
Visible-light-mediated arylation of ortho-hydroxyarylenaminones: direct access to isoflavones†
Satenik
Mkrtchyan
 * and
Viktor O.
Iaroshenko
*
* and
Viktor O.
Iaroshenko
*
Laboratory of Homogeneous Catalysis and Molecular Design at the Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, PL-90-363 Łodź, Poland. E-mail: iva108@gmail.com; viktori@cbmm.lodz.pl
First published on 27th January 2020
Abstract
In this work we highlight two new methods for the synthesis of isoflavones through consecutive domino arylation of ortho-hydroxyarylenaminones with in situ photogenerated aryl radicals. As precursors for aryl radicals we used aryl onium reagents such as diazonium and diaryliodonium salts. Notably, the photo-Meerwein arylation by aryl diazonium tetrafluoroborates demonstrated high efficiency in terms of yields and can be considered as a method of choice for the straightforward assembly of 3-aryl-substituted chromones. Ultimately, 26 compounds were prepared in good to excellent yields using the developed synthetic protocols.
Chromones are important privileged heterocycles that are not only abundant in nature in the form of numerous natural compounds, but have also gained widespread applications in life science, drug discovery, food industry, and health-related aspects of human life.1 Isoflavonoids (3-arylchromones) are compounds with numerous applications in medicines and food production.2
Due to the importance of these heterocycles, research presenting new conceptual syntheses of chromones in general and 3-arylchromones in particular still receives enormous interest. There is a growing demand for cost-effective and efficient methods enabling the rapid and straightforward assembly of chromone frameworks with different substitution patterns. Constantly increasing attention and effort is aimed at the search of new, concise and atom economic routes towards this interesting heterocyclic system. For a long time 3-arylchromones, as well as many other chromone-based drug-like heterocycles, were only available from natural sources; later, however, they became available through a limited set of obsolete cyclisation reactions.1a,3 Nowadays, a bright spectrum of C–C coupling protocols4 including Suzuki,5 Neghishi6 and Stille methods,7 employing 3-halochromones as precursors, are successfully utilised for the facile preparation of those compounds.
The direct C–H functionalisation of 2,3-unsubstituted chromones was also employed as an efficient strategy enabling the construction of diverse 3-substituted chromones, such as 3-acyloxyl, 3-alkyl, 3-perfluoroalkyl, 3-vinyl, 3-alkynyl, and 3-chalcogenyl chromone derevatives.8 Very recently we communicated the arylselenation of 2,3-unsubstituted chromones via a Cu-catalysed direct C–H activation protocol using elemental Se and aryl iodides, which leveraged the corresponding 3-substituted chromones.9 Unfortunately, due to the innate inherent regioselectivity, 3-arylchromones could not be prepared by the direct C-3 C–H arylation of 2,3-unsubstituted chromones using a range of arylation agents; instead, the 2-arylchromones were detected as only isolable products.10 However, due to the C-2 selectivity for the mentioned arylation protocol, currently one of the most prevalent challenges in the field is establishing alternative synthetic routes aimed at the straightforward and concise preparation of 3-arylchromones. Thus, chromone ring construction very often entails consecutive domino reactions initiated by the cyclisation of ortho-hydroxyarylenaminones triggered by numerous hard and soft electrophiles11 as well as radicals.12 Very recently, during the preparation of this manuscript, a study emerged which highlights the use of Pd-promoted oxidative C–H bond arylation of ortho-hydroxyarylenaminones by arylboronic acids.13 The above-mentioned strategies were successfully utilised to prepare myriads of 3-substituted chromone derivatives decorated with a vast range of substituents and functional groups.
As the electrophile-triggered cyclisation of ortho-hydroxyarylenaminones is very well studied, the corresponding radical-mediated processes involving carbon-centred radicals are scarcely presented in the modern literature. To the best of our knowledge there are only four examples, which are depicted in Scheme 1a–d.8d,14 Notably, methods for the efficient radical arylation of ortho-hydroxyarylenaminones are scarce. Since the application of organic radicals in contemporary synthetic organic chemistry has gained new momentum, particularly with the advent of photoredox catalysis in recent years, studies on the application of carbon-centred radicals in the synthesis of 3-arylchromones are of current synthetic interest and urgent importance.15
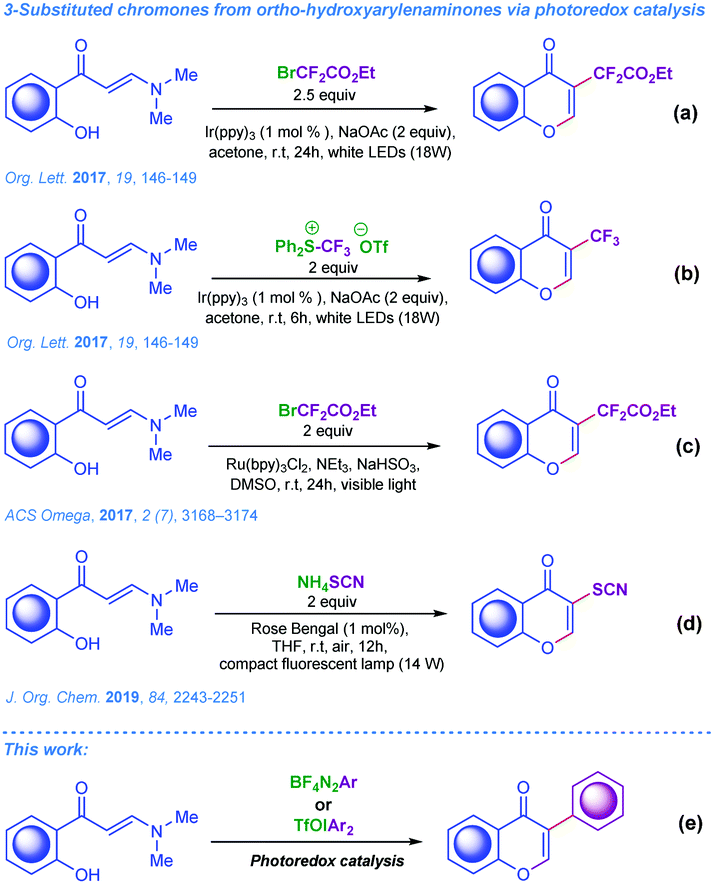 | ||
| Scheme 1 (a–d) Known strategies from the literature for 3-substituted chromones based on photoredox catalysis; (e) our concepts. | ||
Aryl radicals can be generated by different light-driven means from a vast range of substrates. For instance, in recent years diazonium salts, diaryliodonium salts, sulfonium salts, aryl sulfonyl chlorides, etc. have been intensively utilised in the photoredox radical-promoted introduction of aryl substituents.15 In light of the known methods to transform ortho-hydroxyarylenaminones into chromones, we decided to study 3-arylchromone synthesis by the reactions between the in situ photogenerated aryl radicals and ortho-hydroxyarylenaminones. We surmised that this synthetic scenario (1) is in no need of substrate prefunctionalisation, (2) uses readily available reagents, and might visibly improve the preparation of 3-arylchromones.
Herein, in continuation of our work on developing new methods for the preparation of structurally diverse chromone derivatives with different substitution patterns,12,16 we describe, for the first time, two unprecedented domino cyclisation scenarios, wherein the ortho-hydroxyarylenaminone substrates react with aryl radicals photogenerated from aryl onium salts (Scheme 2). The starting point of this study was to identify the appropriate reaction conditions for the efficient preparation of various 3-arylchromones following the appointed synthetic scenarios. For this we set two model reactions and focused our efforts on the elaboration of the optimised reaction conditions (Scheme 2a and b). Tables illustrating the general logic used for optimisation of the reaction conditions are given in the ESI† (Tables S1 and S2). It was revealed that (1) the photo-Meerwein arylation of ortho-hydroxyarylenaminones by aryl diazonium tetrafluoroborates proceeded smoothly under catalysis by Eosin Y I; and (2) the arylation by diaryliodonium triflates catalysed by Ru(bpy)3Cl2·6H2O IV proceeded with high efficiency.
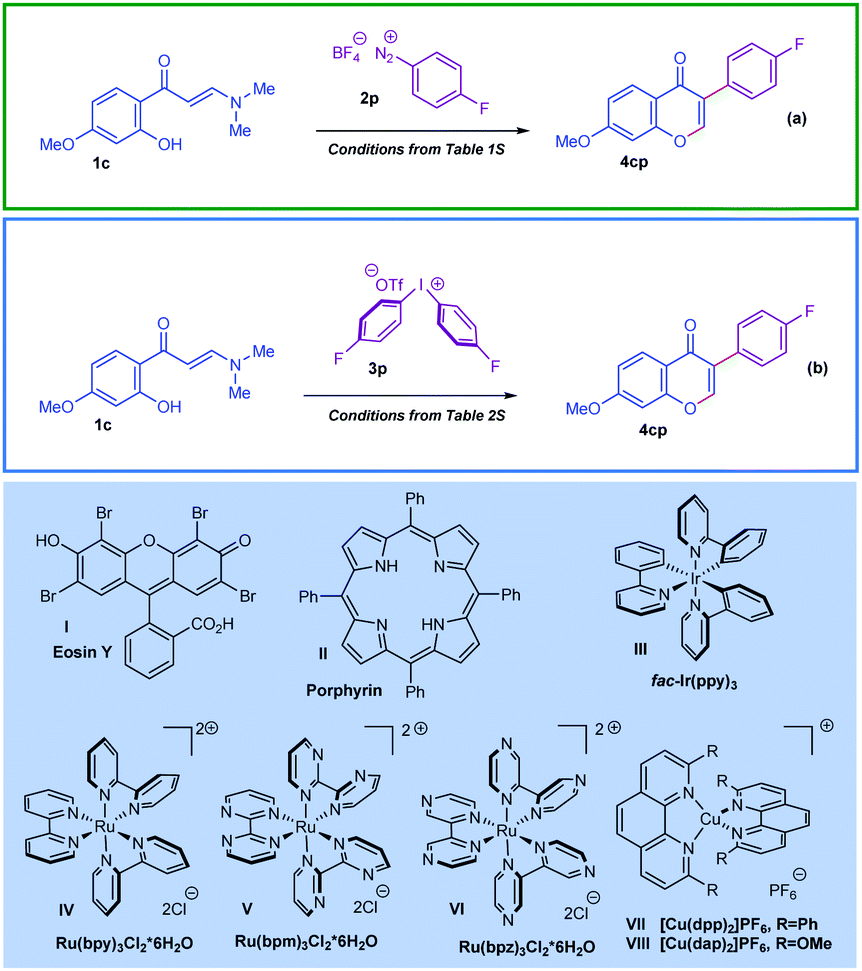 | ||
| Scheme 2 Model reactions for reaction conditions optimization. | ||
As outlined in Scheme 3a, the model ortho-hydroxyarylenaminone reacts smoothly with 4-fluorobenzenediazonium tetrafluoroborate, affording the corresponding chromone product 4cp in 86% yield (Table S1, entry 4, ESI†). The optimised reaction conditions for this transformation consisted of 1.3 equiv. of salt 2p and Eosin Y I photocatalyst (3 mol%) in DMSO. Under intensive irradiation with green light (the best efficiency was observed for 24 W green LED stripes), the reaction reaches completion within 3 hours at room temperature under argon atmosphere. On the other hand, encouragingly, the 3-arylchromone 4cp can also be prepared in 80% yield within 4 hours at room temperature following the route in Scheme 3b by a reaction between ortho-hydroxyarylenaminone and diaryliodonium triflate 3p (1.5 equiv.), applying Ru(bpy)3Cl2·6H2O IV (2 mol%) as a catalyst and CH3CN as a solvent (Table S2, entry 6, ESI†). For this case the best outcome was obtained when we used 34 W Kessil KSH150B Blue LED as a source of blue light. Furthermore, other tested photocatalysts depicted in Scheme 2, like Porphyrin II and fac-Ir(ppy)3III showed catalytic activity for photo-Meerwein arylation as well. For instance, under blue light irradiation fac-Ir(ppy)3 allowed the preparation of chromone 4cp in 79% yield by using a diazonium salt, and in 77% yield by using a diaryliodonium salt (Table S1, entry 10, and Table S2, entry 2, ESI†). Furthermore, a range of common Ru-based photocatalysts was also tested in line with the given transformation. Notably, species V and VI also showed positive outcomes, though not as positive as complex IV (Table S2, entries 12 and 13, ESI†). In terms of light sources, the best yields were observed when the strong 24 W and 34 W irradiation LED sources corresponding to green and blue light were used. In both cases, less powerful light sources were also tested and the model reactions demonstrated high efficiency; however, reducing the power of the light resulted in longer reaction durations (this data is not presented in the ESI†). On the other hand, it should be mentioned that copper complexes VII and VIII, well-defined for such reactions, were rigorously studied as well. Unfortunately, the application of these catalysts to the title arylation protocols failed (Table S2, entries 14–16, ESI†).
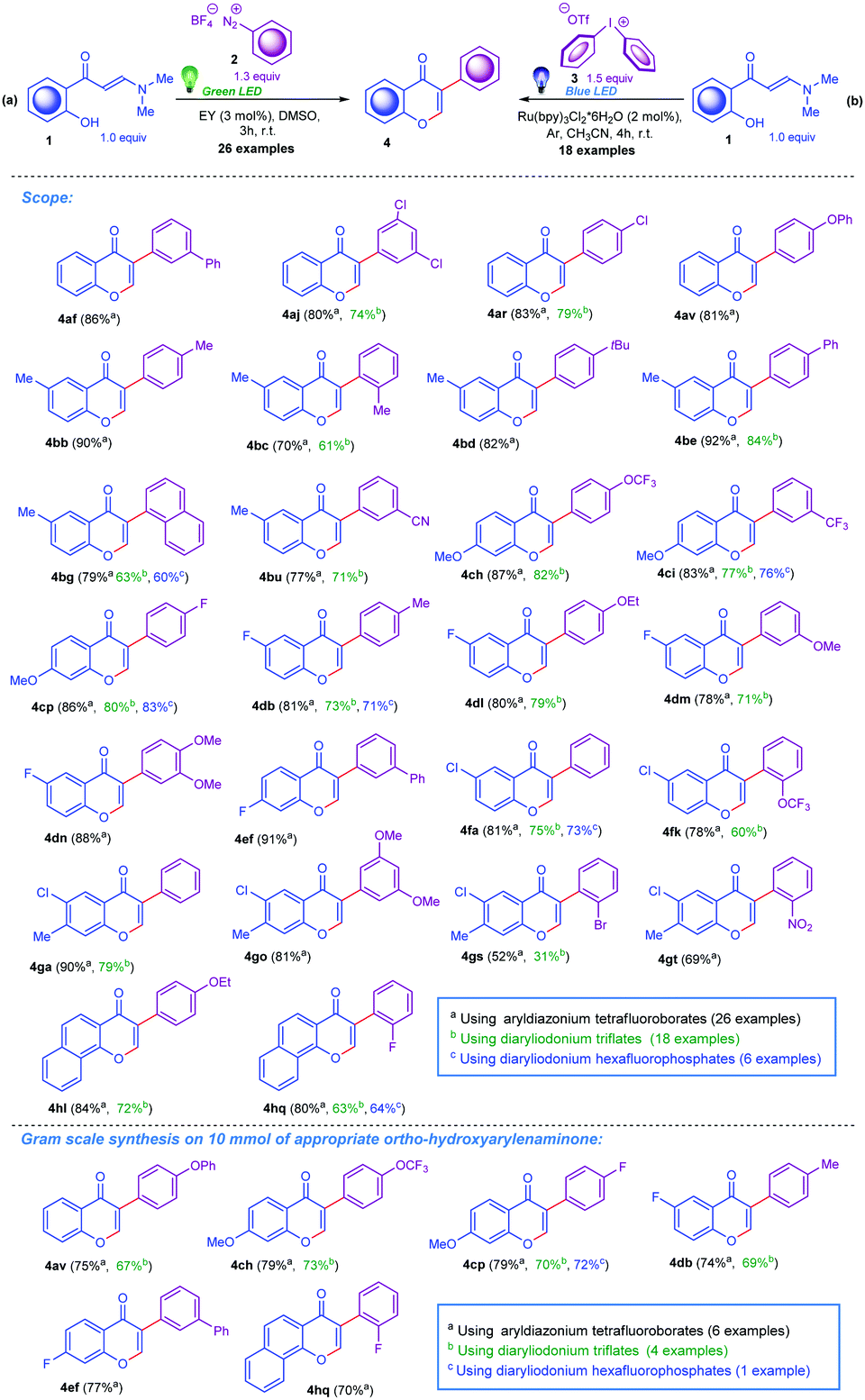 | ||
| Scheme 3 Product scope for the synthesis of 3-arylchromones. | ||
With the inspired optimal reaction conditions for both presented synthetic scenarios in hand, next we decided to explore the substrate scope and evaluated the feasibility of the developed synthetic protocols for the preparation of structurally diverse 3-arylchromones. We selected eight ortho-hydroxyarylenaminones 1, which were reacted with twenty-two aryl diazonium tetrafluoroborates 2 and sixteen diaryliodonium triflates 3. To our delight, various diazonium tetrafluoroborates bearing diverse functional groups including Me, Ph, OPh, OMe, OEt, F, Cl, Br, CN, NO2, CF3, and OCF3 substituents in the ortho-, meta-, and para-positions as well as reagents with a 1-naphthalenyl residue readily reacted with ortho-hydroxyarylenaminones decorated by Me, OMe, OEt, F, or Cl groups on the benzene ring. Generally, this method displayed satisfactory tolerance to both structural components, affording the desired heterocycles in moderate to high yields. Importantly, these methods could be utilised for the preparation of 3-aryl-4H-benzo[h]chromen-4-ones 4hl and 4hq, also in excellent yields (Scheme 3). For the second method involving the diaryliodonium triflates 3, we generally observed slightly diminished overall yields. Furthermore, we illustrated gram scale reactions for both methods to prepare compounds 4av, 4ch, 4cp, 4db, 4ef, and 4hq (Scheme 3). It is important to mention that besides diaryliodonium triflates, the corresponding hexafluorophosphates demonstrated the ability to take part in this reaction, thus six compounds (4bg, 4ci, 4cp, 4db, 4fa, and 4hq) were prepared without significant deviations in the yields (Scheme 3).
To gain insight into the mechanism and prove that the illustrated routes indeed involve photochemically-generated aryl radicals, a set of control experiments were conducted. First, we ran the reactions under our optimised reaction conditions in the dark, as well as without photocatalysts; this led to the failure of the reactions (Table S1, entries 5 and 6, and Table S2, entries 9 and 10, ESI†). Second, the addition of a radical quencher such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 1.5 and 1.7 equiv., respectively) inhibited both reactions by suppressing the formation of product 4cp, most likely by trapping most of the Ar radicals (Table S1, entry 7, and Table S2, entry 11, ESI†). These experiments can be accepted as an indication that photochemically-generated radical intermediates are indeed involved in both transformations.
Finally, according to the results illustrated above and the literature on visible-light photoredox-catalysed reactions available to date,15 a plausible reaction mechanism for this visible-light promoted domino cyclisation was proposed and is outlined in Scheme 4. Initially, under irradiation by an appropriate light source, the Eosin Y and Ru(II) complexes are transferred to their excited states. Under visible-light conditions the generation of aryl radicals (Ar˙) from aryldiazonium tetrafluoroborates and diaryliodonium triflates occurs as the first step in the current mechanism sequences catalysed by exited *Eosin Y and *Ru(II) species, respectively (Scheme 4a and b). To give more detail: the corresponding excited *Eosin Y species undergoes oxidative quenching by the ArN2+ cation, which proceeds according to the major literature data15via a single electron transfer (SET) to afford Ar˙. The excited triplet state of *Ru(II) contains an electron located in the higher singly occupied molecular orbital (SOMO); this electron participates in the SET event, thus making the excited *Ru(II) species a one-electron reductant. Notably, the generation of Ar˙ by visible-light from Ar2I+ follows the typical route for transition-metal photocatalysts,15c,e,17 namely the exited *Ru(II) donates an electron to Ar2I+. This causes its decomposition to ArI and Ar˙. Subsequently, the formed Ar˙ undergoes prompt attack on the enaminone to afford the radical intermediate 5, which afterwards is oxidised to carbocation 6 followed by the pyrone ring formation. Finally, after the elimination of dimethylamine from the 2-(dimethylamino)chroman-4-one 7, the chromone framework 4 forms. A similar type of mechanism was very recently proposed for the visible-light-driven synthesis of 3-thiocyanato and 3-polyfluoroalkyl chromones (Scheme 1a–d).14
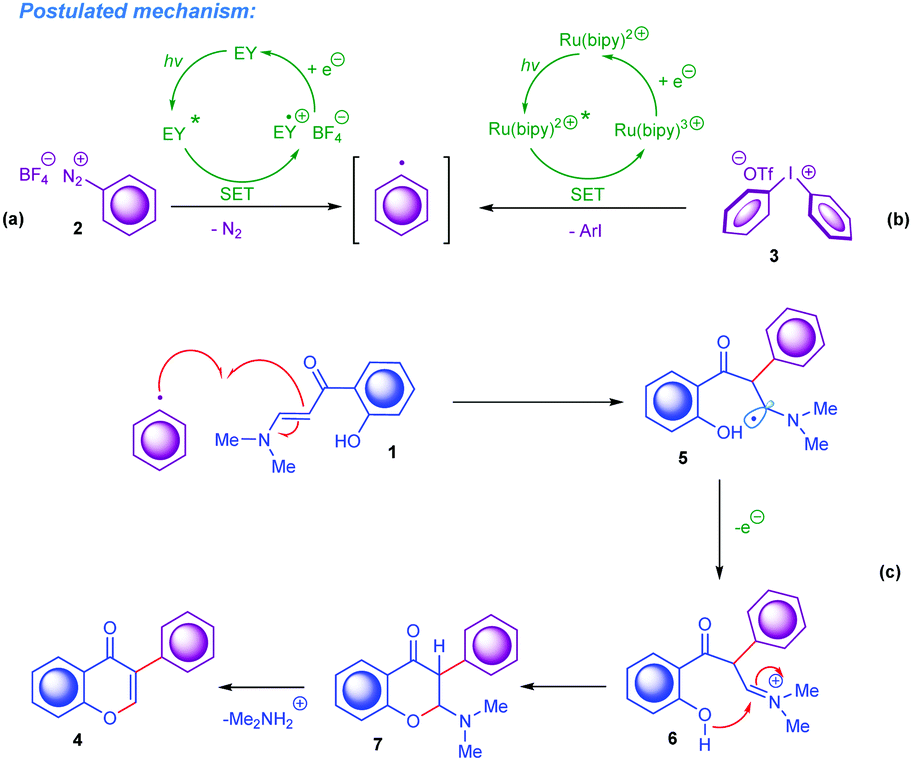 | ||
| Scheme 4 Proposed mechanism for the synthesis of 3-arylchromones by photocatalytic arylation of ortho-hydroxyarylenaminones by aryl onium salts. | ||
In conclusion, we have developed two instrumentally simple, concise and facile routes to structurally diverse 3-arylchromones using photoarylation reactions of ortho-hydroxyarylenaminones by two different reagents, namely aryldiazonium tetrafluoroborates and diaryliodonium triflates, utilising Eosin Y and Ru(bpy)3Cl2·6H2O as photocatalysts, respectively. In a practical context, these two methods provide the desired products with good to excellent yields, with absolute C-3 selectivity and demonstrated excellent levels of functional group tolerance. Comparing these methodologies, it is important to admit that the photo-Meerwein arylation reaction is more efficient as estimated by many parameters: it shows higher yields, utilises less expensive organo photocatalyst; and last but not least, the aryldiazonium salts are more readily available than the corresponding diaryliodonium salts. Preliminary mechanistic studies indicated that the protocols described above involve free-radical pathways. Future work that should focus on the extension of these concepts to other sources of aryl radicals is currently in progress in our laboratories.
This research project is supported by a grant from National Science Centre (NSC) Poland within the framework of the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 665778 (POLONEZ 2 grant, no. 2016/21/P/ST5/00630) obtained by Dr Satenik Mkrtchyan.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a)
“Chromenes, Chromanones, and Chromones”, from The Chemistry of Heterocyclic Compounds, ed. G. P. Ellis, 2009, vol. 31, p. 1196, ISBN: 978-0-470-18852-1 Search PubMed
; (b) A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960–4992 CrossRef CAS
-
(a) J. W. Lampe, J. Nutr., 2003, 133, 956S–964S CrossRef CAS PubMed
-
(a) A. Aitmambetov and V. P. Khilya, Chem. Nat. Compd., 1994, 30(3), 307–311 CrossRef
-
(a) L. Klier, T. Bresser, T. A. Nigst, K. Karaghiosoff and P. Knochel, J. Am. Chem. Soc., 2012, 134, 13584–13587 CrossRef CAS PubMed
-
(a) F.-X. Felpin, J. Org. Chem., 2005, 70, 8575–8578 CrossRef CAS PubMed
- Z. Zhang, J. Qiao, D. Wang, L. Han and R. Ding, Mol. Diversity, 2014, 18, 245–251 CrossRef CAS PubMed
- D. A. Vasselin, A. D. Westwell, C. S. Matthews, T. D. Bradshaw and M. F. G. Stevens, J. Med. Chem., 2006, 49, 3973–3981 CrossRef CAS PubMed
-
(a) Y. Guo, Y. Xiang, L. Wei and J.-P. Wan, Org. Lett., 2018, 20, 3971–3974 CrossRef CAS PubMed
- M. Jakubczyk, S. Mkrtchyan, I. D. Madura, P. H. Marek and V. O. Iaroshenko, RSC Adv., 2019, 9, 25368–25376 RSC
-
(a) M. Khoobi, M. Alipour, S. Zarei, F. Jafarpour and A. Shafiee, Chem. Commun., 2012, 48, 2985–2987 RSC
-
(a) L. Fu and J.-P. Wan, Asian J. Org. Chem., 2019, 8, 767–776 CrossRef CAS
- S. Mkrtchyan and V. O. Iaroshenko, Eur. J. Org. Chem., 2018, 6867–6875 CrossRef CAS
- J.-P. Wan, Z. Tu and Y. Wang, Chem. – Eur. J., 2019, 25, 6907–6910 CrossRef CAS
-
(a) H. Gao, B. Hu, W. Dong, X. Gao, L. Jiang, X. Xie and Z. Zhang, ACS Omega, 2017, 2(7), 3168–3174 CrossRef CAS
-
(a) M. Majek and A. J. von Wangelin, Acc. Chem. Res., 2016, 49, 2316–2327 CrossRef CAS
-
(a) V. O. Iaroshenko, S. Mkrtchyan, A. Gevorgyan, T. Grigoryan, A. Villinger and P. Langer, RSC Adv., 2015, 5, 28717–28724 RSC
- M. K. Bogdos, E. Pinard and J. A. Murphy, Beilstein J. Org. Chem., 2018, 14, 2035–2064 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09945j |
| This journal is © The Royal Society of Chemistry 2020 |