
A general and green fluoroalkylation reaction promoted via noncovalent interactions between acetone and fluoroalkyl iodides†
Ting
Mao‡
a,
Ming-Jian
Ma‡
a,
Liang
Zhao
ab,
De-Pu
Xue
a,
Yanbo
Yu
 c,
Jiwei
Gu
c and
Chun-Yang
He
*ab
c,
Jiwei
Gu
c and
Chun-Yang
He
*ab
aKey Laboratory of Biocatalysis & Chiral Drug Synthesis of Guizhou Province, Generic Drug Research Center of Guizhou Province, School of Pharmacy, Zunyi Medical University, Zunyi, Guizhou, P. R. China. E-mail: hechy2002@163.com
bKey Laboratory of Basic Pharmacology of Ministry of Education and Joint International Research Laboratory of Ethnomedicine of Ministry of Education, School of Pharmacy, Zunyi Medical University, Zunyi, Guizhou, P. R. China
cSchool of Medicine, Washington University in St. Louis, St. Louis, Missouri, USA
First published on 9th January 2020
Abstract
The first example of visible light promoted fluoroalkylation reactions initiated via noncovalent interactions between acetone and fluoroalkyl iodides is presented. The reaction system features synthetic simplicity, mild reaction conditions without any photoredox catalyst, and high functional group tolerance. A wide range of substrate scopes such as alkenes, alkynes and (hetero)arenes were all compatible with the reaction system.
Fluorinated compounds have attracted much attention as candidates for pharmaceuticals and functional materials because of their superior lipophilicity, binding selectivity, bioavailability, and metabolic stability compared to their nonfluoroalkylated analogs.1 Consequently, to meet the increasing demand for these materials in the life sciences and materials science, the development of more practical, economical and environmentally friendly methods enabling the efficient construction of fluorine-containing organic compounds has been one of the most important research topics in organofluorine chemistry.
In the past ten years, numerous methods have been developed to synthesize fluoroalkylated compounds with high efficiency, and major improvements were made via transition-metal catalysts or photo-excited catalysts.2 Very recently, noncovalent interaction (EDA complexes or halogen bond) initiated radical fluoroalkylations have emerged as an attractive and useful strategy to directly introduce fluorinated groups into organic molecules.3 In such transformations, the noncovalent interaction usually occurs between excess amounts of amines or substrates and RFI. We envision that noncovalent interactions might also occur between the solvent and RFI, and a radical intermediate could be generated under the irradiation of visible light if the driving force is strong enough, thus leading to the discovery of unprecedented transformations.
Acetone is one of the most common solvents. We assumed that a noncovalent interaction between the carbonyl group and C–I bond might occur, which could induce radical intermediates under the irradiation of visible light. To test the hypothesis above, we began our investigations by treating alkenes with RFI in acetone to study the atom transfer radical addition (ATRA) reaction. Usually, this protocol was realized by using radical initiators, such as peroxide,4 Na2S2O4,5 Et3B,6 or UV light.7 Recently, transition-metal8 and photoredox catalysis9 have emerged as more effective alternatives, and the utilization of amines,10 phenols11 or phosphines12 as catalysts has also been developed in the past several months (Scheme 1). Herein, we report the first example of visible light promoted fluoroalkylation reactions via noncovalent interactions between the solvent and RFI. A variety of substrate scopes such as alkenes, alkynes and (hetero)arenes were all compatible with the reaction system.
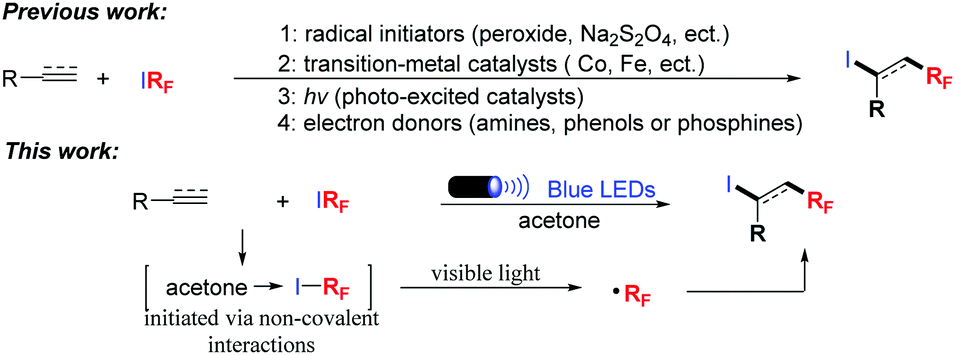 | ||
| Scheme 1 Methods for 1,2-addition of fluoroalkyl iodides to alkenes and alkynes. | ||
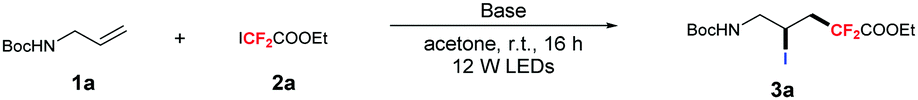
Accordingly, we conducted this reaction by treating tert-butyl allylcarbamate 1a and ethyl iododifluoroacetate 2a (1.5 equiv.) as model substrates. To our delight, 74% yield of the desired product 3a was obtained when the reaction was performed with K2CO3 (1.0 equiv.) in acetone and irradiated by blue LEDs for 16 hours. Further optimization of the inorganic bases demonstrated that other bases were less effective, and the desired product 3a was isolated in 93% yield when 2.0 equiv. of K2CO3 was used (Table 1, entries 1–4). No desired product was observed when control experiments were carried out in the dark, confirming the photochemical nature of the transformation (Table 1, entry 5). To our surprise, the product 3a at 49% yield was still obtained without bases (Table 1, entry 6). Finally, various short-wavelength LEDs were used as light sources to examine the influence of optical wavelength on the reaction, comparable yields were still obtained when LEDs with wavelengths between 425 and 475 nm were used, and the yields decreased with longer wavelengths (Table 1, entries 7–11). For other ketones, 82% yield was still obtained when 3-pentanone was used, and no reaction was observed using ketones with larger substituents (Table 1, entries 12–14). Those results indicate that the interactions between the solvent and fluoroalkyl iodides were decreased with the increase of the steric hindrance. Other solvents were also examined (Table 1, entries 15–18); no reaction occurred when the reaction was performed in dioxane or toluene, and 68% yield was obtained when MeCN was used as the solvent. The major products come from the atom transfer radical addition–elimination (ATRE) reaction when conducted in DMA or DMSO. We reason that noncovalent interactions also occurred between the solvent and RFI (Table 1, entries 17 and 18, for details, see the ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | LEDs (nm) | Solvent | Base (equiv.) | Yieldb (%) |
| a Reaction conditions (unless otherwise specified): 1a (0.3 mmol, 1.0 equiv.), 2a (0.45 mmol, 1.5 equiv.), acetone (2.0 mL), room temperature, 16 h. b NMR yield determined by 19F NMR using fluorobenzene as an internal standard and the number in parentheses is yield of isolated product. | ||||
| 1 | Blue (430–490) | Acetone | K2CO3 (1) | 74 |
| 2 | Blue (430–490) | Acetone | KOAc (1) | 64 |
| 3 | Blue (430–490) | Acetone | K2CO3 (2) | 99 (93) |
| 4 | Blue (430–490) | Acetone | Na2CO3 (2) | 81 |
| 5 | — | Acetone | K2CO3 (2) | 0 |
| 6 | Blue (430–490) | Acetone | — | 49 |
| 7 | Blue (450–455) | Acetone | K2CO3 (2) | 93 |
| 8 | Blue (470–475) | Acetone | K2CO3 (2) | 91 |
| 9 | Blue (490–495) | Acetone | K2CO3 (2) | 72 |
| 10 | Green (510–515) | Acetone | K2CO3 (2) | 67 |
| 11 | Green (545–550) | Acetone | K2CO3 (2) | 52 |
| 12 | Blue (430–490) | S-1 | K2CO3 (2) | 82 |
| 13 | Blue (430–490) | S-2 | K2CO3 (2) | 0 |
| 14 | Blue (430–490) | S-3 | K2CO3 (2) | 0 |
| 15 | Blue (430–490) | Dioxane | K2CO3 (2) | 0 |
| 16 | Blue (430–490) | MeCN | K2CO3 (2) | 68 |
| 17 | Blue (430–490) | DMA | K2CO3 (2) | 26 |
| 18 | Blue (430–490) | DMSO | K2CO3 (2) | 5 |
|
||||
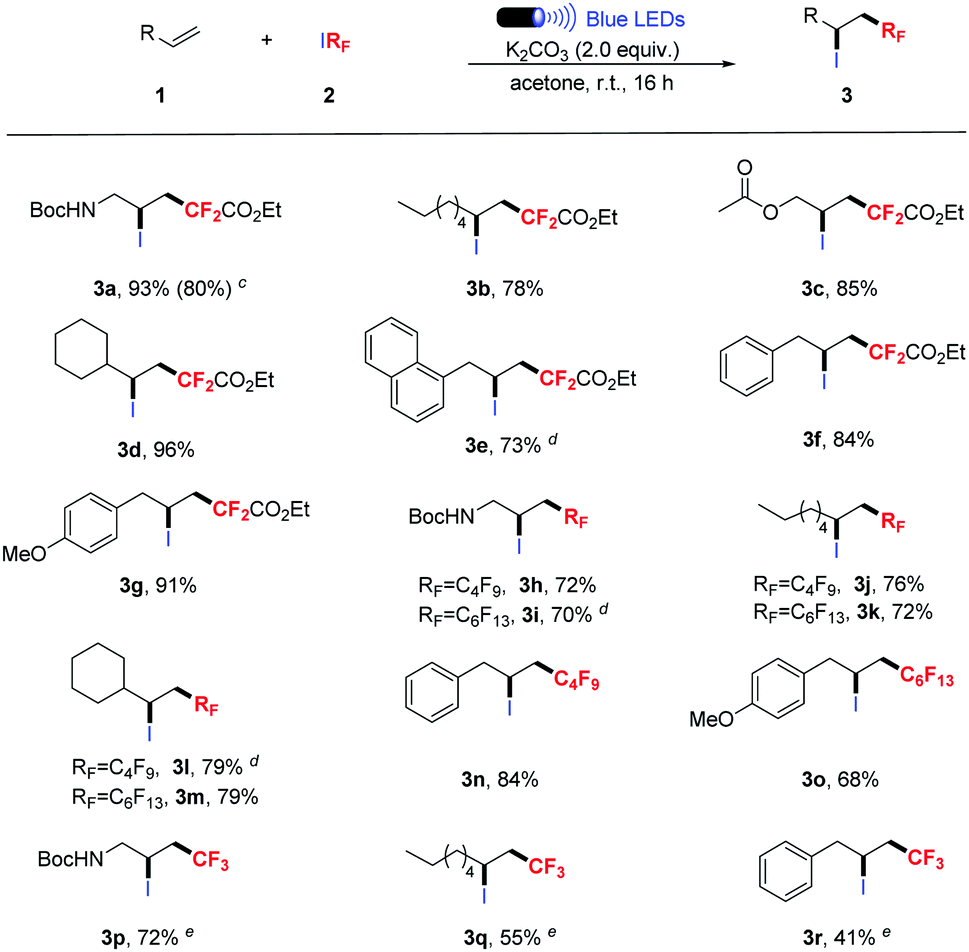
With the optimized reaction conditions in hand, the scope of this photochemical atom transfer radical addition was evaluated using abundant structurally diverse terminal alkenes.10 As shown in Table 2, functional groups such as amine, esters, naphthyl and methoxy were well-tolerated and provided difluoroalkylated ATRA products in good to excellent yields (Table 2, 3a–g). This reaction system is also amenable to the use of other commercial perfluoroalkyl iodides, such as C4F9I and C6F13I, and the corresponding products were obtained in moderate to good yields (Table 2, 3h–o). CF3I was less reactive, and moderate to good yields could be provided when 5.0 equiv. of CF3I was used (Table 2, 3p–r). Remarkably, when the reaction was performed on a gram scale (3a), an 80% yield was still obtained, demonstrating the synthetic utility of the protocol.
| a Reaction conditions: 1 (0.3 mmol, 1.0 equiv.), 2 (0.45 mmol, 1.5 equiv.), K2CO3 (0.6 mmol, 2.0 equiv.) in acetone (2.0 mL), room temperature, 12 W blue LEDs (430–490 nm), 16 h. b Yield of isolated product. c 1a (10 mmol, 1.0 equiv.), 2a (15 mmol, 1.5 equiv.), K2CO3 (2.0 equiv.), acetone (40 mL), room temperature, 24 W blue LED (430–490 nm), 48 h. d 2 (2.0 equiv.) was used. e CF3I (5.0 equiv.) was used. |
|---|
|
Then, we extended the substrate scope to a variety of terminal alkynes. Many important functional groups, such as halide, cyano, methoxy trifluoromethyl and even nitro underwent the process smoothly (Table 3). For aliphatic alkynes, high yield with low stereoselectivity was provided (Table 3, 5a–b). Phenylacetylenes were suitable substrates, and high stereoselectivity could be obtained (Table 3, 5c–g). Moreover, we found that the transformation presented good reactivity when C4F9I and C6F13I were employed, and provided the desired products in good yields (Table 3, 5h–k).
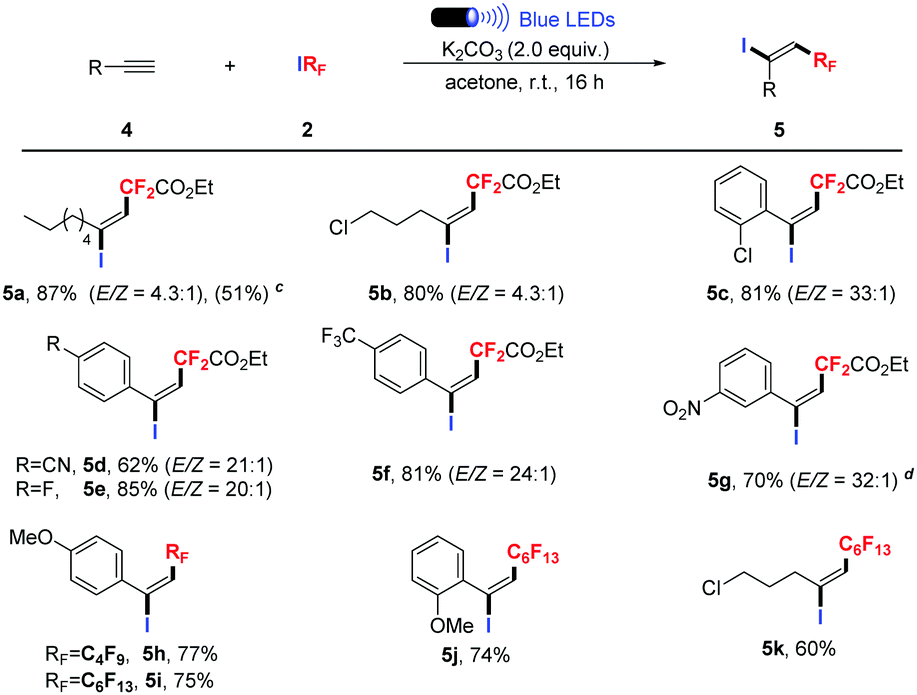
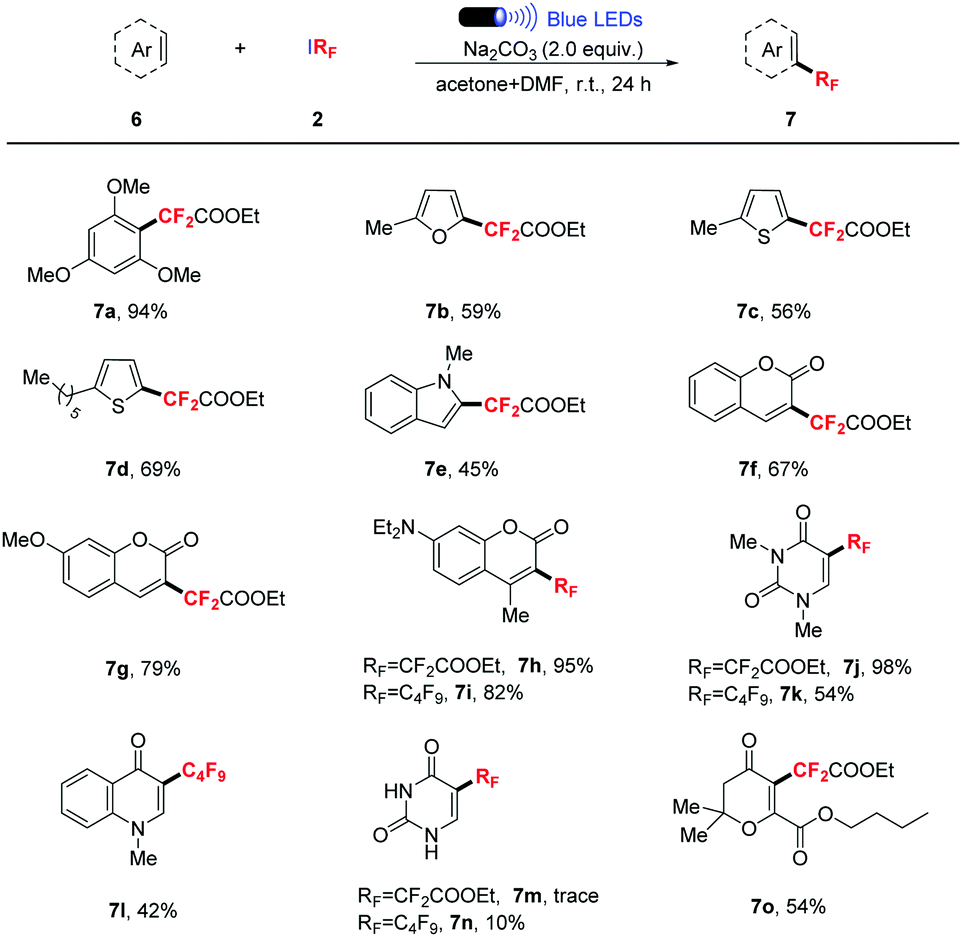
Encouraged by the results of the 1,2-addition of RFI to alkenes and alkynes, we proceeded to investigate direct fluoroalkylation of arenes.13 Pleasingly, by using DMF as a co-solvent, a wide range of arenes and heteroarenes underwent the catalyst-free fluoroalkylation smoothly. 94% yield was obtained when 1,3,5-trimethoxybenzene was treated (Table 4, 7a). Five membered heteroarenes such as furan, thiophene and pyrrole were relatively inert reaction partners, and only moderate yields were given (Table 4, 7b–e). Coumarins and 1,3-dimethyluracil were also suitable substrates for this transformation, delivering the desired products in good to excellent yields (Table 4, 7f–h, 7j). In addition, the C–H perfluoroalkylation of heteroarenes with C4F9I was also investigated, and moderate to good yields were still obtained (Table 4, 7i, 7k–l). Unprotected uracil failed to give satisfactory results (Table 4, 7m–n), which indicated that the mechanism of this transformation is different from our previous report.3f Moreover, butopyronoxyl was also a suitable substrate, and 54% yield was obtained (Table 4, 7o).
To gain insight into the mechanism of this transformation, a series of experiments were conducted. The reaction was suppressed by the addition of a radical scavenger TEMPO (100 mol%) and only 15% yield of 3a was obtained, which suggests that the involvement of radical intermediates is likely during the reaction (for details, see the ESI†). A radical clock experiment employing alpha-cyclopropylstyrene 8 as a substrate produced the ring-opening product 9 in 82% yield (for details, see the ESI†). Optical absorption spectra of the reactants found that the absorption was clearly strengthened when 2a and acetone were mixed, and those results indicate that non-covalent interactions occurred between acetone and 2a (Fig. 1, for details, see the ESI†). In addition, this conclusion was further confirmed by a Job's plot. Finally, the light–dark experiment was performed using alternative intervals of light and dark, and the formation of the product was both feasible during periods of irradiation and dark, which is suggestive of a radical chain process (for details, see the ESI†).
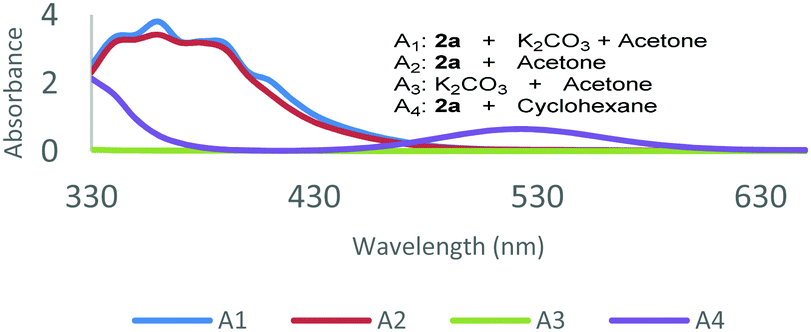 | ||
| Fig. 1 Optical absorption spectra studies. | ||
Based on these preliminary results, a plausible mechanism for this transformation was proposed in Scheme 2. First, non-covalent interactions occurred between acetone and the fluoroalkyl iodides. Then, a fluoroalkyl radical was generated under the irradiation of blue LEDs. Subsequent regioselective addition of ˙RF to alkenes or alkynes (1 or 4) lead to the carbon-radical intermediate (B), which abstracted an iodine atom from RFI to afford the desired product 3 or 5 and regenerated the RF radical. For arenes, cation species were generated via a SET process from an intermediate C, and the fluoroalkylated products (7) were obtained by further deprotonation.
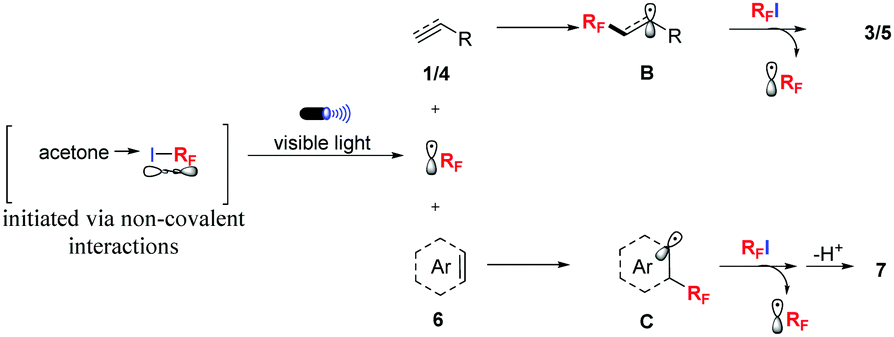 | ||
| Scheme 2 Proposed plausible reaction mechanism. | ||
In summary, we have developed a simple, mild, and efficient protocol for photochemical fluoroalkylation reactions via noncovalent interactions between acetone and fluoroalkyl iodides. The significant advantages of this method are high atom economy, excellent functional group tolerance, and synthetic simplicity, thus providing a facile route for further application in pharmaceuticals and life sciences. Mechanistic studies indicate that the reaction was initiated via non-covalent interactions between acetone and carbon–iodine bonds.
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21702241, and 81760624), and Programs of Guizhou Province (No. 2017-1225, 2018-1427). We thank Dr Jiwei Gu and Dr Yanbo Yu from Washington University for their linguistic assistance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (d) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; (e) D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (f) C. S. Burgey, K. A. Robinson, T. A. Lyle, P. E. J. Sanderson, S. Dale Lewis, B. J. Lucas, J. A. Krueger, R. Singh, C. Miller-Stein, R. B. White, B. Wong, E. A. Lyle, P. D. Williams, C. A. Coburn, B. D. Dorsey, J. C. Barrow, M. T. Stranieri, M. A. Holahan, G. R. Sitko, J. J. Cook, D. R. McMasters, C. M. McDonough, W. M. Sanders, A. A. Wallace, F. C. Clayton, D. Bohn, Y. M. Leonard, T. J. Detwiler, Jr, J. J. Lynch, Jr, Y. Yan, Z. Chen, L. Kuo, S. J. Gardell, J. A. Shafer and J. P. Vacca, J. Med. Chem., 2003, 46, 461 CrossRef CAS; (g) G. L. Grunewald, M. R. Seim, J. Lu, M. Makboul and K. R. Criscione, J. Med. Chem., 2006, 49, 2939 CrossRef CAS; (h) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS.
- For selected reviews, see: (a) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (b) C. Alonso, E. M. Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (d) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed; (e) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (f) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264 CrossRef CAS PubMed; (g) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS; (h) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676 CrossRef CAS PubMed; (i) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718 CrossRef CAS PubMed; (j) X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS; (k) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS.
- For representative papers, see: (a) M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921 CrossRef CAS; (b) V. M. Fernández-Alvarez, M. Nappi, P. Melchiorre and F. Maseras, Org. Lett., 2015, 17, 2676 CrossRef; (c) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485 CrossRef CAS; (d) Q. Guo, M. Wang, H. Liu, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 4747 CrossRef CAS; (e) Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS; (f) Y. Huang, Y.-Y. Lei, L. Zhao, J. Gu, Q. Yao, Z. Wang, X.-F. Li, X. Zhang and C.-Y. He, Chem. Commun., 2018, 54, 13662 RSC; (g) Y. Liu, X.-L. Chen, K. Sun, X.-Y. Li, F.-L. Zeng, X.-C. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Lett., 2019, 21, 4019 CrossRef CAS; (h) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Chem. Commun., 2018, 54, 12710 RSC; (i) G.-R. Park, Y. Choi, M. G. Choi, S.-K. Chang and E. J. Cho, Asian J. Org. Chem., 2017, 6, 436 CrossRef CAS; (j) Y. Cheng, X. Yuan, J. Ma and S. Yu, Chem. – Eur. J., 2015, 21, 8355 CrossRef CAS PubMed; (k) D. Zheng and A. Studer, Org. Lett., 2019, 21, 325 CrossRef CAS ; For review, see: ; (l) A. Postigo, Eur. J. Org. Chem., 2018, 6391 CrossRef CAS.
- X. Fang, X. Yang, X. Yang, S. Mao, Z. Wang, G. Chen and F. Wu, Tetrahedron, 2007, 63, 10684 CrossRef CAS.
- A.-R. Li and Q.-Y. Chen, Synthesis, 1997, 333 CrossRef CAS.
- (a) Y. Takeyama, Y. Ichinose, K. Oshima and K. Utimoto, Tetrahedron Lett., 1989, 30, 3159 CrossRef CAS; (b) Y. Li, H. Li and J. Hu, Tetrahedron, 2009, 65, 478 CrossRef CAS.
- (a) K. Tsuchii, M. Imura, N. Kamada, T. Hirao and A. Ogawa, J. Org. Chem., 2004, 69, 6658 CrossRef CAS; (b) R. Beniazza, L. Remisse, D. Jardel, D. Lastecoueres and J.-M. Vincent, Chem. Commun., 2018, 54, 7451 RSC.
- (a) T. Xu, C. W. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910 CrossRef CAS PubMed; (b) G. Wu and A. J. von Wangelin, Chem. Sci., 1795, 2018, 9 Search PubMed.
- (a) T. Yajima and M. Ikegami, Eur. J. Org. Chem., 2017, 2126 CrossRef CAS; (b) F. Sladojevich, E. McNeill, J. Börgel, S.-L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712 CrossRef CAS PubMed; (c) G. Magagnano, A. Gualandi, M. Marchini, L. Mengozzi, P. Ceroni and P. G. Cozzi, Chem. Commun., 2017, 53, 1591 RSC; (d) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed; (e) Y. Du, R. M. Pearson, C.-H. Lim, S. M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem. – Eur. J., 2017, 23, 10962 CrossRef CAS PubMed; (f) R. Beniazza, R. Atkinson, C. Absalon, F. Castet, S. A. Denisov, N. D. McClenaghan, D. Lastecoueres and J.-M. Vincent, Adv. Synth. Catal., 2016, 358, 2949 CrossRef CAS; (g) S. Kim, G. Park, E. J. Cho and Y. You, J. Org. Chem., 2016, 81, 7072 CrossRef CAS PubMed.
- C. Rosso, J. D. Williams, G. Filippini, M. Prato and C. O. Kappe, Org. Lett., 2019, 21, 5341 CrossRef CAS PubMed.
- E. Zhu, X.-X. Liu, A.-J. Wang, T. Mao, L. Zhao, X. Zhang and C.-Y. He, Chem. Commun., 2019, 55, 12259 RSC.
- (a) L. Zhao, Y. Huang, Z. Wang, E. Zhu, T. Mao, J. Jia, J. Gu, X.-F. Li and C.-Y. He, Org. Lett., 2019, 21, 6705 CrossRef CAS; (b) L. Helmecke, L. Spittler, K. Baumgarten and C. Czekelius, Org. Lett., 2019, 21, 7823 CrossRef CAS PubMed.
- For recent examples on direct fluoroalkylation of arenes and heteroarenes, see: (a) M. J. Hossain, T. Ono, K. Wakiya and Y. Hisaeda, Chem. Commun., 2017, 53, 10878 RSC; (b) L. Wang, X.-J. Wei, W.-L. Jia, J.-J. Zhong, L.-Z. Wu and Q. Liu, Org. Lett., 2014, 16, 5842 CrossRef CAS PubMed; (c) W. Yu, X.-H. Xu and F.-L. Qing, Org. Lett., 2016, 18, 5130 CrossRef CAS; (d) J. Jung, E. Kim, Y. You and E. J. Cho, Adv. Synth. Catal., 2014, 356, 2741 CrossRef CAS; (e) X. Wang, S. Zhao, J. Liu, D. Zhu, M. Guo, X. Tang and G. Wang, Org. Lett., 2017, 19, 4187 CrossRef CAS PubMed; (f) C.-H. Qu, G.-T. Song, J. Xu, W. Yan, C.-H. Zhou, H.-Y. Li, Z.-Z. Chen and Z.-G. Xu, Org. Lett., 2019, 21, 8169 CrossRef CAS PubMed; (g) S. Zhang, N. Rotta-Loria, F. Weniger, J. Rabeah, H. Neumann, C. Taeschler and M. Beller, Chem. Commun., 2019, 55, 6723 RSC; (h) B. Yang, D. Yu, X.-H. Xu and F.-L. Qing, ACS Catal., 2018, 8, 2839 CrossRef CAS; (i) C. Yuan, L. Zhu, Y. Lan and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1277 CrossRef CAS PubMed; (j) C.-Y. He, J. Kong, X. Li, X. Li, Q. Yao and F.-M. Yuan, J. Org. Chem., 2017, 82, 910 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09517a |
| ‡ T. Mao and M.-J. Ma contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |