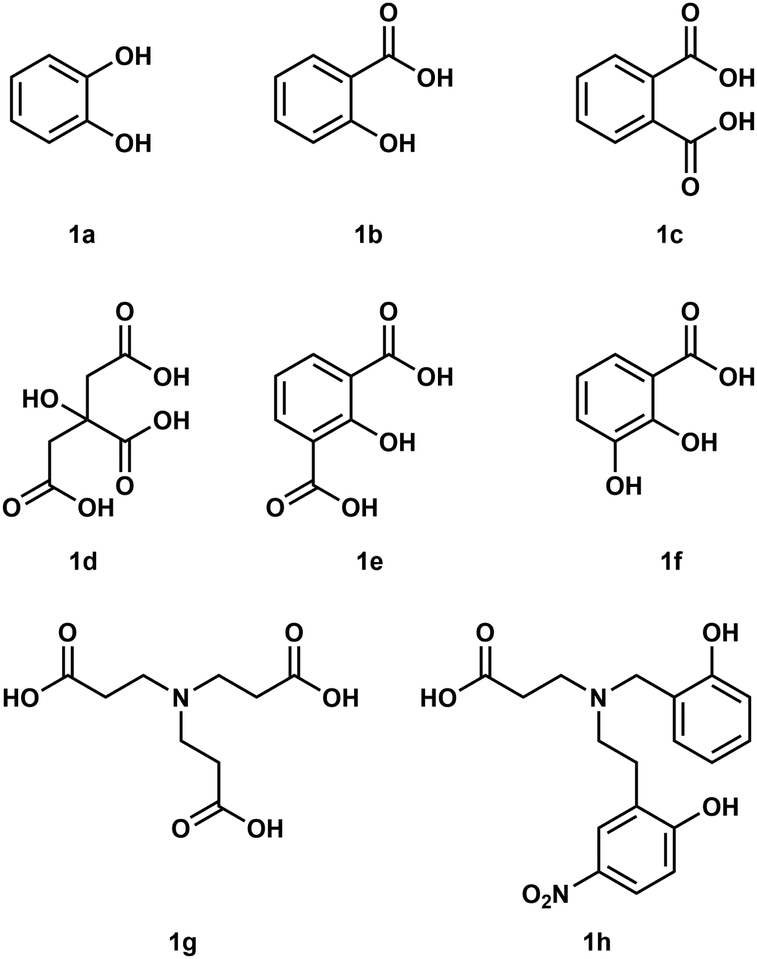 | ||
| Fig. 1 Polydentate ligands used in aqueous beryllium chemistry.36,39,40,42–45 | ||
All these ligands are able to compete with H2O for beryllium coordination and give important insights into stable binding geometries. However, these ligands cannot be easily compared. Therefore, it was not possible to obtain directly comparable beryllium binding affinities of different functional groups or between subtle variations in binding geometries. However, this would be immensely important to evaluate potential Be2+ binding sites in peptides and polysaccharides. Also, only very recently the interaction of beryllium cations with small bioorganic ligands was investigated computationally.48
The main problem with the above mentioned ligands is that these have to be able to compete with the massive excess of water molecules. Therefore, we decided to use non-aqueous and non-coordinating solvents to obtain reliable data on the relative binding affinity of Be2+ to different biologically relevant functional groups. This eliminates the need of chelate assistance and simplifies the system since no solvent coordination has to be considered. The obvious choice was the use of phenyl derivatives due to their relation with 1a–c, e, f and their high solubility in aromatic and chlorinated solvents. Benzoic acid and benzyl alcohol were used as model for carboxy and hydroxy side chains of peptides respectively, while ethyl benzoate and benzaldehyde were used as mimic for the peptide bond (Fig. 2).49 It should be mentioned that carboxylic acid amides could not be studied this way due to solubility issues. These systems are currently investigated in water-related inorganic solvents.
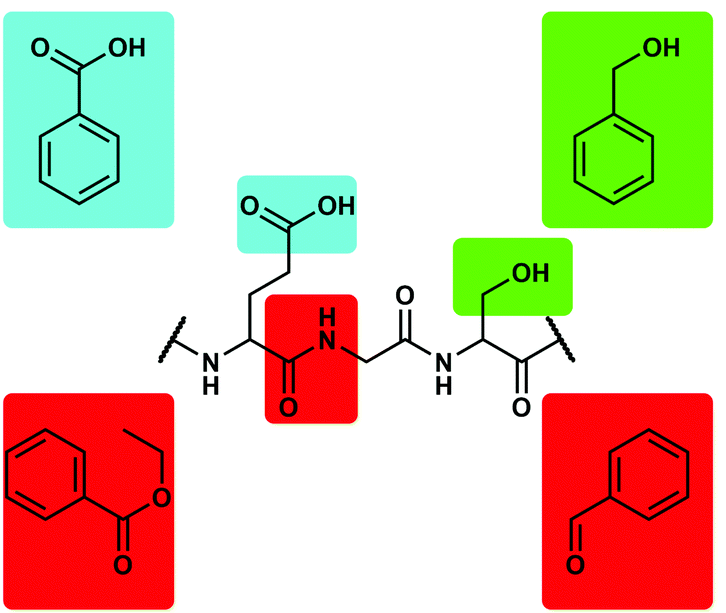 | ||
| Fig. 2 Potential binding sites for Be2+ in peptides and corresponding model ligands.49 | ||
Reaction of BeCl2 with benzyl alcohol, benzaldehyde and ethyl benzoate in benzene led to formation of the simple adducts 3, 4 and 5, respectively (Scheme 2). All these complexes feature a pseudo tetrahedrally coordinated beryllium atom in the centre, which is coordinated by two chlorido and two of the respective model ligands. The shorter Be–O and longer Be–Cl distances in 3 compared to 4 and 5 suggest that the alcohol is more strongly bound than the aldehyde and ester. In contrast to the alcohol, aldehyde and ester, benzoic acid reacts with BeCl2 under HCl evolution. Therefore, the acidity of the carboxy function is increased beyond that of hydrogen chloride upon beryllium coordination. The resulting binary beryllium benzoate (6) comprises of an unprecedented dodecanuclear macro cycle (Fig. 3). In 6 two beryllium atoms are bridged by three benzoate ligands, which results in the formation of dinuclear [Be2(PhCO2)3]+ subunits. These are bridged by an additional benzoate to the next one.496 is the only known example of a neutral homoleptic beryllium monocarboxylate. This is presumably caused by high moisture sensitivity of these compounds, which leads to the formation of the oxo-carboxylates. These have been known for decades, are highly stable and feature an oxygen centred [Be4O]6+ core. This is exemplified for μ4-oxo-hexakis(μ2-benzoato-O,O′)tetraberyllium ([(Be4O)(PhCO2)6], 7) in Fig. 4.50 The influence of the acidity and steric bulk of the carboxylic acid and the electron density of the carboxylate on the formation of homoleptic and oxo-beryllium carboxylates is currently studied in our group.
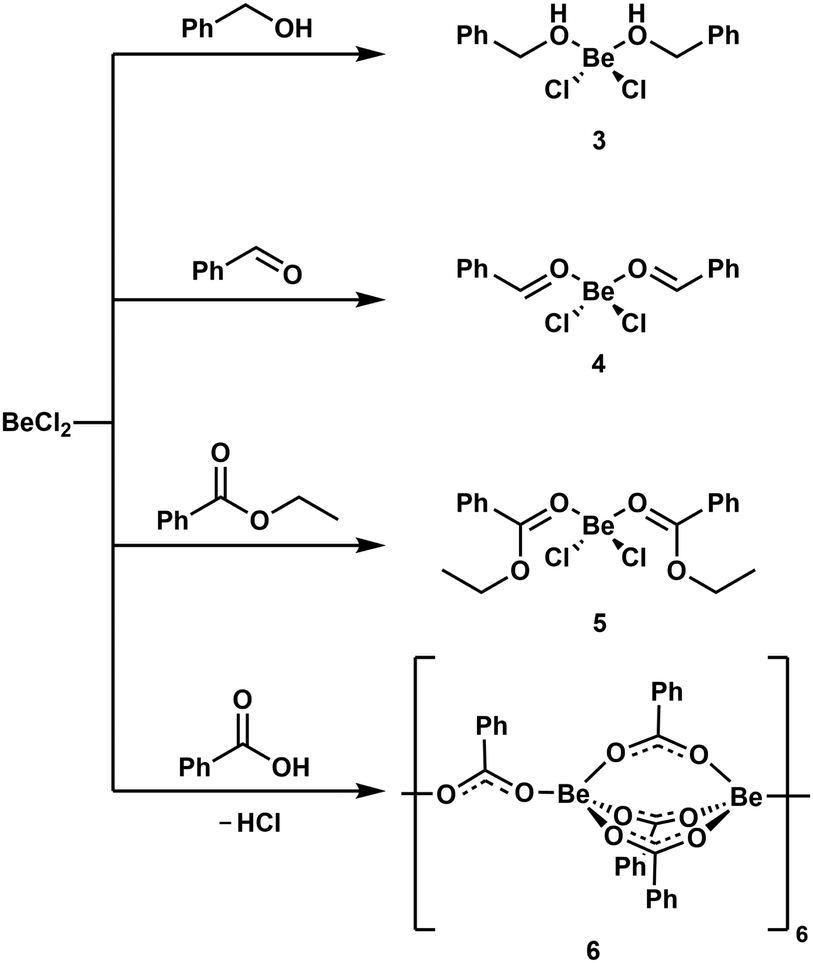 | ||
| Scheme 2 Reaction of BeCl2 with benzyl alcohol, benzaldehyde, ethyl benzoate and benzoic acid.49 | ||
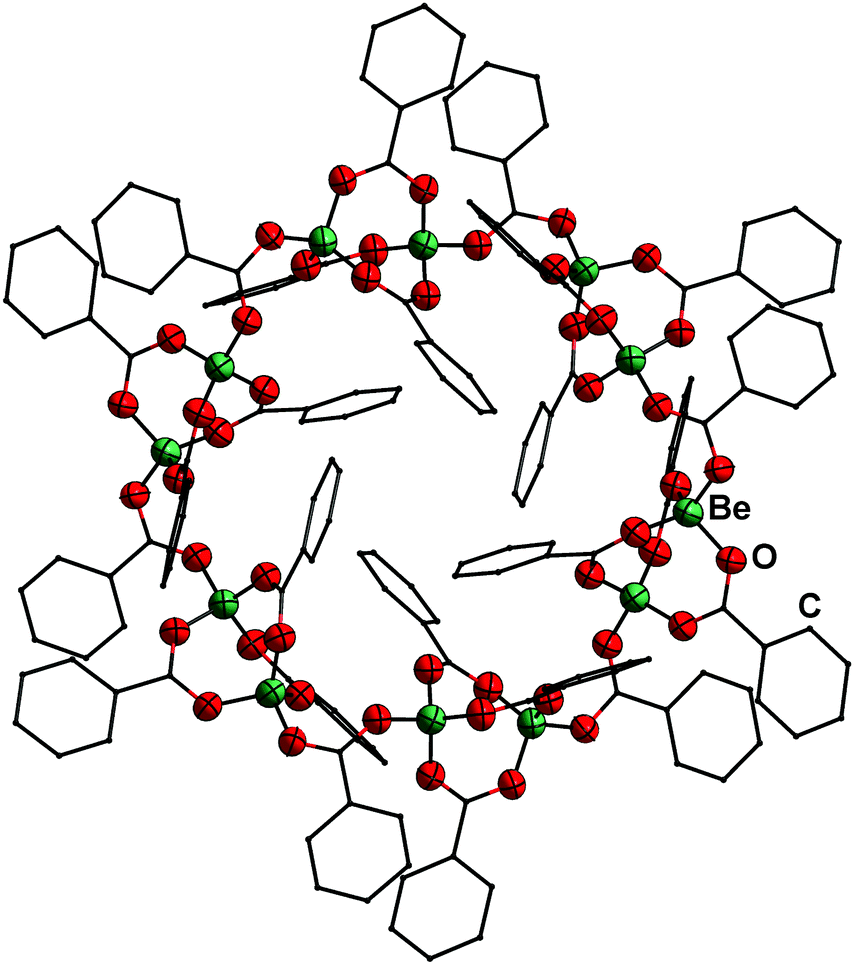 | ||
| Fig. 3 Molecular structure of [Be(PhCO2)2]12 (6) in the solid state. Ellipsoids are depicted at 70% probability at 100 K. Carbon atoms are depicted as wire frame and hydrogen atoms are omitted for clarity.49 | ||
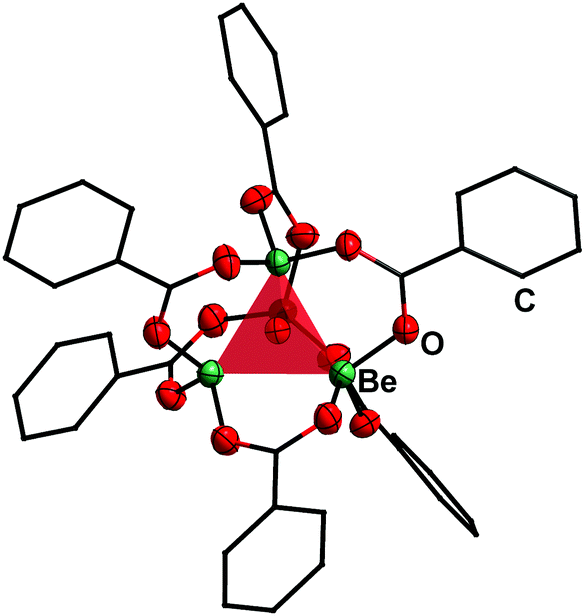 | ||
| Fig. 4 Molecular structure of [(Be4O)(PhCO2)6] (7) in the solid state.50 The tetrahedron of the central [Be4O]6+ core is highlighted in red. Ellipsoids are depicted at 70% probability at 143 K. Carbon atoms are depicted as wire frame and hydrogen atoms are omitted for clarity. | ||
Competition reactions of two different ligands with BeCl2 in non-coordinating solvents gave a distinct trend for the binding affinity of the investigated ligands. This revealed that the Be2+ affinity increased from ester via aldehyde and alcohol to carboxylate (Fig. 5). This is in line with the assumption that Be2+ is bound by glutamic and aspartic acid side chains of the peptides. It also suggests that the interaction of beryllium ions with the peptide backbone might be negligible.49 However, this can only be said definitively when competition reactions have been performed with carboxylic acid amides in a suitable solvent.
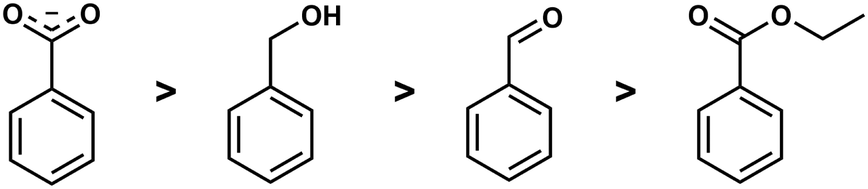 | ||
| Fig. 5 Relative binding affinity of biologically relevant functional groups to Be2+.49 | ||
Besides proteins also polysaccharides possess a high number of O-donor sites, which should be good ligating groups for Be2+, due to its high oxophilicity. Polysaccharides are found on the outer layer of cell membranes and play a vital role for immune recognition. Therefore, it was plausible that the interaction of beryllium ions with polysaccharides might play a role in its action as an adjuvant. Also the interaction of Be2+ with the cell membrane is likely involved in the induction of cell death, which poses the beginning of beryllium sensitisation. However, there was no research on the coordination chemistry of beryllium with saccharides or related biomimetic ligands. Thus, we investigated the reactivity of beryllium halides with glucose, fructose and their derivatives. Additionally simple cyclic polyols, hemiacetals as well as tetrahydropyranols and -furanols were investigated as model ligands.51Fig. 6 depicts some of these ligands and their relation with glucose.
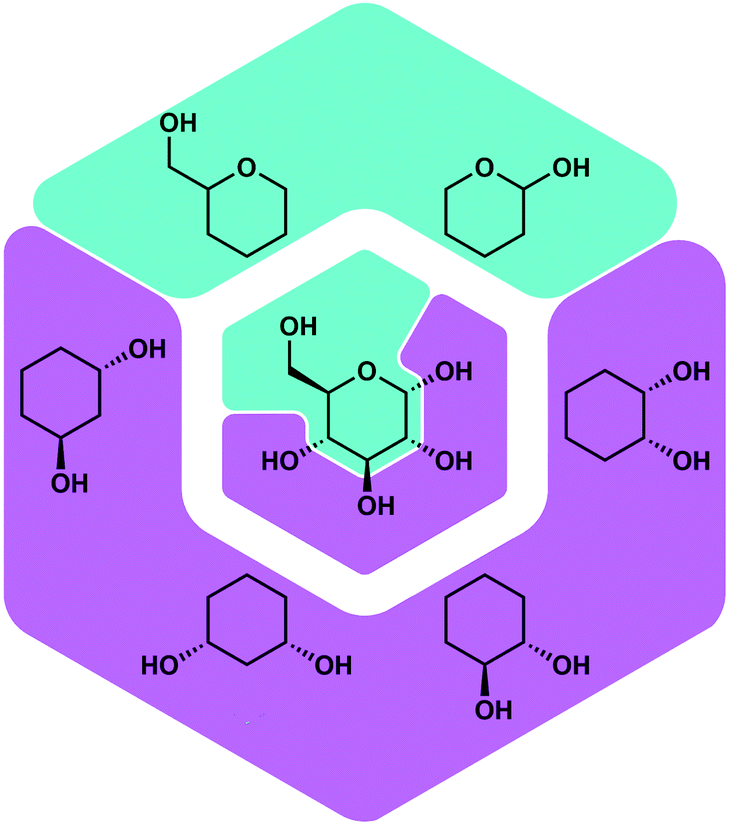 | ||
| Fig. 6 Potential binding sites for Be2+ in saccharides and corresponding model ligands.51 | ||
Reaction of one equivalent of cis-1,2-cyclopentanediol with BeCl2 initially results in the formation of the simple adduct 8a shown in Scheme 3. This complex is closely related to benzyl alcohol adduct 3. 8a slowly cleaves off HCl under the formation of tetranuclear complex 8b, which features an eight-membered Be–O-heterocycle. Such heterocycles have also been observed for beryllium siloxanato compounds.52 The release of HCl from 8a indicates that upon coordination of the cyclic diol to the beryllium atom its OH-groups are acidified to an extent, that they are able to protonate Cl−. If 8a is reacted instantaneously with a second equivalent of the diol, cationic beryllium complex 8c is formed. The same reactivity is also observed for cis-1,3-cyclopentanediol as well as for cis-1,2- and cis-1,3-cyclohexanediol. While all these reactions could be carried out in dichloromethane, good quality crystals of cationic complexes like 8c could only be obtained with BeBr2 in liquid SO2.51,53
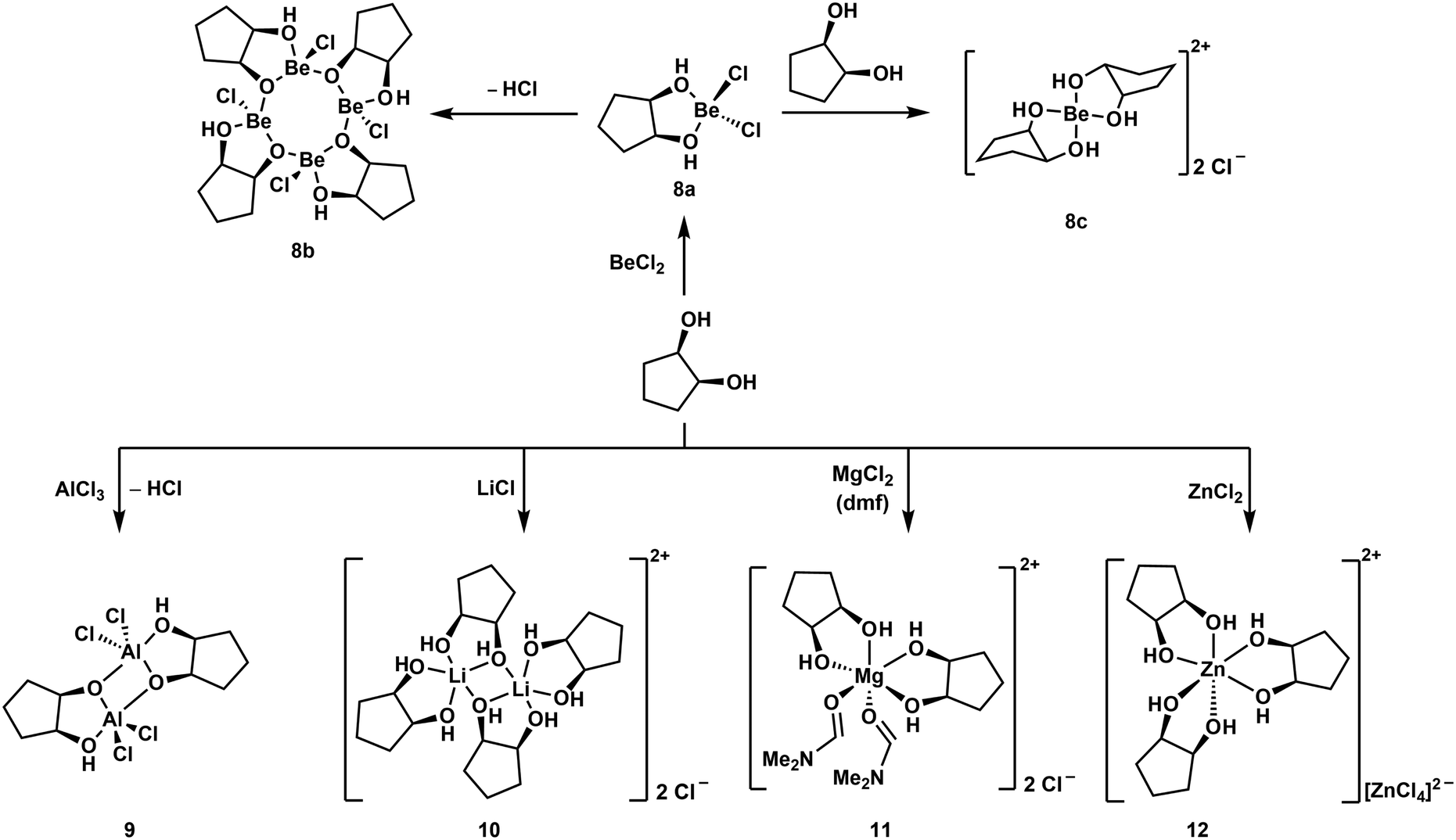 | ||
| Scheme 3 Reaction of a cyclic diol with (pseudo) main group metal chlorides, yielding different coordination environments around the metals.51 | ||
Similar complexes were also obtained with tetrahydropyran-2-yl- and tetrahydrofuran-2-ylmethanol. However, BeCl2 decomposes hemiacetals and sugars, which was attributed to their open chain aldehyde isomers. Accordingly α-D-methylglucoside does not decompose in the presence of Be2+. This supports the assumption that Lewis acid-induced alterations of the polysaccharides might start the immune response to beryllium.51
In all these compounds the beryllium atom is exclusively four-coordinated and adopts a pseudo tetrahedral coordination geometry. This was never the case, when related hard main and pseudo main group metal cations were reacted with cis-1,2-diols under similar conditions. Reactions with AlCl3 and LiCl yielded dinuclear complexes 9 and 10 respectively. In these the metal atoms are coordinated in a trigonal bipyramidal fashion. MgCl2 and ZnCl2 gave octahedral compounds 11 and 12, respectively. This is presumably due to the significantly smaller size of the Be2+ ion (Table 1) and shows the limitations of the substitution of beryllium by other elements to study its coordination chemistry.51 However, we could show that boron and beryllium atoms can adopt comparable coordination environments with chelating ligands.54
To further evaluate the stability of cyclic polyol complexes of beryllium cis,cis-1,3,5-cyclohexanetriol was investigated (Scheme 4). In contrast to the bidentate ligands mentioned above, beryllium complexes with this triol were significantly less soluble and only dimethylformamide (dmf) proved to be suitable. Reaction of the triol with BeCl2 in dmf leads to the dissociation of both chlorido ligands and the formation of cation 13a. This only releases HCl at elevated temperatures and subsequently trimerises to complex cation 13b, which itself dimerises, presumably under decomposition of one of the ligands and further HCl loss, to hexanuclear compound 13c.51
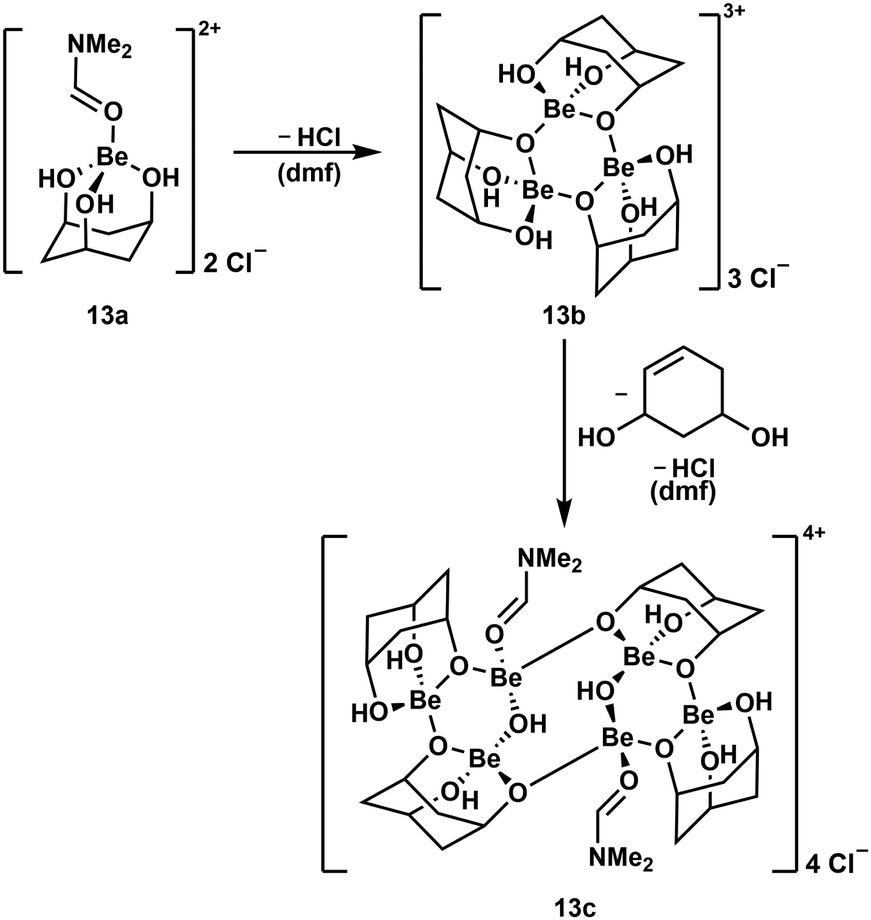 | ||
| Scheme 4 Consecutive HCl loss and supposed ligand decomposition during the formation of multinuclear beryllium complex cations.51 | ||
With the help of competition reactions and the evaluation of the complex stabilities and their solid state properties a clear trend in the binding affinity of the investigated chelating alcohols could be derived (Fig. 7). This suggests that only binding sites which at least offer a κ3-coordination and a tetrahedral coordination environment may irreversibly bind Be2+ in the body.51 This is further supported by the observation of high beryllium mobility in the BeCl2 crown ether system.55,56
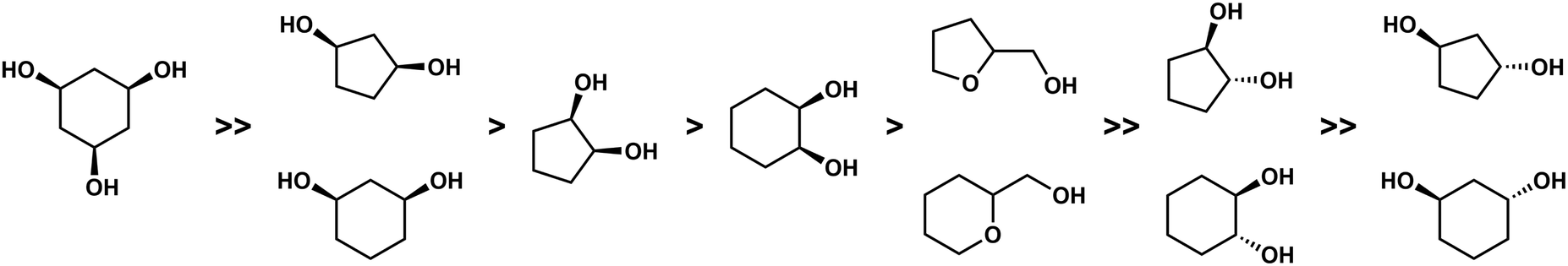 | ||
| Fig. 7 Relative binding affinity of polydentate O-donor ligands to Be2+.51 | ||
As mentioned above, multinuclear beryllium complexes, which comprise of Be–O-heterocycles, are frequently formed with various O-donor ligands. This is also known from other examples like dinuclear 14,39 trinuclear 15,57 tetranuclear 1658 and hexanuclear 2,41 which are illustrated in Fig. 8. Therefore, multinuclear beryllium complexes should be considered in the search for the beryllium species liable for CBD.51
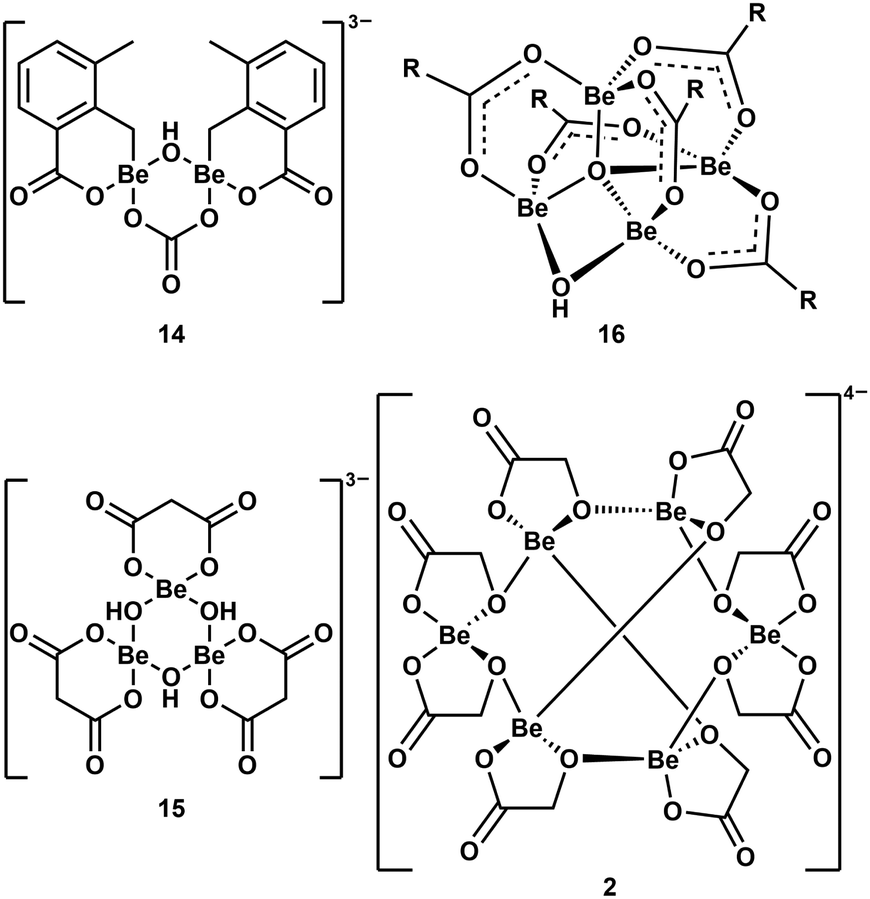 | ||
| Fig. 8 Multinuclear beryllium compounds with bio-relevant ligands, which all contain six- or eight-membered beryllium heterocycles (R = 2-F-C6H4).39,41,57,58 | ||
While the described approach with non-coordinating organic solvents works for simple ligands, it fails when ionic complexes are formed or more complex biomolecules are used. This necessitates the use of coordinating organic-solvents or of water-related inorganic ones. However, there is so little knowledge on the solution behaviour of simple beryllium compounds that this has to be studied before these solvent systems can be applied for more advanced studies. Also it became evident that a larger pool of beryllium precursor compounds was needed to synthesise complexes more directly.
3 Expanding the spectrum of beryllium precursors
Due to the limited fields of industrial application for beryllium, the only commercially available compounds are beryllium metal and BeO.3 However, these are of very limited use for the coordination chemistry of beryllium. Therefore, suitable beryllium precursors have to be synthesised from these. The most commonly used starting material is BeCl2,13 which was traditionally prepared from the reaction of beryllium metal in a stream of chlorine at elevated temperature according to eqn (1).59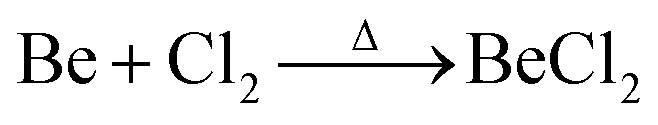 | (1) |
However, this preparation is slow and tedious when done with appropriate safety measures, while short reaction times can only be achieved under high chlorine streams and pressure above one atmosphere in the reaction vessel. Therefore, safer methods have been developed, which allow for the direct synthesis of BeBr2 and BeI2 from the elements in coordinating organic solvents. But, this only allows the preparation of the solvent adducts L2BeX2 (L = Et2O, MeCN; X = Br, I).60–62 While these are good starting materials if consecutive reactions are also performed in strong donor solvents, they cannot be used if weakly coordinating ligands or exchange equilibria shall be studied. Thus, based on the preparation of (Me2S)2BeBr2,63 we developed an improved synthesis for binary BeCl2, BeBr2 and BeI2, which uses defined halogen vapour pressures. This allows for the fast and safe synthesis of large amounts of these versatile starting materials.64
In recent years most of beryllium chemistry was based on beryllium halides.13 However, the halides form relatively strong Be–X bonds and therefore tend to compete with other ligands for beryllium coordination. To have access to another beryllium salt with a more weakly coordinating counter ion, we conducted experiments on the synthesis of beryllium triflate. Beryllium metal as well as BeCl2 can be dissolved in triflic acid under the formation of a colourless oil. However, defined products have only been obtained so far, when this oil was extracted with O-donor solvents. This yields solvent adducts L2Be(OTf)2 (L = H2O, thf) as shown in eqn (2) and (3).65
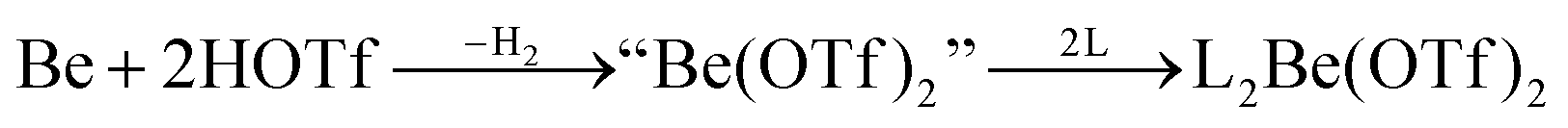 | (2) |
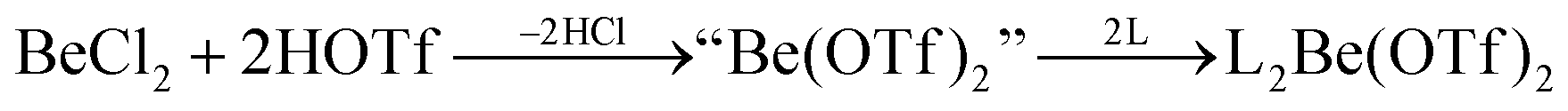 | (3) |
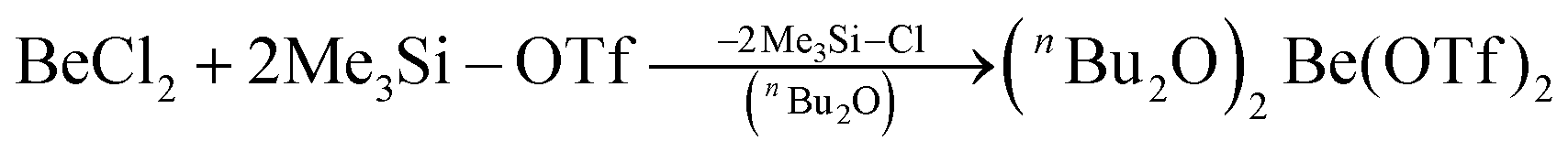 | (4) |
As an alternative the access to beryllium pseudo halides was explored. These together with chloride, bromide and iodide can be synthesised from metallic beryllium and the corresponding ammonium (pseudo) halide in liquid ammonia (eqn (5)) and are obtained as the tetraammine complexes: [Be(NH3)4]X2 (X = Cl, Br, I, CN, SCN, N3).66
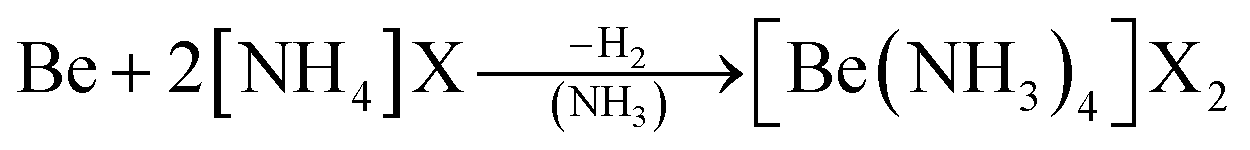 | (5) |
Volatile ammonium cyanide and azide were generated in situ from the corresponding trimethylsilyl compounds according to eqn (6).
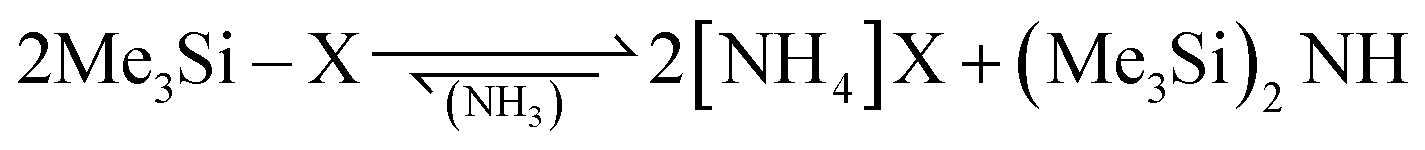 | (6) |
In case of ammonium fluoride only diammine (H3N)2BeF2 can be accessed in liquid ammonia due to the strong Be–F bonds. Analogous (H3N)2BeX2 (X = Cl, Br, I) can be made by directly reacting beryllium metal with two equivalents ammonium halide at elevated temperature according to eqn (7).66
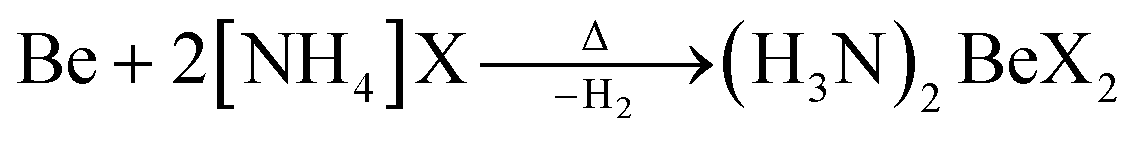 | (7) |
Organic beryllium compounds are, in theory, ideal precursors for complexes with anionic ligands. However, their synthesis in etheral solutions requires multiple vacuum distillations and the obtained products are not ether free.67 Because of this, a synthetic route which avoids the use of O-donor solvents was developed. Trimethylphosphine was used as a solubilising agent to enable the alkylation of BeCl2 in aromatic solvents according to eqn (8).
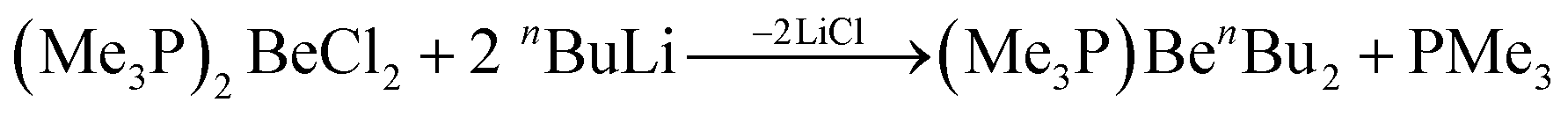 | (8) |
The remaining phosphine in (Me3P)BenBu2 can be removed in vacuo to generate ether-free nBu2Be as shown in eqn (9).67
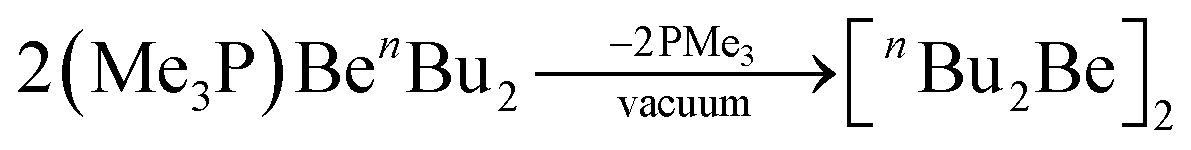 | (9) |
Even though this procedure allows for the synthesis of ether-free beryllium organyles it has several drawbacks. On the one hand, O-donor solvents can only be completely avoided with lithium organyles that are soluble in aromatic and aliphatic solvents. Thus only butyl derivatives can be obtained, if commercially available lithium organyles are used. All butyl beryllium compounds are liquids at ambient temperature, which severely hampers their use for micro scale reactions. On the other hand, this synthesis still requires BeCl2, which has to be prepared beforehand. To solve these issues, we reinvestigated the synthesis of BePh2 (17). 17 can easily be prepared in relatively large quantities from beryllium metal and diphenylmercury according to eqn (10).68–70
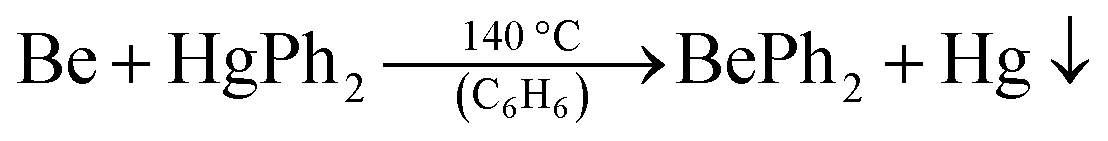 | (10) |
17 is a solid at ambient temperature and has a linear, trinuclear structure in the solid state and in solution (Fig. 9). It is stable against chlorinated solvents and shows a remarkably low ether affinity, while the Be–C bonds are rather stable.71 Therefore, diphenylberyllium is one of the most promising precursors for the investigation of the coordination chemistry of beryllium with protic ligands. However, considering the toxicity of mercury and its compounds, the carcinogenicity of benzene and the health issues associated with beryllium, appropriate safety precautions are strongly advised.72
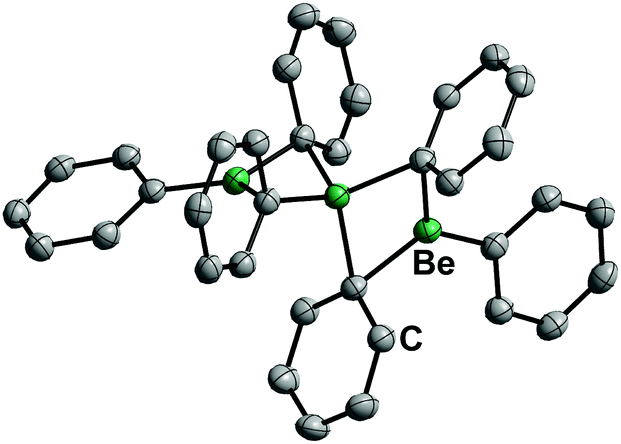 | ||
| Fig. 9 Molecular structure of BePh2 (17) in the solid state. Ellipsoids are depicted at 70% probability at 100 K. Hydrogen atoms are omitted for clarity.71 | ||
4 Speciation in solution
As mentioned above non-coordinating solvents are not able to solubilise most ionic beryllium compounds or biomolecules. For this task strong donor solvents are necessary. However, as soon as these are used, the huge excess of solvent leads to competition for beryllium coordination between ligands an solvent. Therefore, the species formed in these solvents have to be studied first. 9Be NMR spectroscopy is a valuable tool for this, since the chemical shift and line width of the quadrupolar 9Be nucleus is indicative for its coordination environment.73,74 Astonishingly, there were virtually no current studies on non-aqueous systems. Therefore, we started to investigate solvents, which were promising to dissolve beryllium compounds as well as biomolecules.Liquid ammonia is a solvent with properties, which are closely related to water. It readily forms hydrogen bonds and undergoes autoprotolysis. Beryllium metal dissolves under dihydrogen formation in water, if the medium is either acidic or basic.35 This can also be observed in ammono-acidic or ammono-basic NH3.66,75 Due to these properties liquid NH3 was intensively investigated to understand which pH dependent beryllium species are present in solution (Fig. 10).76
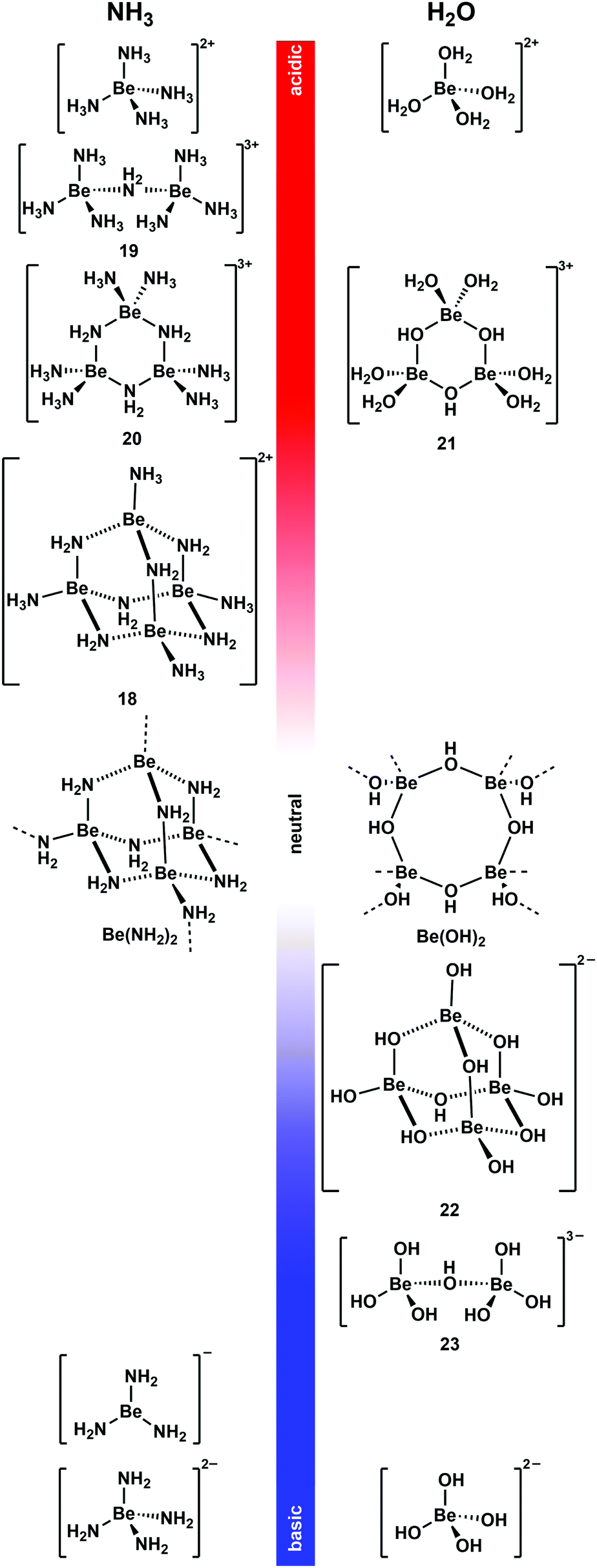 | ||
| Fig. 10 pH-Dependent beryllium complexes observed in NH3 (left)66,75–78 and H2O (right)79–83 solutions. | ||
Beryllium metal dissolves in the presence of ammono acids like ammonium halides and pseudo halides ([NH4]X) in liquid NH3 (vide supra).66 Also beryllium halides acidify NH3 to an extent that metallic beryllium dissolves.76 This is presumably due to the strong Lewis acidity of the Be2+ ion, which makes [Be(NH3)4]2+ a potent Brønsted acid. [Be(NH3)4]2+ is the only species present in solution when beryllium halides are dissolved in liquid NH3 or if Be is reacted with two equivalents [NH4]X.76,77 Up to two equivalent of Be per [NH4]X can be dissolved. This results in the formation of tetranuclear complex cation [Be4(NH2)6(NH3)4]2+ (18).76 This molecule adopts an adamantane-shaped structure, which is closely related to the solid state structure of homoleptic beryllium amide (Be(NH2)2).78 A combination of 1H, 9Be and 15N NMR spectroscopy in 14NH3 and 15NH3 and computational chemistry revealed dinuclear 19 and trinuclear 20 to be intermediates in the formation of 18 from [Be(NH3)4]2+. 18 is the final product from the reaction of beryllium metal in ammono-acidic NH3 and addition of more Be does not lead to further aggregation to compounds of higher nuclearity.76
Metallic beryllium can also be dissolved in ammono-basic NH3. This can be achieved through the addition of alkali metals. In this system the beryllium metal catalyses the formation of the alkali metal amides, which in turn promote the oxidative dissolution of Be. However, the reaction stops as soon as the electride solutions have discoloured. This indicates that solvated electrons are necessary to facilitate the reduction of protons to dihydrogen. Depending on the alkali metal used, different compounds were isolated. Potassium, rubidium and caesium lead to the formation of M[Be(NH2)3] (M = K, Rb, Cs). Here the only beryllium species detectable in solution and the solid sate is trigonal planar [Be(NH2)3]−. The reaction with sodium yields [Na4(NH2)2][Be(NH2)4]. However, it is not clear if the species in solution is actually tetrahedral beryllate [Be(NH2)4]2−. If lithium is used, an almost insoluble mixture of LiNH2 and Be(NH2)2 is obtained.75
From these results it is evident that the beryllium species found in liquid NH3 and H2O are closely related. At low pH values the dominating species are [Be(NH3)4]2+ and [Be(H2O)4]2+, respectively.76,79 When the pH is increased condensation reactions lead to the formation of NH2- and OH-bridged multinuclear complex cations35,76,84 and trinuclear 20 and 21 show similar connectivities.76,80 When a neutral pH value is approached practically insoluble Be(NH2)2 and Be(OH)2 are formed.78,81 A further increase of the basicity of the solution leads to the formation of beryllates.35 From basic, aqueous solutions di- and tetranuclear, OH-bridged complex anions [Be2(OH)7]3− (23) and [Be4(OH)10]2− (22) were isolated.82,85 Their structures are closely related to cations 19 and 18, respectively.76 Under very basic conditions only mononuclear beryllates [Be(NH2)3]−, [Be(NH2)4]2− and [Be(OH)4]2− are present in NH3 and H2O respectively.75,83 Because of these closely related beryllium compounds in water and liquid ammonia, NH3 seems a promising solvent to perform water-like beryllium coordination chemistry in an oxygen-atom-free solvent. This was also shown recently by computational means.86
While NH3 readily displaces halides and pseudo halides under the formation of [Be(NH3)4]2+,66 this is not the case for organic amines. These generally give only the simple adducts (R3N)2BeCl2 or in case of pyrrolidine (pyrr) monocationic [(pyrr)3BeCl]+.87 A higher sterical demand of the amine leads to the formation of mono adducts, with no indication of the dissociation of the halides. In case of NEt3 and BeCl2 dinuclear complex [(Et3N)BeCl2]2 (24a) is formed, where the pseudo tetrahedrally coordinated beryllium atoms are μ2-bridged by two chlorine atoms.88 The complex geometry is closely related to phosphine adduct [(Cy3P)BeCl2]2.89 The reaction of NEt3 with BeI2 gives, in contrast to BeCl2, trigonal planar and monomeric (Et3N)BeI2 (25a). The analogous reaction with BeBr2 yields monomeric (Et3N)BeBr2 (25b) in solution, which dimerises in the solid state to [(Et3N)BeBr2]2 (24b). The difference in the coordination environment of the beryllium atoms is caused by their decreasing positive partial charge from chloride via bromide to iodide. This was quantified by population analysis.88
As described above dmf is a good solvent for cationic beryllium complexes.51 However, dmf is a strong O-donor solvent and readily coordinates to beryllium atoms. To comprehensively understand NMR spectra measured in dmf-d7, the solution behaviour of BeF2, BeCl2, BeBr2 and BeI2 was investigated. Additionally stoichiometric reactions with dmf in non-coordinating solvents were performed to understand the species present in solution.90 BeF2 is neither soluble in dmf nor can it be reacted with dmf in other solvents. In contrast to this, the other halides react readily with one equivalent of dmf in chlorinated solvents and give well soluble complexes. In case of BeCl2 and BeBr2 μ2-halido-bridged dinuclear complexes [(dmf)BeCl2]2 (26a) and [(dmf)BeBr2]2 (26b) are formed, respectively (Scheme 5).90 These complexes show a similar connectivity to the NEt3 compounds 24a and 24b.88 The reaction product of BeI2 with one equivalent dmf is also a dinuclear complex [(dmf)BeI2]2 (27), however here the two beryllium atoms are μ2-bridged by the oxygen atoms of the two dmf molecules. The dmf adducts 26a, 26b and 27 retain the same structure in solution and the solid state, which is in contrast to some of the NEt3 adducts.88,90 While 26a and 26b are stable in solution even at elevated temperatures the iodo complex 27 exhibits selfionisation under the formation of [(dmf)4Be2I2][Be2I6] (28). The different molecular constitution and reactivity of 27 compared to 26a and 26b is caused by the weak Be–I bond.90 When an additional equivalent dmf is added to 26a, 26b or 27, mononuclear adducts of two dmf molecules to the respective beryllium halides are formed (29; X = Cl (a), Br (b), I (c)). However, the 9Be NMR spectrum of iodo complex 29c shows extremely broad signals, which is indicative for fast exchange between different compounds in solution. Addition of a third equivalent of dmf to 29a leads to the formation of complex cation [(dmf)3BeCl]+ (30a), which reacts with one more dmf molecule to dication [(dmf)4Be]2+ (31). Solutions of these complexes show mixtures of 29a, 30a and 31 in the NMR spectra, with monocation 30a being the dominant species. 30a is also the major compound in dmf solutions of BeCl2, while the dominant species for BeBr2 and BeI2 is dication 31. While [(dmf)3BeI]+ (30c) could also be isolated in the solid state and there is evidence that this species also exists in solution, [(dmf)3BeBr]+ (30b) seems not to be stable.90
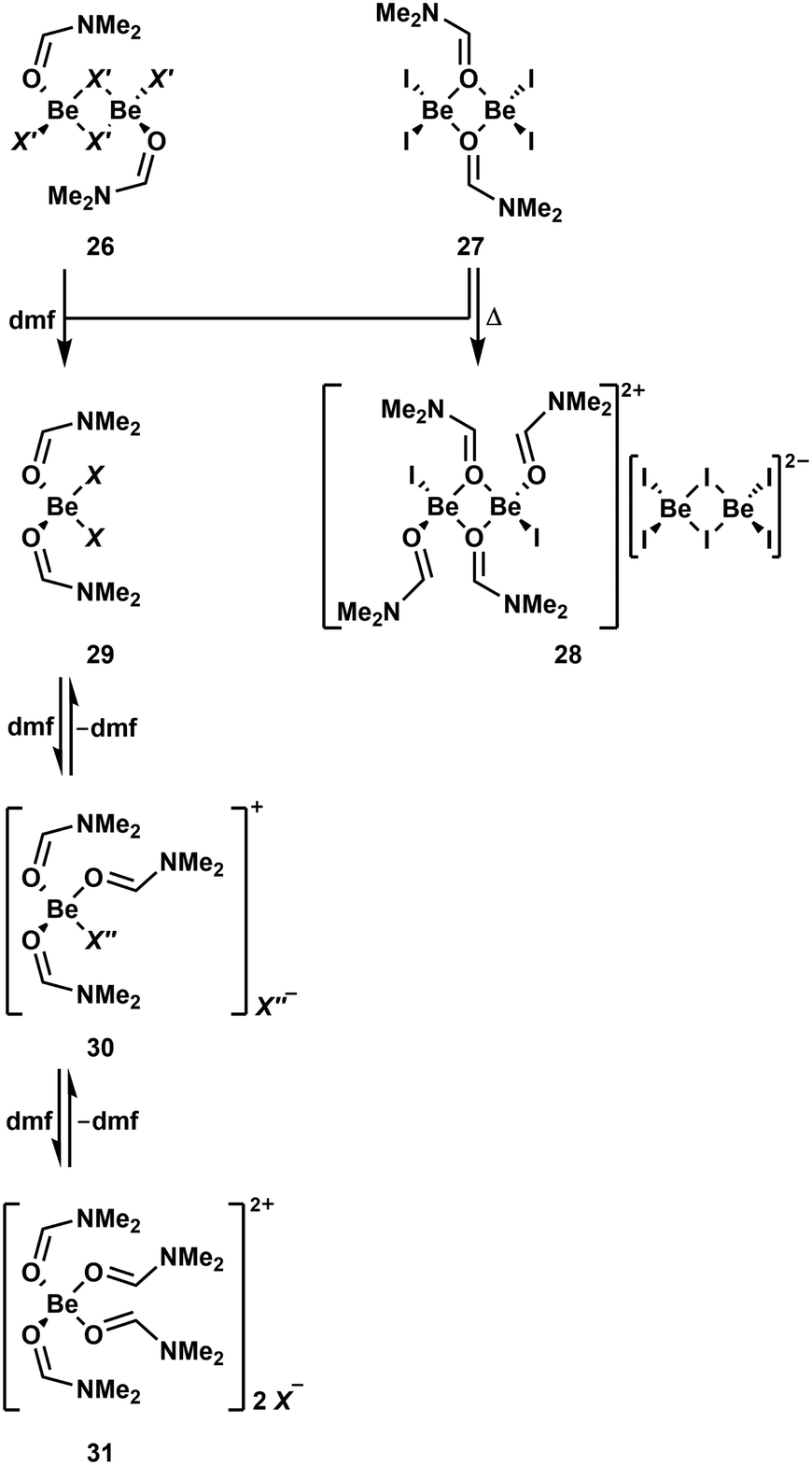 | ||
| Scheme 5 Various N,N-dimethylformamide (dmf) adducts of the beryllium halides. X = Cl (a), Br (b), I (c); X′ = Cl (a), Br (b); X′′ = Cl (a), I (c).90,91 | ||
5 Beryllium-induced reactions
To understand the action of Be2+ on cells and especially how the beryllium-induced cell death is initiated, it is important to understand what reactions occur upon beryllium coordination. Astonishingly, there were virtually no studies on this topic.13 Because of this, we began the investigation of beryllium-induced reactions at ligands with various functional groups.An understanding of simple ligand substitution reactions is necessary to evaluate what species can be formed. However, in case of beryllium this was mainly investigated computationally.93–97 To change this we investigated the electronic and steric influence of carboxylic acid ester ligands in compounds 32a on the substitution of chlorido by nitrato ligands (Scheme 6). This showed that the higher the steric demand of the ligands is, the lower is the reaction rate, while the electronic properties of the ligands made little differences.92 An in depth study on ligand exchange mechanisms at beryllium atoms is currently performed.
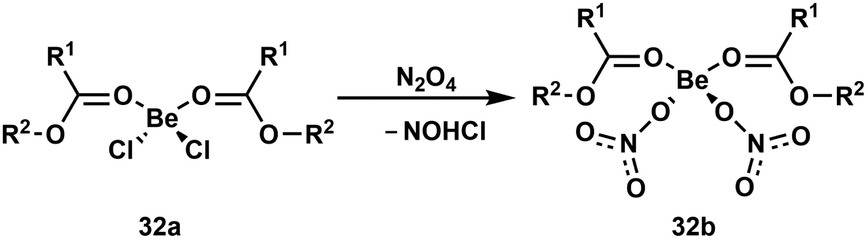 | ||
| Scheme 6 Displacement of chlorido by nitrato ligands in beryllium esterates. R1 = H, Me; R2 = Me, Et, iPr.92 | ||
As discussed above, hemiacetals and saccharides decompose in the presence of Be2+, due to their open chain isomer with an aldehyde function.51 To understand this reactivity better, we investigated the action of the beryllium halides on various aldehydes.98 While BeF2 again shows no reactivity due to its low solubility, the other halides readily catalyse the trimerisation of pivaldehyde and isobutyraldehyde to trioxanes. Butyraldehyde on the other hand is catalytically decomposed. If only two equivalents butyraldehyde are used, an aldol condensation reaction is induced and the formed product acts as a ligand to the beryllium atom. However, this complex is not stable for long periods and decomposes. Also stoichiometric reactions with isobutyraldehyde lead to aldehyde decomposition and polymerisation. In contrast to this, pivaldehyde forms stable complexes with BeCl2 and BeBr2. This is due to the absence of hydrogen atoms in β-position. This gives the simple adducts (tBuCHO)2BeCl2 (33a) and (tBuCHO)2BeBr2 (33b),98 which are comparable to benzaldehyde adduct 4.49 The corresponding iodo complex 33c can also be synthesised from BeI2 and two equivalents pivaldehyde (Scheme 7). However, this compound is not stable in solution and the isomerisation of one pivaldehyde into 3-methyl-2-butanone is induced and yields (tBuCHO)(iPrCOMe)BeI2 (34). The isomerisation of pivaldehyde into 3-methyl-2-butanone can also be performed with BeCl2 and BeBr2. However, to achieve this only one equivalent of aldehyde has to be used. This indicates that BeI2 is a stronger Lewis acid, which is presumable caused by the weak Be–I bond. Accordingly, 34 is further converted into dinuclear complex 35 through the beryllium-induced aldol reaction of the coordinated pivaldehyde and 3-methyl-2-butanone and subsequent HI cleavage. The weak Be–I bond and the resulting higher reactivity of the beryllium iodo compounds is also highlighted by the fact that the reaction of three equivalents pivaldehyde with BeI2 leads to the formation of cationic [(tBuCHO)3BeI]+ (36). The analogous reaction with BeCl2 or BeBr2 only leads to adducts 33a and 33b. One of the carbonyl groups of 36 is then nucleophilically attacked by I− to give mononuclear alcoholate 37, which dimerises into 38.98 These reaction pathways are summarised in Scheme 7.
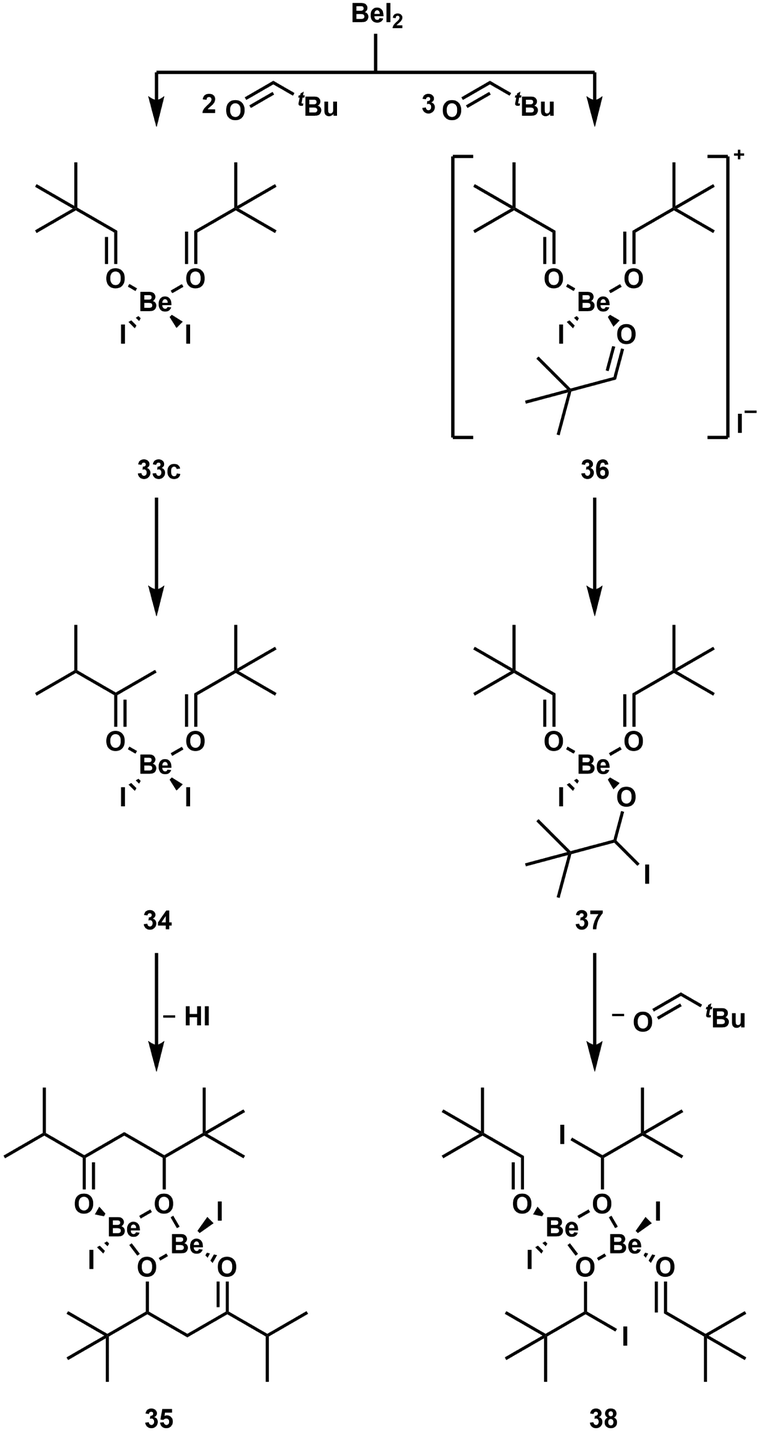 | ||
| Scheme 7 BeI2 induced conversions of pivaldehyde.98 | ||
Since C–H and C–O bond cleavage in cis,cis-1,3,5-cyclohexanetriol was observed (vide supra),51 we studied the coordination behaviour of partially silicon based crown ethers towards BeCl2. In these ligands Si–O and C–O bond cleavage was induced upon beryllium coordination. The obtained hexanuclear complexes like 39 (Scheme 8, top) comprise of one eight-membered Be–O-heterocycle, which is annulated by two six-membered Be–O-cycles. Also, in these compounds the beryllium atoms are exclusively tetrahedrally coordinated.52 Alkali metals, however, do not decompose the sila crown ethers.99 This shows that even very strong bonds are broken to facilitate tetrahedral coordination environments around Be2+.
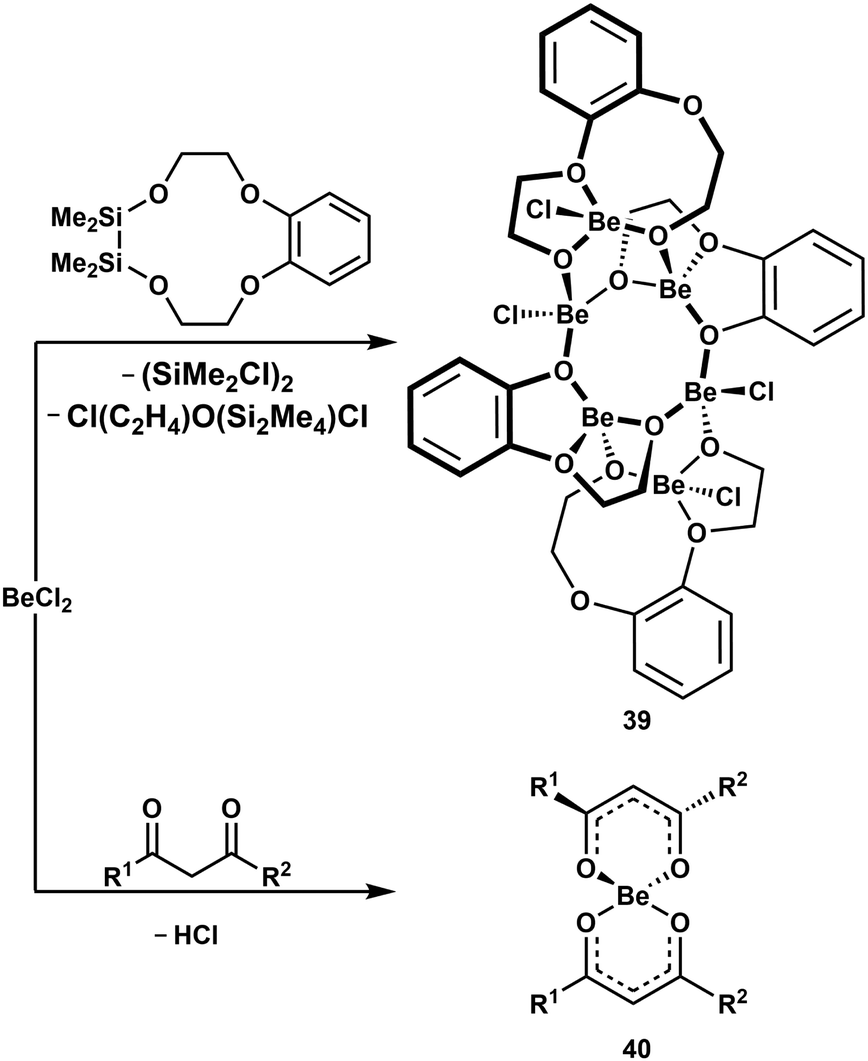 | ||
| Scheme 8 Beryllium-initiated Si–O, C–O and C–H bond cleavage.38,52 R1 = R2 = Ph; R1 = 2-C4H3S, R2 = CF3. | ||
The coordination of chelating diols to Be2+ increases the acidity of the hydroxy groups to an extent that exceeds anhydrous HCl, as described above.51 We could also show, that the CH-acidity of chelating 1,3-diketone is increased significantly upon coordination to beryllium. This leads to Cl− protonation and subsequent formation of ketylketonates like 40 under HCl evolution (Scheme 8, bottom).38
Due to the high oxophilicity of beryllium, its compounds react with water under the formation of beryllium hydroxo species and the generation of protons. These can be trapped with Lewis bases. In case of NEt3 adduct 24a this reaction gives amonium chloroberyllate 41 (Scheme 9, top).88 Lewis base adduct 24a is also able to activate less reactive C–Cl bonds, which leads to the formation of beryllates like 42 (Scheme 9, bottom).88 Similar reactions can also be performed with beryllium complexes of other Lewis bases like phosphines.100 A driving force for these reactions is the formation of strong Be–Cl bonds. This is also the reason why some coordination compounds of BeBr2 and BeI2 exhibit halide exchange with chlorinated solvents under formation of the respective BeCl2 complexes.88 The Be–F bond is even stronger than the Be–Cl bond. Thus, it is not surprising that the C–F bonds of α,α,α-trifluorotoluene can be cleaved with BeBr2 or BeI2 under the formation of BeF2 and α,α,α-tribromo- or α,α,α-triiodotoluene, respectively.101
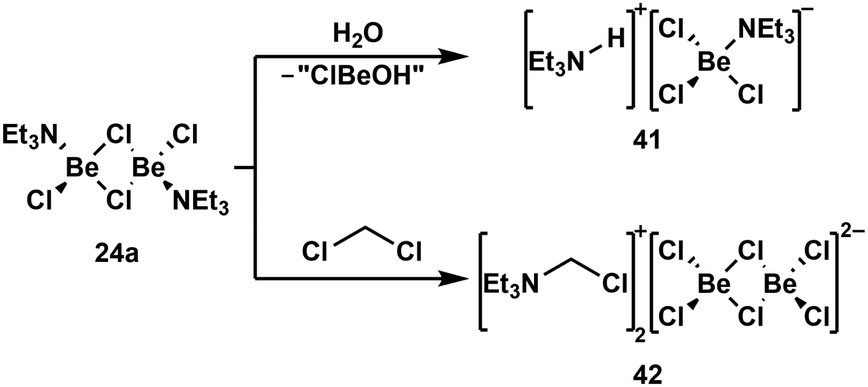 | ||
| Scheme 9 Lewis acid base adduct induced O–H and C–Cl bond activation.88 | ||
The high oxophilicity of beryllium is again highlighted by the fact that amido bridged tetranuclear cation 18 reacts with traces of H2O in pyridine (py) under the formation of oxygen centred octanuclear complex-cation [(Be4O){(py)Be(NH2)3}4]2+ (43) according to eqn (11).76
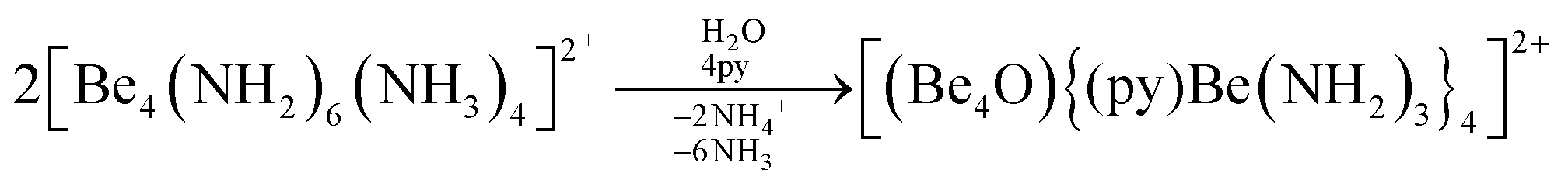 | (11) |
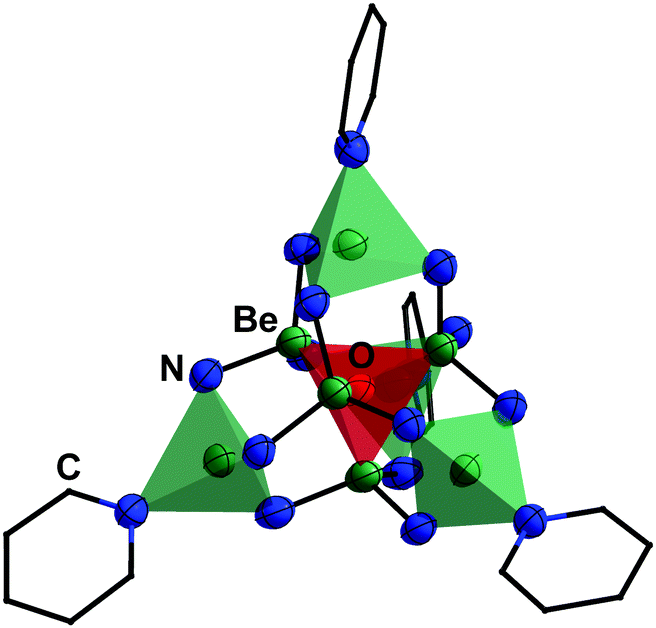 | ||
| Fig. 11 Molecular structure of [(Be4O){(py)Be(NH2)3}4]2+ (43, py = pyridine) in the solid state. The tetrahedron of the central [Be4O]6+ core is highlighted in red and of the [(py)Be(NH2)3]− units in green. Ellipsoids are depicted at 70% probability at 100 K. Carbon atoms are depicted as wire frame and hydrogen atoms are omitted for clarity.76 | ||
6 Conclusions
Beryllium coordination complexes with biomimetic ligands suggest that it is very likely that the Be2+ ion is always tetrahedrally coordinated in vivo. Even though the solubility of beryllium species is very low at biological pH values, the beryllium ions in solution should be highly mobile. Furthermore, only multidentate (κ3 or κ4) ligating sites in peptides and polysaccharides seem to be able to bind Be2+ irreversibly. Among the functional groups studied thus far the highest beryllium binding affinities are observed for carboxylates followed by alcohols. However, it should be mentioned that carboxylic acid amides are yet to be studied. Also, multinuclear beryllium complexes are formed very frequently. Therefore, these should be considered in the search for the actual species which binds inside the MHCII/peptide complex. Thus, it is plausible that the long latency times for the onset of CBD are caused by the necessity to form this multinuclear species at the low Be2+ concentrations in the body. However, to definitely answer the question how Be2+ is bound inside the body the directed synthesis of complexes with di- and oligopeptides will be necessary. These reactions will most likely be only possible in water-related inorganic solvents.Due to its strong Lewis acidity the beryllium ion causes the isomerisation or decomposition of O-donor ligands or the cleavage of C–H and C–O bonds. Additionally, beryllium coordination increases the acidity of carboxy and hydroxy as well as CH-acidic groups significantly. These groups might then act as potent Brønsted acids, which can in turn react with their surroundings. These reactions, which denature biomolecules might be the cause for the toxicity of beryllium ions and the origin of the beryllium-induced cell death.
Due to the small size of the Be2+ ion its coordination chemistry is unique. Therefore, other metal ions cannot be used to mimic its behaviour in the body. However, the charge density of Be2+ and Al3+ are comparable. Therefore, the reactivity they induce in functional groups and accordingly in peptides and polysaccharides should be comparable. This might explain why both metal ions act as adjuvants for the immune system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author expresses his gratitude to M. Müller and N. Spang for discussions over the manuscript. Prof. F. Kraus is thanked for moral and financial support as well as the provision of laboratory space. The DFG is gratefully acknowledged for financial support (BU2725/8-1).References
- R. Puchta, Nat. Chem., 2011, 3, 416 CrossRef CAS PubMed.
- S. Freeman, Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd., Hoboken, 2015 Search PubMed.
- P. Fröhlich, T. Lorenz, G. Martin, B. Brett and M. Bertau, Angew. Chem., Int. Ed., 2017, 56, 2544–2580 CrossRef PubMed.
- A. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, 1960 Search PubMed.
- L. C. Perera, O. Raymond, W. Henderson, P. J. Brothers and P. G. Plieger, Coord. Chem. Rev., 2017, 352, 264–290 CrossRef CAS.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nat. Catal., 2018, 1, 40–47 CrossRef CAS.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- G. Wang, J. E. Walley, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, J. Am. Chem. Soc., 2020, 142, 4560–4564 CrossRef CAS PubMed.
- A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed.
- M. R. Buchner, Chem. – Eur. J., 2019, 25, 12018–12036 CrossRef CAS PubMed.
- M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 405–412 CAS.
- T. M. McCleskey, V. Buchner, R. W. Field and B. L. Scott, Rev. Environ. Health, 2009, 24, 75–115 CAS.
- B. L. Scott, T. M. McCleskey, A. Chaudhary, E. Hong-Geller and S. Gnanakaran, Chem. Commun., 2008, 2837–2847 RSC.
- J. R. Balmes, J. L. Abraham, R. A. Dweik, E. Fireman, A. P. Fontenot, L. A. Maier, J. Muller-Quernheim, G. Ostiguy, L. D. Pepper, C. Saltini, C. R. Schuler, T. K. Takaro and P. F. Wambach, Am. J. Respir. Crit. Care Med., 2014, 190, e3–e59 CrossRef PubMed.
- A. E. Wyman and S. E. Hines, Curr. Opin. Allergy Clin. Immunol., 2018, 18, 73–79 CrossRef CAS PubMed.
- A. S. McKee, D. G. Mack, F. Crawford and A. P. Fontenot, Mucosal Immunol., 2015, 8, 1237–1247 CrossRef CAS.
- A. S. McKee and A. P. Fontenot, Curr. Opin. Immunol., 2016, 42, 25–30 CrossRef CAS.
- A. P. Fontenot, Ann. Am. Thorac. Soc., 2018, 15, S81–S85 CrossRef PubMed.
- D. G. Mack, A. M. Lanham, M. T. Falta, B. E. Palmer, L. A. Maier and A. P. Fontenot, Am. J. Respir. Crit. Care Med., 2010, 181, 1241–1249 CrossRef CAS PubMed.
- L. S. Newman, Science, 1993, 262, 197–198 CrossRef CAS PubMed.
- A. P. Fontenot, M. Torres, W. H. Marshall, L. S. Newman and B. L. Kotzin, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12717–12722 CrossRef CAS PubMed.
- A. Fontenot and B. Kotzin, Tissue Antigens, 2003, 62, 449–458 CrossRef CAS PubMed.
- M. Amicosante, F. Berretta, R. Dweik and C. Saltini, Immunology, 2009, 128, e462–e470 CrossRef.
- L. J. Silveira, E. C. McCanlies, T. E. Fingerlin, M. V. Van Dyke, M. M. Mroz, M. Strand, A. P. Fontenot, N. Bowerman, D. M. Dabelea, C. R. Schuler, A. Weston and L. A. Maier, J. Immunol., 2012, 189, 4014–4023 CrossRef CAS PubMed.
- M. T. Falta, C. Pinilla, D. G. Mack, A. N. Tinega, F. Crawford, M. Giulianotti, R. Santos, G. M. Clayton, Y. Wang, X. Zhang, L. A. Maier, P. Marrack, J. W. Kappler and A. P. Fontenot, J. Exp. Med., 2013, 210, 1403–1418 CrossRef CAS PubMed.
- N. A. Bowerman, M. T. Falta, D. G. Mack, F. Wehrmann, F. Crawford, M. M. Mroz, L. A. Maier, J. W. Kappler and A. P. Fontenot, J. Immunol., 2014, 192, 4571–4580 CrossRef CAS PubMed.
- G. Clayton, Y. Wang, F. Crawford, A. Novikov, B. Wimberly, J. Kieft, M. Falta, N. Bowerman, P. Marrack, A. Fontenot, S. Dai and J. Kappler, Cell, 2014, 158, 132–142 CrossRef CAS PubMed.
- S. De, G. Sabu and M. Zacharias, Phys. Chem. Chem. Phys., 2020, 22, 799–810 RSC.
- B. L. Scott, Z. Wang, B. L. Marrone and N. N. Sauer, J. Inorg. Biochem., 2003, 94, 5–13 CrossRef CAS.
- R. J. F. Berger and R. Mera-Adasme, Z. Naturforsch., B: J. Chem. Sci., 2016, 71, 71–75 CAS.
- R. J. Berger, P. Håkansson and R. Mera-Adasme, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 413–419 CAS.
- H. Schmidbaur, Coord. Chem. Rev., 2001, 215, 223–242 CrossRef CAS.
- O. Kumberger, J. Riede and H. Schmidbaur, Chem. Ber., 1992, 125, 2701–2703 CrossRef CAS.
- P. Klüfers, P. Mayer and J. Schuhmacher, Z. Anorg. Allg. Chem., 1995, 621, 1373–1379 CrossRef.
- O. Raymond, P. J. Brothers, M. R. Buchner, J. R. Lane, M. Müller, N. Spang, W. Henderson and P. G. Plieger, Inorg. Chem., 2019, 58, 6388–6398 CrossRef CAS PubMed.
- H. Schmidbaur and O. Kumberger, Chem. Ber., 1993, 126, 3–9 CrossRef CAS.
- M. Schmidt, A. Bauer and H. Schmidbaur, Inorg. Chem., 1997, 36, 2040–2043 CrossRef CAS PubMed.
- O. Kumberger, J. Riede and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 1992, 47, 1717–1720 CAS.
- T. S. Keizer, N. N. Sauer and T. M. McCleskey, J. Inorg. Biochem., 2005, 99, 1174–1181 CrossRef CAS PubMed.
- T. S. Keizer, N. N. Sauer and T. M. McCleskey, J. Am. Chem. Soc., 2004, 126, 9484–9485 CrossRef CAS PubMed.
- E. Chinea, S. Dominguez, A. Mederos, F. Brito, J. M. Arrieta, A. Sanchez and G. Germain, Inorg. Chem., 1995, 34, 1579–1587 CrossRef CAS.
- K. J. Shaffer, R. J. Davidson, A. K. Burrell, T. M. McCleskey and P. G. Plieger, Inorg. Chem., 2013, 52, 3969–3975 CrossRef CAS PubMed.
- D. J. Nixon, L. C. Perera, T. N. Dais, P. J. Brothers, W. Henderson and P. G. Plieger, Phys. Chem. Chem. Phys., 2019, 21, 19660–19666 RSC.
- O. Raymond, W. Henderson, P. J. Brothers and P. G. Plieger, Eur. J. Inorg. Chem., 2018, 1120–1130 CrossRef CAS.
- R. López, N. Díaz and D. Suárez, ChemPhysChem, 2020, 21, 99–112 CrossRef PubMed.
- M. Müller and M. R. Buchner, Angew. Chem., Int. Ed., 2018, 57, 9180–9184 CrossRef PubMed.
- R. J. F. Berger, M. A. Schmidt, J. Jusélius, D. Sundholm, P. Sirsch and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 979–989 CAS.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2019, 25, 16257–16269 CrossRef PubMed.
- M. R. Buchner, M. Müller, F. Dankert, K. Reuter and C. von Hänisch, Dalton Trans., 2018, 47, 16393–16397 RSC.
- K. Reuter, S. S. Rudel, M. R. Buchner, F. Kraus and C. von Hänisch, Chem. – Eur. J., 2017, 23, 9607–9617 CrossRef CAS PubMed.
- M. R. Buchner, M. Müller, O. Raymond, R. J. Severinsen, D. J. Nixon, W. Henderson, P. J. Brothers, G. J. Rowlands and P. G. Plieger, Eur. J. Inorg. Chem., 2019, 3863–3868 CrossRef CAS.
- M. R. Buchner and M. Müller, Z. Anorg. Allg. Chem., 2018, 644, 1186–1189 CrossRef CAS.
- O. Raymond, W. Henderson, J. R. Lane, P. J. Brothers and P. G. Plieger, J. Coord. Chem., 2020, 73, 1–16 CrossRef CAS.
- P. Barbaro, F. Cecconi, C. A. Ghilardi, S. Midollini, A. Orlandini, L. Alderighi, D. Peters, A. Vacca, E. Chinea and A. Mederos, Inorg. Chim. Acta, 1997, 262, 187–194 CrossRef CAS.
- R. J. F. Berger, S. Jana, R. Fröhlich and N. W. Mitzel, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 1131–1135 CAS.
- O. Hönigschmidt and T. Johannsen, Z. Naturforsch., 1946, 1, 650–655 Search PubMed.
- C. Jones and A. Stasch, Anal. Sci.: X-Ray Struct. Anal. Online, 2007, 23, x115–x116 CrossRef CAS PubMed.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2010, 636, 1438–1440 CrossRef.
- A. Paparo and C. Jones, Chem. – Asian J., 2019, 14, 486–490 CrossRef CAS PubMed.
- D. Himmel and I. Krossing, Z. Anorg. Allg. Chem., 2006, 632, 2021–2023 CrossRef CAS.
- M. Müller, F. Pielnhofer and M. R. Buchner, Dalton Trans., 2018, 47, 12506–12510 RSC.
- M. Müller and M. R. Buchner, Z. Kristallogr., 2020 DOI:10.1515/zkri-2020-0016.
- M. Müller and M. R. Buchner, Chem. Commun., 2019, 55, 13649–13652 RSC.
- M. R. Buchner, M. Müller and S. S. Rudel, Angew. Chem., Int. Ed., 2017, 56, 1130–1134 CrossRef CAS PubMed.
- H. Gilman and F. Schulze, J. Chem. Soc., 1927, 2663–2669 RSC.
- G. Wittig, F. J. Meyer and G. Lange, Justus Liebigs Ann. Chem., 1951, 571, 167–201 CrossRef CAS.
- G. E. Coates and M. Tranah, J. Chem. Soc. A, 1967, 236–239 RSC.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2020 DOI:10.1002/chem.202000259.
- D. Naglav, M. R. Buchner, G. Bendt, F. Kraus and S. Schulz, Angew. Chem., Int. Ed., 2016, 55, 10562–10576 CrossRef CAS PubMed.
- P. G. Plieger, K. D. John, T. S. Keizer, T. M. McCleskey, A. K. Burrell and R. L. Martin, J. Am. Chem. Soc., 2004, 126, 14651–14658 CrossRef CAS PubMed.
- J. K. Buchanan and P. G. Plieger, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 459–472 CAS.
- M. Müller and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 483–489 Search PubMed.
- M. Müller, A. J. Karttunen and M. R. Buchner, Chem. Sci., 2020, 11, 5414–5422 RSC.
- F. Kraus, S. A. Baer, M. R. Buchner and A. J. Karttunen, Chem. – Eur. J., 2012, 18, 2131–2142 CrossRef CAS PubMed.
- H. Jacobs, Z. Anorg. Allg. Chem., 1976, 427, 1–7 CrossRef CAS.
- I. G. Dance and H. C. Freeman, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 304–310 CrossRef CAS.
- F. Cecconi, C. A. Ghilardi, S. Midollini, A. Orlandini and A. Mederos, Inorg. Chem., 1998, 37, 146–148 CrossRef CAS PubMed.
- R. Stahl, C. Jung, H. D. Lutz, W. Kockelmann and H. Jacobs, Z. Anorg. Allg. Chem., 1998, 624, 1130–1136 CrossRef CAS.
- H. Schmidbaur, M. Schmidt, A. Schier, J. Riede, T. Tamm and P. Pyykkö, J. Am. Chem. Soc., 1998, 120, 2967–2968 CrossRef CAS.
- B. T. R. Littlefield, C. Hinde and M. T. Weller, Dalton Trans., 2011, 40, 782–784 RSC.
- O. Raymond, W. Henderson, P. J. Brothers and P. G. Plieger, Eur. J. Inorg. Chem., 2017, 2691–2699 CrossRef CAS.
- M. Schmidt, A. Schier, J. Riede and H. Schmidbaur, Inorg. Chem., 1998, 37, 3452–3453 CrossRef CAS PubMed.
- O. Raymond, M. Bühl, J. R. Lane, W. Henderson, P. J. Brothers and P. G. Plieger, Inorg. Chem., 2020, 59, 2413–2425 CrossRef CAS PubMed.
- M. P. Dressel, S. Nogai, R. J. F. Berger and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 173–182 CAS.
- M. R. Buchner, M. Müller and N. Spang, Dalton Trans., 2020, 49, 7708–7712 RSC.
- H. Braunschweig and K. Gruß, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 55–57 CAS.
- M. Müller and M. R. Buchner, Inorg. Chem., 2019, 58, 13276–13284 CrossRef PubMed.
- T. Hohl, T. Sinn and C. Hoch, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 509–516 CAS.
- B. Scheibe and M. R. Buchner, Eur. J. Inorg. Chem., 2018, 2300–2308 CrossRef CAS.
- R. Puchta, N. van Eikema Hommes and R. van Eldik, Helv. Chim. Acta, 2005, 88, 911–922 CrossRef CAS.
- R. Puchta and R. van Eldik, Z. Anorg. Allg. Chem., 2008, 634, 735–739 CrossRef CAS.
- R. Puchta and R. van Eldik, Helv. Chim. Acta, 2008, 91, 1063–1071 CrossRef CAS.
- A. Budimir, M. Walther, R. Puchta and R. van Eldik, Z. Anorg. Allg. Chem., 2011, 637, 515–522 CrossRef CAS.
- M. Walther and R. Puchta, RSC Adv., 2012, 2, 5815–5821 RSC.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2019, 25, 11147–11156 CrossRef PubMed.
- K. Reuter, M. R. Buchner, G. Thiele and C. von Hänisch, Inorg. Chem., 2016, 55, 4441–4447 CrossRef CAS PubMed.
- M. R. Buchner, N. Spang, M. Müller and S. S. Rudel, Inorg. Chem., 2018, 57, 11314–11317 CrossRef CAS PubMed.
- F. Dankert, H. L. Deubner, M. Müller, M. R. Buchner, F. Kraus and C. von Hänisch, Z. Anorg. Allg. Chem., 2020 DOI:10.1002/zaac.201900297.
| This journal is © The Royal Society of Chemistry 2020 |