
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The partial dehydrogenation of aluminium dihydrides†
Thomas N.
Hooper
 a,
Samantha
Lau
a,
Wenyi
Chen
a,
Ryan K.
Brown
a,
Martí
Garçon
a,
Karen
Luong
a,
Nathan S.
Barrow
b,
Andrew S.
Tatton‡
c,
George A.
Sackman
df,
Christopher
Richardson
e,
Andrew J. P.
White
a,
Richard I.
Cooper
f,
Alison J.
Edwards
d,
Ian J.
Casely
b and
Mark R.
Crimmin
*a
a,
Samantha
Lau
a,
Wenyi
Chen
a,
Ryan K.
Brown
a,
Martí
Garçon
a,
Karen
Luong
a,
Nathan S.
Barrow
b,
Andrew S.
Tatton‡
c,
George A.
Sackman
df,
Christopher
Richardson
e,
Andrew J. P.
White
a,
Richard I.
Cooper
f,
Alison J.
Edwards
d,
Ian J.
Casely
b and
Mark R.
Crimmin
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, 80 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
bJohnson Matthey Technology Centre, Blounts Court, Sonning Common, Reading, RG4 9NH, UK
cDepartment of Materials, University of Oxford, OX1 3PH, UK
dAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation, Australia
eUniversity of Wollongong, Wollongong, NSW 2522, Australia
fChemical Crystallography, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK
First published on 13th August 2019
Abstract
The reactions of a series of β-diketiminate stabilised aluminium dihydrides with ruthenium bis(phosphine), palladium bis(phosphine) and palladium cyclopentadienyl complexes is reported. In the case of ruthenium, alane coordination occurs with no evidence for hydrogen loss resulting in the formation of ruthenium complexes with a pseudo–octahedral geometry and cis-relation of phosphine ligands. These new ruthenium complexes have been characterised by multinuclear and variable temperature NMR spectroscopy, IR spectroscopy and single crystal X-ray diffraction. In the case of palladium, a series of structural snapshots of alane dehydrogenation have been isolated and crystallographically characterised. Variation of the palladium precursor and ligand on aluminium allows kinetic control over reactivity and isolation of intermetallic complexes that contain new Pd–Al and Pd–Pd interactions. These complexes differ by the ratio of H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al (2:1, 1.5:1 and 1:1) with lower hydride content species forming with dihydrogen loss. A combination of X-ray and neutron diffraction studies have been used to interrogate the structures and provide confidence in the assignment of the number and position of hydride ligands. 27Al MAS NMR spectroscopy and calculations (DFT, QTAIM) have been used to gain an understanding of the dehydrogenation processes. The latter provide evidence for dehydrogenation being accompanied by metal–metal bond formation and an increased negative charge on Al due to the covalency of the new metal–metal bonds. To the best of our knowledge, we present the first structural information for intermediate species in alane dehydrogenation including a rare neutron diffraction study of a palladium–aluminium hydride complex. Furthermore, as part of these studies we have obtained the first SS 27Al NMR data on an aluminium(I) complex. Our findings are relevant to hydrogen storage, materials chemistry and catalysis.
:Al (2:1, 1.5:1 and 1:1) with lower hydride content species forming with dihydrogen loss. A combination of X-ray and neutron diffraction studies have been used to interrogate the structures and provide confidence in the assignment of the number and position of hydride ligands. 27Al MAS NMR spectroscopy and calculations (DFT, QTAIM) have been used to gain an understanding of the dehydrogenation processes. The latter provide evidence for dehydrogenation being accompanied by metal–metal bond formation and an increased negative charge on Al due to the covalency of the new metal–metal bonds. To the best of our knowledge, we present the first structural information for intermediate species in alane dehydrogenation including a rare neutron diffraction study of a palladium–aluminium hydride complex. Furthermore, as part of these studies we have obtained the first SS 27Al NMR data on an aluminium(I) complex. Our findings are relevant to hydrogen storage, materials chemistry and catalysis.
Introduction
Metal aluminium hydrides (e.g. MAlH4, M = Li, Na, K) have been proposed as hydrogen storage materials.1 For example, LiAlH4 contains a high weight percentage of hydrogen and can undergo fast thermal dehydrogenation when doped with a transition metal, such as TiCl3, to form LiH and Al(s).2,3 While the practical use of LiAlH4 for hydrogen storage remains a point of debate, the dehydrogenation step itself is of significant interest to materials and synthetic chemists alike. For example, very recently it has been shown that both the shape and size of aluminium nanocrystals formed during the dehydrogenation of AlH3·L can be controlled through careful selection of the ligand, L, on aluminium.4The dehydrogenation of main group hydride compounds holds promise as a route to low-oxidation state intermediates. This approach has several advantages over current methods. The dehydrogenation of group 13 hydrides is potentially reversible through controlling the pressure of dihydrogen. It does not rely on the use of harsh reagents such as K or KC8 typical for metal dihalide reduction. Furthermore, many of the dihydride precursors are readily accessible on large scales through reactions of inexpensive and commercial hydrides such as LiAlH4 with suitable ligand precursors.
Although surprisingly little is known about the dehydrogenation of molecular aluminium hydrides,5–7 the microscopic reverse, the hydrogenation of low oxidation state aluminium compounds, is increasingly common. Dihydrogen undergoes oxidative addition to a handful of aluminium(I) compounds under mild conditions in solution.8–10 In a more extreme environment, Al4H6 has been generated by rapidly vaporising aluminium metal in the presence of dihydrogen. This cluster has been characterised as its anion in the gas-phase by photoelectron spectroscopy and shown to be just one of a broader series of molecules including Al4H4 and AlnHn+2 (n = 4–8).11,12
As stated, precedent for the dehydrogenation step itself is limited. The formation of complexes containing {Al2H4} and {Al6H6} fragments following reduction of aluminium(III) hydrides with a magnesium reagent has been reported.13,14 We have invoked the dehydrogenation of β-diketiminate stabilised aluminium(III) dihydrides to form aluminium(I) ligands during the palladium catalysed transformation of C–H into C–Al bonds.15,16 The hypothesis is supported by the observation that analogous gallium(III) dihydrides react with transition metal complexes by dehydrogenation (Fig. 1).17–19 In related studies, mesityl borane (MesBH2) has been shown to undergo reversible dehydrogenation on a ruthenium bis(phosphine) fragment to form a ruthenium borylene complex,20 while the aminoborane i-Pr2N–BH2 can be dehydrogenated with similar transition metal complexes provided a hydrogen acceptor (tert-butylethylene) is present.21
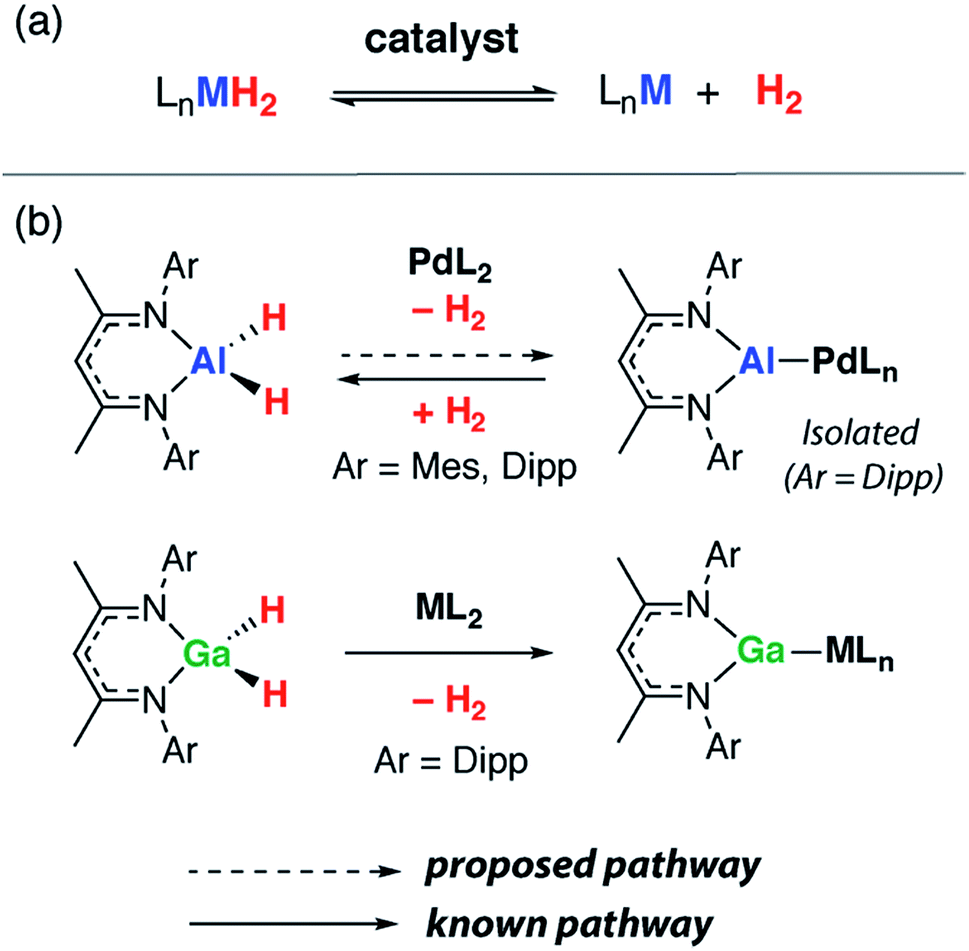 | ||
| Fig. 1 (a) Reversible dehydrogenation to form low-valent main group complexes. (b) Dehydrogenation of β-diketiminate Ga and Al dihydrides. (Mes = 2,4,6-trimethylphenyl, Dipp = 2,6-di-iso-propylphenyl). | ||
In this paper, we report a series of reactions that result in the partial dehydrogenation of the β-diketiminate stabilised aluminium dihydrides using transition metal complexes. While Ru—Al bimetallics involve expected three-dimensional geometries and show little sign of dihydrogen loss, related palladium compounds contain densely packed two-dimensional arrays of metal atoms bridged by hydride ligands. Although the complete dehydrogenation to form an aluminium(I) intermediate (or coordination complex thereof) is yet to be observed, we have isolated a series of complexes that contain different H:Al ratios (2:1 → 1:1) due to partial dehydrogenation. DFT calculations suggest that dehydrogenation is accompanied by the formation of covalent metal–metal bonds along with an associated increase in the negative charge on the Al centres; an effect that could be interpreted as lowering the formal oxidation state due to electron transfer to Al.
Results and discussion
Reactions of alanes with ruthenium complexes
The reactions of two aluminium dihydrides (1a–b) with a ruthenium bis(phosphine) complex were conducted. The choice of the metal fragment was dictated by the observation that closely related fragments have been shown to effect the reversible dehydrogenation of boranes.20 Addition of 1 equiv. of [Ru(H)2(N2)2(PCy3)2] to 1 equiv. of 1a proceeded cleanly at 25 °C to generate the aluminium ruthenium heterobimetallic hydride complex 2 (Scheme 1).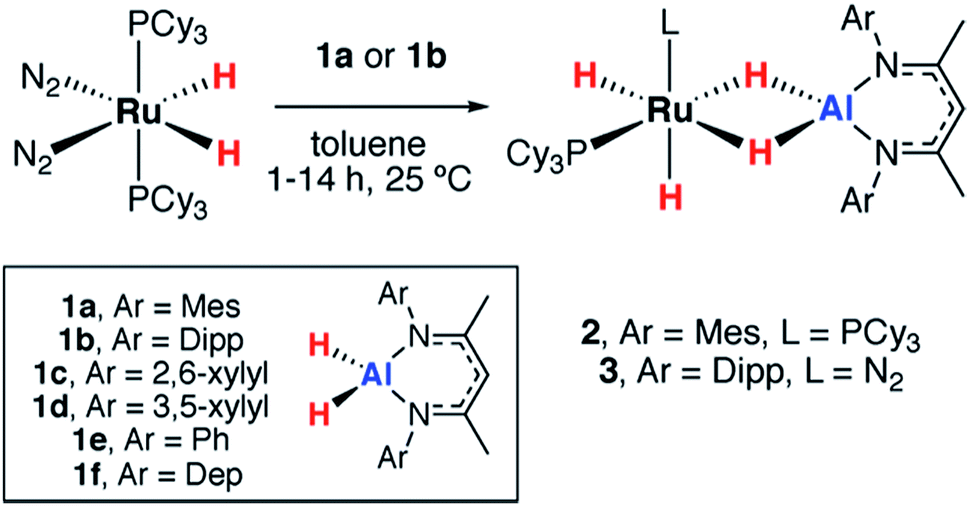 | ||
| Scheme 1 Reactions of ruthenium complexes with aluminium dihydrides. (Dep = 2,6-diethylphenyl). | ||
2 was characterised by a broad peak at δH = −11.77 ppm (fwhm = 1021 Hz) for all four hydrides in the 1H NMR spectrum at 293 K. Upon cooling to 273 K the hydride resonance decoalesced into two resonances at δH = −9.69 (fwhm = 77 Hz) and −13.49 (fwhm = 101 Hz) ppm assigned to the terminal and bridging hydrides respectively. At lower temperatures, a second fluxional process resolved at 213 K with four broad peaks observed at δH = −9.32 (fwhm = 84 Hz), −9.91 (fwhm = 77 Hz), −12.45 (fwhm = 217 Hz) and −13.86 (fwhm = 198 Hz) ppm in the 1H NMR spectrum for the four magnetically inequivalent hydrides. Long T1(min) = 247 ms were measured for the hydride resonances at 313 K at 400 MHz, indicative of classical hydride behavior. At 293 K only one resonance was observed in the 31P{1H} NMR at δP = 69.9 ppm which upon cooling the reaction to 213 K decoalesced into two broad peaks observed at δP = 70.7 and 66.2 ppm. No 2JP–H coupling could be resolved.
The data are consistent with at least one fluxional process operating which results in exchange of the hydride positions.22 The low temperature NMR spectroscopy data support the assignment of 2 in which the PCy3 ligands adopt a cis-arrangement. Although single crystals suitable for X-ray diffraction could not be obtained, the structure can confidently be assigned based on the multinuclear NMR data and comparison to the literature. Related σ-silane complexes have been reported by Sabo-Etienne and coworkers,23–25 while we recently disclosed a series of Ru–M (M = Al, Zn, Mg) complexes with similar coordination geometries.26
A further reaction of 1 equiv. of [Ru(H)2(N2)2(PCy3)2] with 1 equiv. of 1b proceeded cleanly at 25 °C to generate 3 (Scheme 1). This complex demonstrated three resonances for the hydride environments at 25 °C in the 1H NMR spectrum, δH = −9.05 (br s), −9.73 (d, 2JP–H = 41.6 Hz), −16.11 (d, 2JP–H = 15.8 Hz) ppm. At 233 K, the resonance at δH = −9.05 ppm decoalesced into two peaks at δH = −7.83 and −10.49 ppm. T1 measurements were taken across the 333–193 K range at 400 MHz and the four hydride signals exhibited a T1(min) between 200–250 ms consistent with their assignment as classical hydrides.27,28 Monitoring the reaction between [Ru(H)2(N2)2(PCy3)2] and 1b by 31P{1H} NMR spectroscopy revealed the formation of PCy3, δP = 9.9 ppm. The structure of 3 was ultimately confirmed by X-ray crystallography (Fig. 2). While the single crystal X-ray data are discussed in detail below, it is pertinent to note that, in contrast to 2, 3 contains only a single coordinated PCy3 ligand. Dissociation of 1 equiv. of PCy3 is accompanied by coordination of N2 to Ru. The presence of a dinitrogen ligand in 3 was confirmed by IR spectroscopy with a strong absorbance at νN–N = 2135 cm−1 and the corresponding hydride stretches at νRu–H = 1908 and 1872 cm−1.
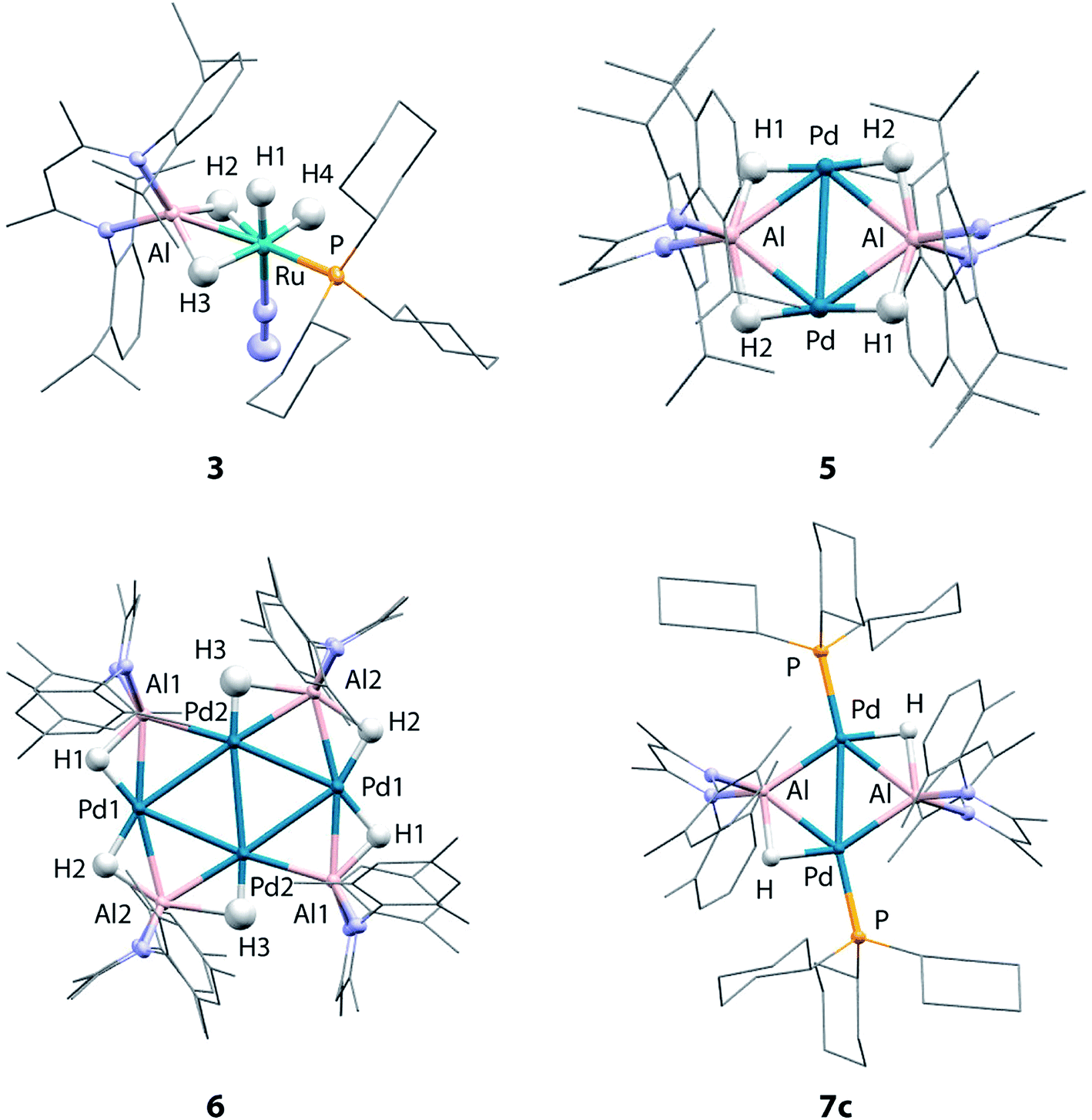 | ||
| Fig. 2 Crystal structures of 3, 5, 6 and 7c. | ||
Reactions of alanes with palladium complexes
The reaction of [Pd(PCy3)2] with 1a in toluene-d8 at 25 °C quickly forms an equilibrium mixture due to reversible exchange of the phosphine and alane ligands (Scheme 2). The reaction was monitored using variable temperature NMR spectroscopy between 193 and 373 K. In the low temperature regime (below 203 K) two broad hydride resonances are observed at δ = −0.62 (d, 2JP–H = 84 Hz) and +4.90 ppm attributed to bridging and terminal hydrides respectively in the σ-complex 4 (see ESI† for spectra). A ROESY experiment conducted at 193 K revealed chemical exchange between these two resonances, while their assignment as hydrides was unambiguously confirmed by repeating the experiment with the deuteride 1a-d2. The large 2JP–H coupling was confirmed by not only obtaining the 1H{31P} spectrum which shows selective decoupling but also the 31P NMR spectrum which shows a resonance at δ = 37.9 ppm (d, 2JP–H = 84 Hz). In the high temperature regime (above 223 K), the diagnostic transition metal hydride resonance is no longer observed, suggesting a shift of the equilibrium back toward the starting materials. The 2JP–H coupling constant of 84 Hz is significantly larger than the typical range of 0–20 Hz expected for Pd complexes with a cis relationship between the hydrogen and phosphorus atoms.29,30 Nevertheless, this J value is still smaller than the range of 150–200 Hz established for crystallographically characterised square-planar palladium complexes involving a trans2JP–H coupling.31–33 An equivalent reaction using 1f also gave a low intensity broad doublet resonance at δ = −0.61 ppm (2JH–P ≈ 85 Hz) in the low temperature 1H NMR spectrum at 193 K which resolved to a broad singlet in the 1H{31P} NMR spectrum (see ESI†). Mixing [Pd(PCy3)2] with 1b did not give an observable reaction by NMR spectroscopy, even at low temperatures, likely due to the increased steric bulk around the alane fragment.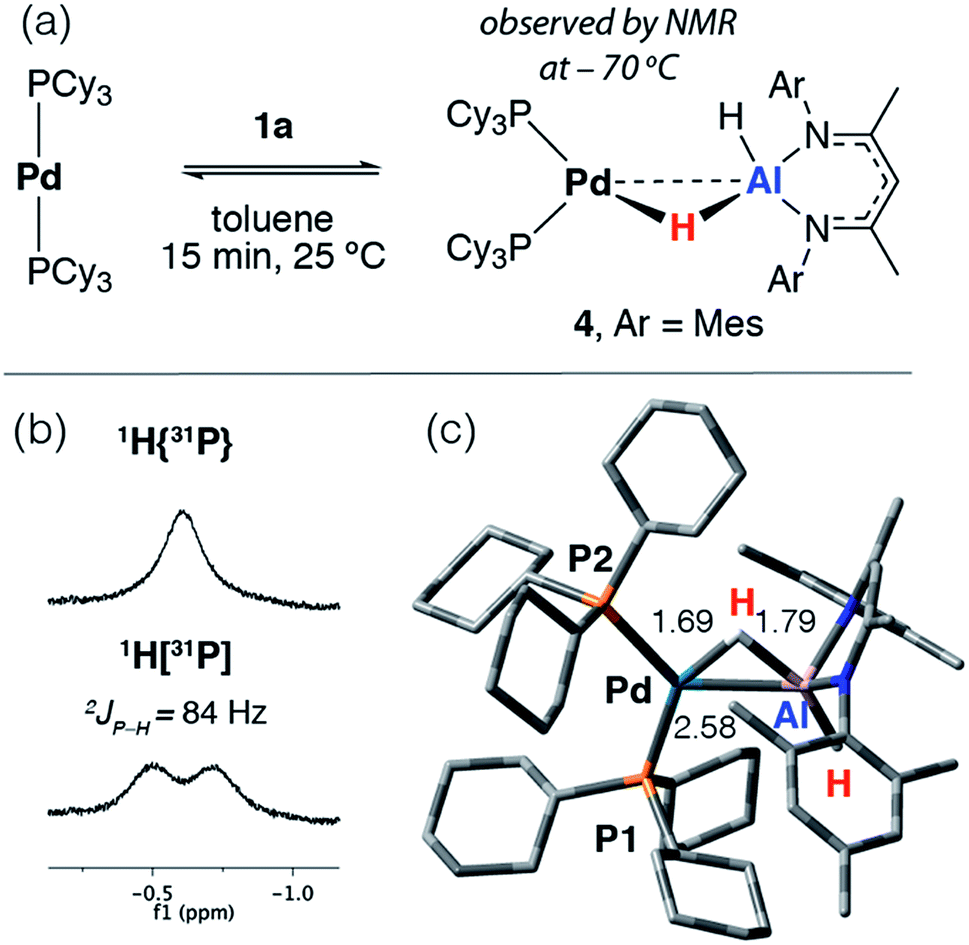 | ||
| Scheme 2 (a) Reversible reaction of 1a with [Pd(PCy3)2]. (b) Hydride region of the 1H NMR spectrum at −70 °C in toluene-d8. (c) Calculated structure of 4 with selected bond lengths in Å. | ||
The simplest explanation for these data is an associative process that forms the three-coordinate σ-alane complex 4. We have isolated σ-alane complexes based on d10 Cu(I) fragments that are isoelectronic to [Pd(PCy3)2] and provided a detailed analysis of their electronic structure.34,35 In the current case, it appears that the σ-alane species is only stable at low temperature due to a weak binding event combined with an expected unfavourable reaction entropy. DFT calculations were used to interrogate the thermodynamics of ligand exchange and the formation of 4 was found to be modestly exergonic, consistent with the reversible process described above (see ESI† for details). The nature and magnitude of the 2JP–H coupling in 4 are best described by considering the calculated geometry in which the bond angles approach those associated with trans (P1–Pd–H = 150°) and cis (P2–Pd–H = 87°) geometries. Due to line broadening effects it appears that only the larger of 2JP–H couplings, associated with the trans-like relation between the hydride atom and a single PCy3 ligand, is resolved in the low temperature NMR data.
Related reversible processes involving the addition of silanes and germanes to [Pd2(dcpe)2] (dcpe = 1,2-bis(dicyclohexylphosphino)ethane) have been reported to form [Pd(dcpe)(H)(ER3)] (E = Si, Ge).36,37 In these instances, σ-silane or σ-germane complexes are invoked only as unstable intermediates in reversible redox processes that involve the oxidative addition and reductive elimination of the E–H bond to the palladium centre. Despite the precedent for 4, at this time we cannot unambiguously rule out its assignment as a linear two coordinate complex formed from phosphine dissociation from Pd. Indeed, based on the calculated thermodynamics of phosphine dissociation (see ESI†) both the latter species and 4 may be formed as part of an equilibrium mixture.
Further evidence for the interaction of Al–H bonds with the Pd centre derives from H/D exchange reactions. The Al–H bonds of 1a and 1c were observed to undergo H/D scrambling when in the presence of [Pd(PCy3)2] and C6D6 under ambient conditions. Storage of a C6D6 solution of 1c with a catalytic amount (5 mol%) of [Pd(PCy3)2] resulted in loss of the broad Al–H resonance at δ = 4.59 ppm in the 1H NMR spectrum and a corresponding increase (compared to a ferrocene standard in a capillary) of the residual solvent peak for C6D5H after 5 days. Removal of the solvent and dissolving in C6H12 (to prevent further reaction) allowed collection of the 2H NMR spectrum which showed the largest peak to be a broad resonance at δ = 4.39 ppm, corresponding with the Al–D resonance of 1c-d2.
We have previously reported that, upon standing at 25 °C solutions of [Pd(PCy3)2] and 1a gradually yield red crystals of 7a complex (Scheme 3) as a result of the non-reversible dimerization and dehydrogenation of 4.157a is insoluble in hydrocarbon solution and it appears the crystallisation may play a role in driving the equilibria towards this product. Modification of the palladium and alane precursors allowed identification of similar non-reversible reactivity and isolation of a series of remarkable intermetallic complexes all involving Pd–H–Al interactions but differing in the ratio of H:Al due to partial dehydrogenation. Hence, the reaction between 1b and [Pd(η5-C5H5)(η3-C3H4Ph)] yielded the tetrametallic complex 5 with a 2:1 ratio of H:Al, while addition of 1a to [Pd(η5-C5H5)(η1-C3H4Ph)(IMes)] (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene) led to the isolation of the octametallic cluster 6 with a 1.5:1 ratio of H:Al (Scheme 3). Dihydrogen was observed as a low intensity resonance at δ = 4.47 ppm in the 1H NMR spectra of the reaction mixtures of 6 and 7c which disappeared upon degassing of the sample.
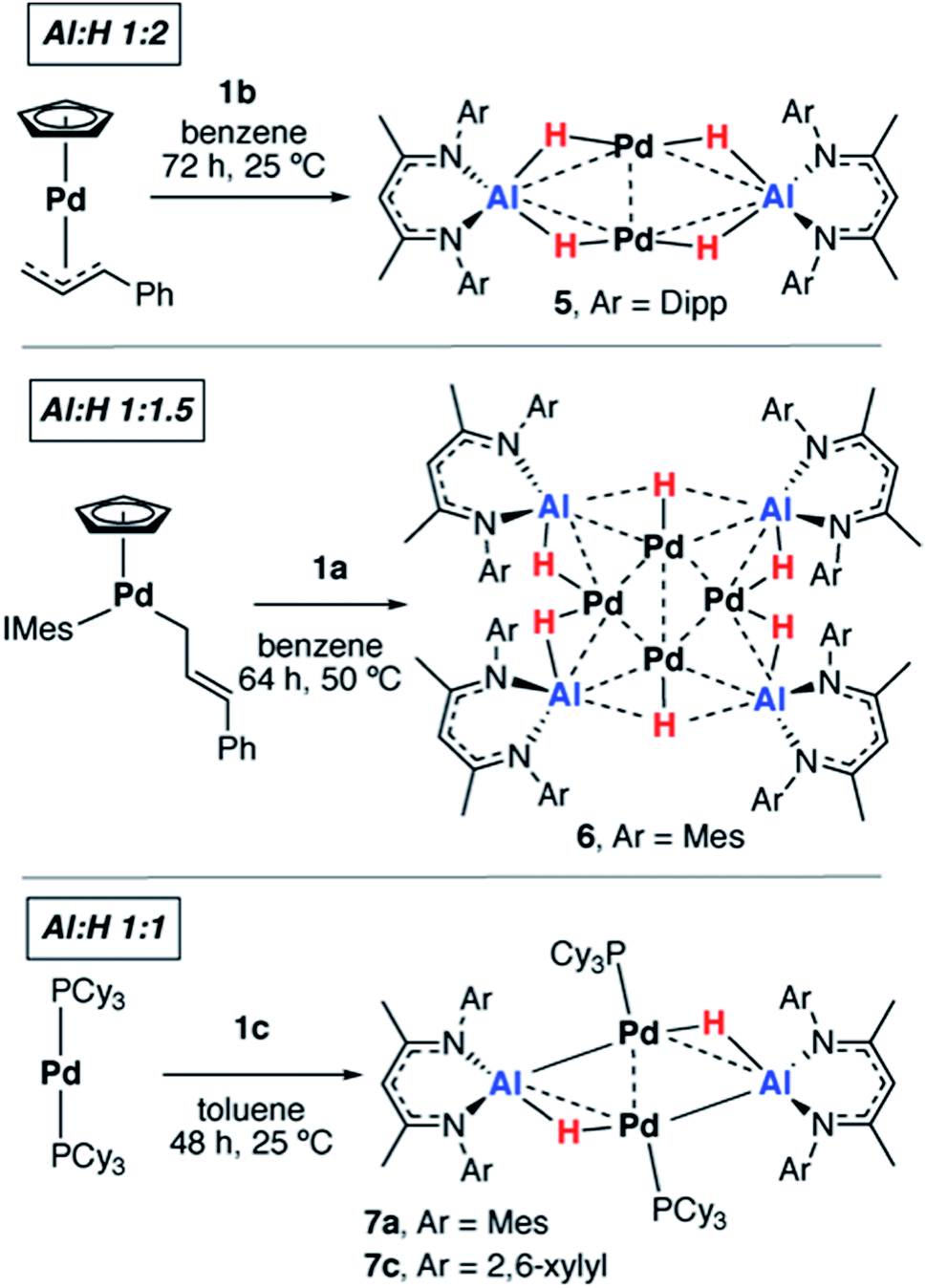 | ||
| Scheme 3 Non-reversible reactions of 1a–c with palladium precursors. | ||
A systematic investigation of the alane (1a–f, Scheme 1) in the reactions involving [Pd(PCy3)2] showed that the dehydrogenation to form complexes with a 1:1 ratio of H:Al was highly dependent on the steric demands of the ligand (Scheme 3). 1c reacted with [Pd(PCy3]2 to give red crystals of 7c, an exact analogue of the mesityl substituted derivative 7a. 1f reacted with [Pd(PCy3)2] at 60 °C (160 h) to give an orange solution with a 31P{1H} NMR spectrum displaying broad peaks at δ = 38.9 and 9.8 ppm for [Pd(PCy3)2] and free PCy3 respectively and a new resonance at δ = 40.1 ppm. Dihydrogen was observed in the 1H NMR spectrum which along with free PCy3 suggests a product similar to 7c but we were unable to isolate a pure solid or single-crystals from this reaction, preventing characterisation of this product. Further reactions of 1a–b with [Pd(PtBu3)2] or [Pd(PPh3)4] led to the formation of non-crystalline insoluble material and complex reaction mixtures respectively. There was no apparent reaction between 1b and [Pd(PCy3)2] after heating to 80 °C for 120 h. Addition of 1d and 1e with [Pd(PCy3)2] led to the formation of complex mixtures. Although the complete product distribution could not be determined, a few single crystals obtained from the reaction of 1e with [Pd(PCy3)2] allowed identification of a decomposition pathway involving hydride transfer from the metals to the electrophilic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N position of the backbone of the ligand system (see ESI†). It appears that ortho-substituents on the aryl groups of the ligand are a necessary requirement to block the electrophilic site and prevent this mode of decomposition.
N position of the backbone of the ligand system (see ESI†). It appears that ortho-substituents on the aryl groups of the ligand are a necessary requirement to block the electrophilic site and prevent this mode of decomposition.
The new Pd–Al species are insoluble in common laboratory solvents (benzene, toluene, hexane, THF, Et2O) preventing acquisition of complete NMR data. Nevertheless, the partial solubility of 5 allowed the hydride resonances to be located at δ = –2.17 ppm (fwhm = 50 Hz) in C6D6 at 298 K provided the 1H{27Al} spectrum was obtained. All new species possess the expected complex set of IR stretches below 1600 cm−1 and, consistent with previous observations,7 the bridging nature of the Al–H–Pd interaction shifts the hydride vibrations into the fingerprint region of the spectra.
Solid-state structures
Although the low solubility of 5, 6 and 7c in standard solvents precluded solution characterisation, these complexes have been characterised by single crystal X-ray diffraction (Fig. 2). In all four structures, the hydride ligands were located in the difference map and the positions refined freely. Where possible their presence has been confirmed by other spectroscopic or chemical methods. In the Pd/Al complexes 5, 6 and 7c the only hydride ligands were found to bridge Pd–Al edges and to lie in the plane of the metal centres. Crystallographically characterised structures of Pd dimers and cluster complexes containing Pd⋯H⋯Pd bridging interactions have Pd–Pd distances similar to those found in 5, 6 and 7c,38–40 but we do not find evidence of edge-bridging or face-capping Pd⋯H⋯Pd hydrides in the complexes reported herein. DFT studies have been carried out to confirm the presence and position of the crystallographically assigned hydride ligands (vide infra) with structural optimisation showing good correlation between the experimental and calculated structures.41The structure of 3 confirms the loss of one PCy3 ligand from the starting material as indicated by the solution studies. The Ru(II) centre is in a distorted octahedral environment with one PCy3 ligand, one terminally bound dinitrogen, and the alane bound through both hydrides. Two terminal hydrides complete the coordination sphere. The PCy3 ligand is trans to one of the Al bound hydrides. The Ru–Al distance of 2.3037(9) Å is within the sum of the single covalent bond radii42,43 despite the two bridging hydrides. The majority of previously reported Ru–Al complexes contain more than two metal centres and the Ru–Al distance in 3 is amongst the shortest reported.26,44–47 The examples of complexes with shorter Ru–Al distances result from reaction of low-valent [Cp*Al]4 with Ru precursors.46,47
5 contains a rhomboidal core of {Pd2Al2} with Pd and Al in alternate positions and hydride ligands bridging each edge of the square. The Pd–Pd distance (3.0655(3) Å) is shorter than the Al–Al (3.934 Å) distance. The molecule contains an inversion centre with an asymmetric unit of half the {Pd2Al2H4} complex and the {Pd2Al2H4} unit is found to be almost planar. The plane created by Al and the two nitrogen atoms in the supporting ligand is almost perpendicular (89.8°) to the {Pd2Al2} plane, also indicating that the hydride ligands lie in the {Pd2Al2} plane. The alane is the sole ligand for the Pd–Pd unit and is bound slightly asymmetrically with Pd–Al distances of 2.4663(6) and 2.5203(6) Å. The Pd–Pd and Pd–Al distances are slightly longer than those found in a previously reported analogue 7a (Pd–Pd 2.8717(4) Å; Pd–Al 2.4308(7) and 2.4538(8) Å) which shares the same metal connectivity.15
The structure of 6 features an octametallic {Pd4Al4} core, double that of 5, 7a and 7c. The metal centres lie in a plane around an inversion centre with the Pd centres forming a diamond shape and Al centres bridging each edge of this diamond. The structure is found to contain 6 hydride ligands (H:Al = 1.5:1) which also lie in the plane of the metals. Al2 is still nominally bound to 2 hydrides while Al1 only bonds to one, although the uncertainty in the position of H3 means it is possible H3 is shared between Al1 and Al2. Pd1 forms two hydride bridges while Pd2 has only one. The Pd–Pd distances fall in a 0.1 Å range with the Pd2–Pd2′ distance the shortest (2.8883(5) Å) and the Pd–Al distances also fall in a 0.1 Å range of one another. The planes created by Al and the two nitrogen atoms in the supporting ligands are again almost perpendicular (89.3 and 87.6°) to the plane of the metal centres. There does not appear to be literature precedent for this type of planar Pd containing structure. Complexes with four or more Pd centres in this diamond configuration are generally not planar, those close to planarity are forced so by being sandwiched between planar aromatic ligands.48–52 A few high nuclearity clusters supported by main group ligands are known, including triangular (tris)platinum complexes and planar (tetrakis)palladium complexes. In the latter a single Pd centre is connected to three Pd and Si or Ge atoms.53–55 To the best of our knowledge, there are no reported structurally characterised examples of Pd4Al4 species or of Al complexes with any transition metal with the configuration observed in 6. Furthermore, complexes 5 and 6 represent rare examples of palladium clusters free from addition phosphine ligands on the group 10 metal.
Two crystal forms of 7c were identified by diffraction experiments. Single crystal X-ray diffraction of red crystals of form 1 (triclinic, P-1, V = 1597 Å3) obtained from benzene solution revealed that the metallic core of 7c is virtually identical to that of 7a with the same planar {Pd2Al2H2} motif and H:Al ratio of 1:1.15 The change in the supporting ligand for Al from containing mesityl groups in 7a to 2,6-xylyl groups in 7c appears to have little effect. The Pd–Pd (7c: 2.8760(3) Å; 7a: 2.8717(4) Å) and Pd–Al (7c: 2.4360(5) and 2.4504(5) Å; 7a: 2.4308(7) and 2.4538(8) Å) distances are similar in both cases leading to topologically identical structures. Form 2 (triclinic, P-1, V = 3717 Å3) formed as large red blocks from a C6D6 solution and was subject to single crystal neutron diffraction. This experiment revealed that H/D isotope exchange between the hydrides and the solvent had occurred during the crystallisation (see above) providing an isotopically enriched sample of 7c-d2. Neutron diffraction of 7c-d2 confirmed the position and number of the deuteride ligands and hence the corresponding hydrides in 7c. Two molecules are present in the asymmetric unit of this form. The Pd–D bond lengths of 1.80(2) Å is similar to the Al–D bond lengths of 1.77(3) Å and are consistent with the proposed bridging interaction. This rare example of a neutron structure of a palladium hydride species validates a general structural motif that has been invoked numerous times for related Pd and Pt complexes with group 14 hydrides (Si and Ge) (Table 1).56–61
| Compound | M–Al | M–M | M–P | M–D | Al–D |
|---|---|---|---|---|---|
| 3 | 2.3037(9) | — | 2.3149(8) | — | — |
| 5 | 2.4663(6) | 3.0655(3) | — | — | — |
| 2.5203(6) | |||||
| 6 | 2.4137(10) | 2.9197(4) | — | — | — |
| 2.5089(10) | 2.9720(4) | ||||
| 2.4320(10) | 2.8883(5) | ||||
| 2.4222(11) | |||||
| 7c (X-ray-form 1) | 2.4360(5) | 2.8760(3) | 2.3397(5) | — | — |
| 2.4504(5) | |||||
| 7c (X-ray-form 2) | 2.4332 (9) | 2.9329(5) | 2.3744(8) | — | — |
| 2.4496(9) | |||||
| 2.4384(9) | 2.8879(4) | 2.3358(8) | |||
| 2.4419(9) | |||||
| 7c (Neutron-form 2) | 2.43(3) | 2.90(3) | 2.31(2) | 1.77(2) | 1.81(3) |
| 2.43(3) | |||||
| 2.43(3) | 2.86(3) | 2.39(2) | 1.80(2) | 1.77(3) | |
| 2.42(3) |
Solid-state 27Al NMR data
27Al NMR spectroscopy was used as a means to gain insight into the Al environment upon coordination and dehydrogenation. In our experience, acquisition of satisfactory solution 27Al NMR data on a broad range of aluminium complexes with or without metal coordination is challenging because of the line-broadening effects inherent with the quadrupolar I = 5/2 27Al nucleus. Solid state (SS) NMR spectroscopy offers an alternative approach to study broad signals, by accessing higher-power pulses enabling a greater excitation bandwidth, and by employing magic angle spinning (MAS) to reduce the effects of chemical shift anisotropy and quadrupolar interactions. In combination with computational methods, this provides a powerful approach.62–65In order to validate the approach, a series of reference samples were investigated by 27Al MAS NMR spectroscopy. Considering the aluminium fragments in isolation, two extremes for the extent of dehydrogenation are clearly represented by the aluminium dihydrides (1a–f) and the corresponding monomeric aluminium(I) complex 8, originally reported by Roesky and co-workers.66 Further points of comparison, the aluminium dichloride complexes, 9a–b were considered.67 Isotropic chemical shift values obtained from SS 27Al NMR spectroscopy were confirmed by comparison to the available solution state data and showed good agreement, validating the approach (Table 2).
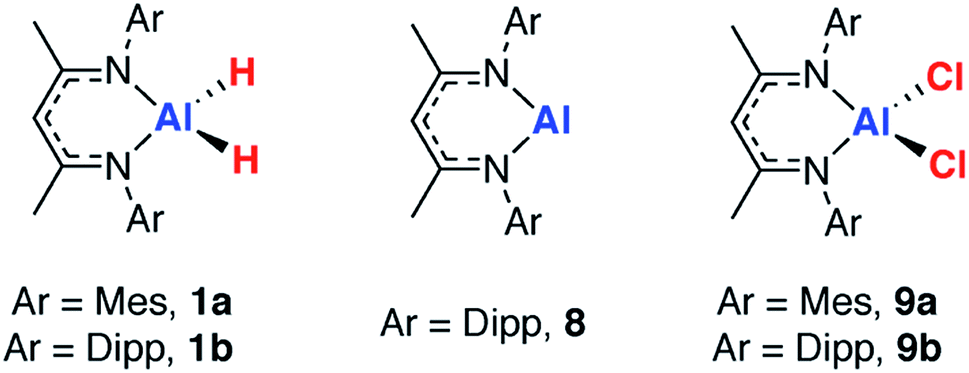
In our hands, 27Al MAS NMR spectroscopy of 8 showed a remarkably broad spectrum with isotropic shift δAl = 120 ppm and a series of features seen out to +1000 and −1000 ppm. 27Al NMR data for a series of aluminium(I) compounds bearing cyclopentadienyl ligands have been calculated to range from δAl = +850 to −170 ppm.69 While in solution it has been suggested that 8 possesses a 27Al NMR chemical shift of δAl = 590 ± 40 ppm (fwhm = 30000 Hz),68 it appears that this only represents part of the spectral features apparent in the solid-state. To support the new assignment DFT calculations were performed, which agreed very well with experiment, and gave NMR parameters of δAl = 115 ppm with CQ = 23.0 MHz and CSA = 175 ppm (Fig. 3 – red line). At 9.4 T and 30 kHz MAS, this results in a very wide central transition peak, which is strong evidence for the highly asymmetric Al(I) environment. The same spectral features were obtained from three independently synthesised samples of 8. In all samples, a peak consistent with a major AlO4 impurity were observed. Upon exposure of samples of 8 to air a visible change in colour from red to colourless was observed and 27Al MAS NMR confirmed the loss of the resonances associated with 8. For comparison, δAl and CQ values for 1a–b, and 9a–b are listed alongside those of 8 in Table 2. This is an exceptionally rare example of a successful 27Al MAS NMR spectroscopy experiment on an aluminium centre in the +1 oxidation state.
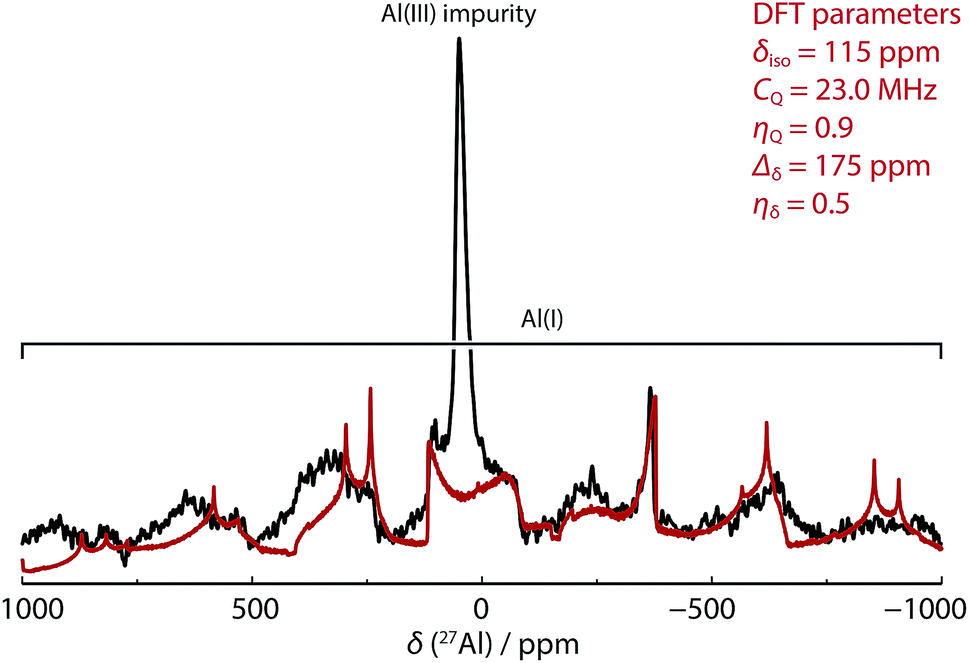 | ||
| Fig. 3 SS 27Al MAS NMR spectrum of 8 (black line, 52464 scans with 1 s relaxation delay) overlaid with simulated lineshape of just the central transition from DFT parameters (red line). | ||
Comparison of the data reveals that absolute values of the isotropic chemical shift are not a good indicator of formal oxidation state. Schnöckel and co-workers have already commented that the ligand environment and coordination number at aluminium has a large impact on chemical shift values.69 Further, Arnold and co-workers have compared the electronic structure of 1, 8 and 9 using a combination of polarised aluminium K-edge absorption near edge structure (XANES) spectroscopy and first-principles calculations.70 They concluded that, despite the difference in formal oxidation state of aluminium, the charge distributions about aluminium are similar throughout the series. The CQ values are an indicator of asymmetry in the electric field gradient across the molecule and show a rational trend. Complex 8 has the highest CQ value (23.9 MHz DFT calculations), indicative of the two-coordinate environment at aluminium. As the crystal field environment about Al becomes more symmetric with introduction of hydride, alkyl and chloride ligands values of CQ decrease. The CQ values are less sensitive than δAl to changes of the steric profile of the β-diketiminate ligand (Table 2).
27Al MAS NMR spectra were recorded on a series of transition metal alane complexes with different H:Al ratios. These included Ru and Pd complexes reported herein along with some previously isolated Rh complexes from our group.71 As expected from the reference samples, there is no clear correlation of the δAl(iso) or CQ values with the extent of dehydrogenation (see ESI, Table S6.1†), suggesting that an alternative approach is required to interrogate the dehydrogenation step.
DFT and QTAIM calculations
In contrast to the 27Al NMR experiments, DFT calculations provided a good indicator for the extent of on-metal dehydrogenation. Both NBO and QTAIM calculations are consistent with dehydrogenation being a process in which dihydrogen production is accompanied by metal–metal bond formation and transfer of electron-density to the Al centres.Analysis of the NPA charges from NBO calculations on geometry optimised structures were performed on both the aforementioned reference compounds and a comprehensive series of aluminium containing heterometallic complexes reported by our group and others (Fig. 4), including σ-alane complexes,34,72 metal aluminyls bearing an X-type aluminium ligand,71 and metal aluminylenes involving an L-type aluminium ligand.15 There is a trend in the charge on aluminium as the structures proceed from σ-alane to aluminyl to aluminylene ligands. As the ratio of H:Al decreases across the series 3 > 5 > 6 > 7c the charges on the aluminium centres decrease in the same order +1.6 > +1.4 > +1.2 = +1.2. These charges parallel the range established for the reference complex categories in Fig. 4 and the calculated NPA charges on the genuine Al(III) and Al(I) complexes 1b and 8 of +1.5 and +0.8 respectively.
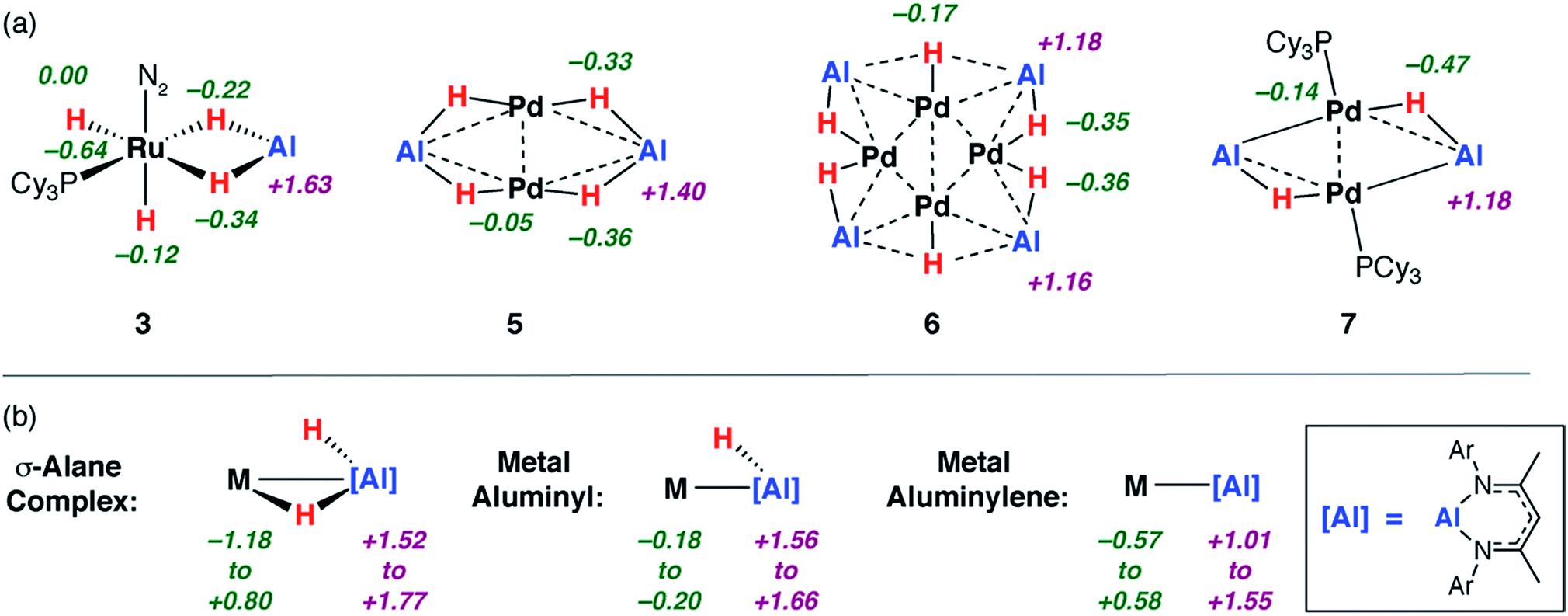 | ||
| Fig. 4 Calculated NPA charges for (a) 3, 5–7c alongside (b) a series of metal σ-alane, aluminyl and aluminylene complexes. | ||
Charge transfer to Al appears to be a result of the formation of covalent metal–metal bonds. The nature of the metal–metal interactions can be considered more deeply by comparing results from DFT and QTAIM calculations. While the Pd–Pd and Pd–Al distances in 5 and 7c vary little regardless of structure, calculations provide more insight. Comparison of the QTAIM molecular graphs (Fig. 5) shows that where hydride ligands are present the bonding is dominated by 3c,2e− interactions involving Pd–H–Al bridges. Critical paths are found between Al and H atoms and Pd and H atoms but not between Pd and Al. NBO calculations provide a similar picture, characterising a high degree of covalency and a bridging interaction that is dominated by metal–hydride bonding. Pd–Al bonds form as hydride ligands are removed. Hence, in 7c QTAIM data return bond paths between Pd and Al for the edges without hydride ligands but not those with. NBO calculations support a much larger degree of Pd–Al covalent bonding along these open edges and a bonding situation reminiscent of that found in metal aluminyl complexes.71 While the nature of the Pd–Pd bonding in these complexes is less clear, based on NBO calculations and the low covalent character in both 5 and 7c, it is likely dominated by closed shell interactions.
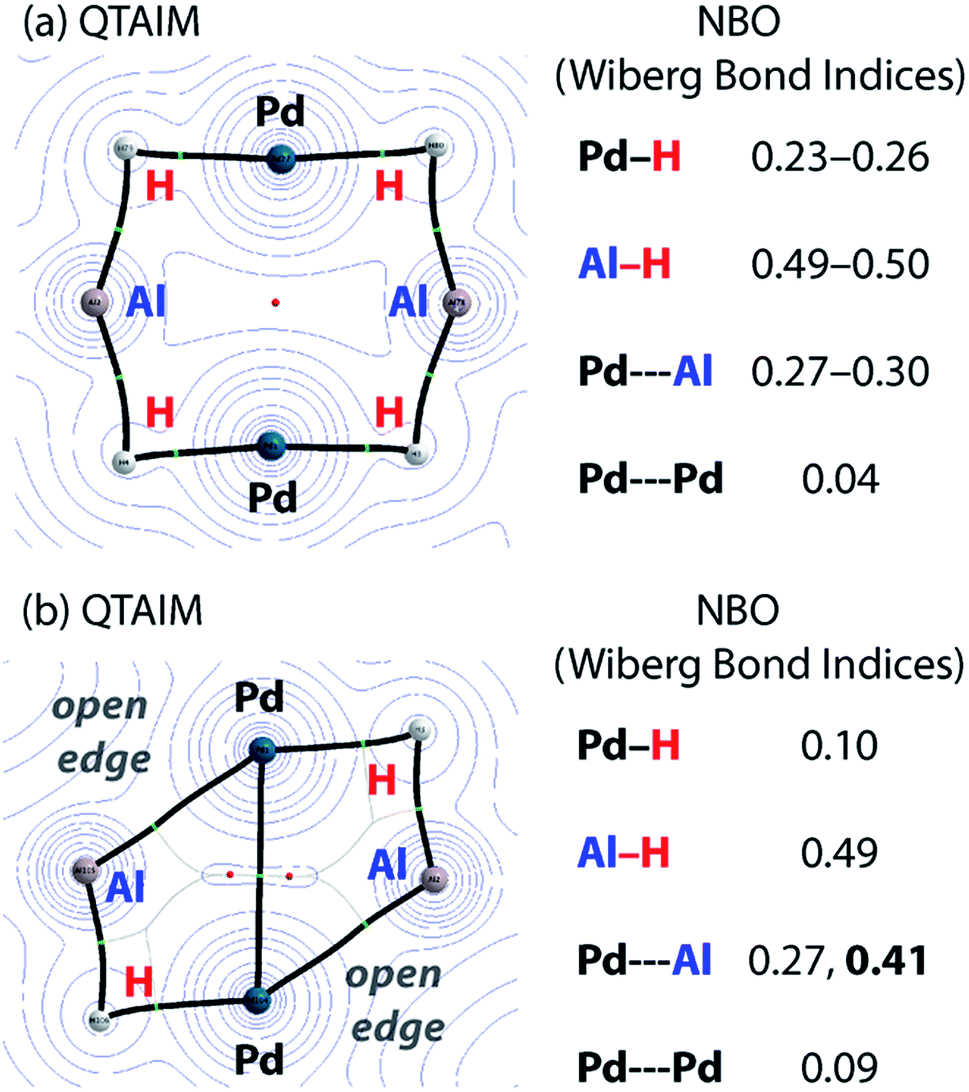 | ||
| Fig. 5 Comparison of QTAIM molecular graphs for (a) 5 and (b) 7c. | ||
The data imply that on-metal dehydrogenation is accompanied by metal–metal bond formation. Clusters with lower hydride content form metal–metal bonds where bridging 3c,2e– M–H–M interactions are not possible. Due to the large covalent character of the Pd–Al metal bonds dehydrogenation occurs with charge accumulation on aluminium. While this finding could be interpreted in terms of a decrease of the formal oxidation state of aluminium due to dehydrogenation, care must be taken with the assignment of oxidation states to the Al and transition metal centres in these molecules. Values of formal oxidation state are often of little meaning in molecules with a high covalent character to the bonding. Nevertheless, the calculations show a clear change in the electrostatic component to the bonding as hydride ligands are lost and metal–metal interactions are formed.
Conclusions
In summary, we report a series of reactions of structurally related aluminium dihydrides with Ru and Pd transition metal fragments. While the resulting ruthenium–aluminium heterobimetallics are 3-dimensional coordination compounds involving ligation of the aluminium dihydride to Ru, the isolated palladium–aluminium complexes all incorporate a 2-dimensional array of metal atoms supported by auxiliary ligands. These latter intermetallic complexes contain Pd–Al and Pd–Pd interactions and differ in terms of the H:Al ratio. Those with lower hydride content form with liberation of dihydrogen. Despite observation of palladium–aluminium intermetallics with 2:1, 1.5:1 and 1:1 ratios of H:Al, the complete dehydrogenation of the molecular aluminium dihydrides is yet to be observed. Spectroscopic methods and calculations were used to gain insight into the on-palladium dehydrogenation process. 27Al MAS NMR spectroscopy parameters did not correlate with the level of dehydrogenation but did allow the unambiguous characterisation of an Al(I) complex by 27Al MAS NMR. DFT and QTAIM calculations showed that the extent of alane dehydrogenation was accompanied by a decrease in the charge on the aluminium centres: along with Pd–Al bond formation in molecular regions in which hydride ligands had been removed.
Despite increased interest in aluminium hydrides as hydrogen storage materials little is known about the intermediates in the transition metal mediated dehydrogenation process. This study provides some of the first structural snapshots of on-metal dehydrogenation of alanes by using sterically demanding ligands to access kinetic products of hydrogen loss. The data we report begins to inform not only the mechanism of interconversion of aluminium(III) hydride precursors and aluminium(0) materials in hydrogen storage applications, but also recently reported catalytic methods that involve dehydrocoupling of molecular aluminium(III) hydrides with organic substrates.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful to the European Research Council (FluoroFix:677367) and the Royal Society (UF090149). Johnson Matthey are thanked for generous donation of PdCl2. AST acknowledges support from the Collaborative Computing Project for NMR Crystallography (CCP-NC) funded by EPSRC (EP/M022501/1). ANSTO is thanked for allocation of neutron beam-time on KOALA to proposal P6932.References
- S.-I. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- J. A. Dilts and E. C. Ashby, Inorg. Chem., 1972, 11, 1230–1236 CrossRef CAS.
- X. Liu, G. S. Mcgrady, H. W. Langmi and C. M. Jensen, J. Am. Chem. Soc., 2009, 131, 5032–5033 CrossRef CAS PubMed.
- B. D. Clark, C. J. DeSantis, G. Wu, D. Renard, M. J. McClain, L. Bursi, A.-L. Tsai, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2019, 141, 1716–1724 CrossRef CAS PubMed.
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305–3366 CrossRef CAS PubMed.
- A. J. Downs and C. R. Pulham, Chem. Soc. Rev., 1994, 23, 175–184 RSC.
- M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- K. Nagata, T. Murosaki, T. Agou, T. Sasamori, T. Matsuo and N. Tokitoh, Angew. Chem., Int. Ed., 2016, 128, 13069–13072 CrossRef.
- X. Li, A. Grubisic, S. T. Stokes, J. Cordes, G. F. Ganteför, K. H. Bowen, B. Kiran, M. Willis, P. Jena, R. Burgert and H. Schnöckel, Science, 2007, 315, 356–358 CrossRef CAS PubMed.
- A. Grubisic, X. Li, S. T. Stokes, J. Cordes, G. F. Ganteför, K. H. Bowen, B. Kiran, P. Jena, R. Burgert and H. Schnöckel, J. Am. Chem. Soc., 2007, 129, 5969–5975 CrossRef CAS PubMed.
- S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865–869 CrossRef CAS PubMed.
- S. J. Bonyhady, D. Collis, N. Holzmann, A. J. Edwards, R. O. Piltz, G. Frenking, A. Stasch and C. Jones, Nat. Commun., 2018, 9, 3079 CrossRef PubMed.
- T. N. Hooper, M. Garçon, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 5435–5440 RSC.
- W. Chen, T. N. Hooper, J. Ng, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2017, 56, 12687–12691 CrossRef CAS PubMed.
- J. A. B. Abdalla, A. Caise, C. P. Sindlinger, R. Tirfoin, A. L. Thompson, A. J. Edwards and S. Aldridge, Nat. Chem., 2017, 9, 1256–1262 CrossRef CAS PubMed.
- J. A. B. Abdalla, I. M. Riddlestone, J. Turner, P. A. Kaufman, R. Tirfoin, N. Phillips and S. Aldridge, Chem.–Eur. J., 2014, 20, 17624–17634 CrossRef CAS.
- J. Turner, J. A. B. Abdalla, J. I. Bates, R. Tirfoin, M. J. Kelly, N. Phillips and S. Aldridge, Chem. Sci., 2013, 4, 4245–4250 RSC.
- G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2008, 130, 12878–12879 CrossRef CAS.
- D. A. Addy, J. I. Bates, M. J. Kelly, I. M. Riddlestone and S. Aldridge, Organometallics, 2013, 32, 1583–1586 CrossRef CAS.
- The activation parameters for the exchange process between the axial and equatorial PCy3 ligands were modelled using peak-fitting to give ΔG‡298K = 8.8 ± 2.7 kcal mol−1, ΔH‡298K = 10.8 ± 1.1 kcal mol−1 and ΔS‡ = +6.6 ± 5.2 cal K−1 mol−1. Similar values could be extracted from the 1H NMR data (ESI†). The positive entropy of activation suggests that the mechanism of exchange may proceed through a dissociative process.
- K. Hussein, C. J. Marsden, J.-C. Barthelat, V. Rodriguez, S. Conejero, S. Sabo-Etienne, B. Donnadieu and B. Chaudret, Chem. Commun., 1999, 1315–1316 RSC.
- T. Ayed, J.-C. Barthelat, B. Tangour, C. Pradère, B. Donnadieu, M. Grellier and S. Sabo-Etienne, Organometallics, 2005, 24, 3824–3826 CrossRef CAS.
- R. B. Said, K. Hussein, J.-C. Barthelat, I. Atheaux, S. Sabo-Etienne, M. Grellier, B. Donnadieu and B. Chaudret, Dalton Trans., 2003, 4139–4146 RSC.
- S. Lau, A. J. P. White, I. J. Casely and M. R. Crimmin, Organometallics, 2018, 37, 4521–4526 CrossRef CAS.
- D. G. Hamilton and R. H. Crabtree, J. Am. Chem. Soc., 1988, 110, 4126–4133 CrossRef CAS.
- P. J. Desrosiers, L. Cai, Z. Lin, R. Richards and J. Halpern, J. Am. Chem. Soc., 1991, 113, 4173–4184 CrossRef CAS.
- K. Kudo, M. Hidai and Y. Uchida, J. Organomet. Chem., 1973, 56, 413–418 CrossRef CAS.
- S. Fantasia, J. D. Egbert, V. Jurčík, C. S. J. Cazin, H. Jacobsen, L. Cavallo, D. M. Heinekey and S. P. Nolan, Angew. Chem., Int. Ed., 2009, 48, 5182–5186 CrossRef CAS PubMed.
- P. J. Perez, J. C. Calabrese and E. E. Bunel, Organometallics, 2001, 20, 337–345 CrossRef CAS.
- S. A. Wander, A. Miedanar, B. C. Noll, R. M. Barkley and D. L. DuBois, Organometallics, 1996, 15, 3360–3373 CrossRef CAS.
- T. Fanjul, G. Eastham, N. Fey, A. Hamilton, A. G. Orpen, P. G. Pringle and M. Waugh, Organometallics, 2010, 29, 2292–2305 CrossRef CAS.
- A. E. Nako, Q. W. Tan, A. J. P. White and M. R. Crimmin, Organometallics, 2014, 33, 2685–2688 CrossRef CAS.
- A. Hicken, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2017, 56, 8669–8682 CrossRef CAS PubMed.
- R. C. Boyle, J. T. Mague and M. J. Fink, J. Am. Chem. Soc., 2003, 125, 3228–3229 CrossRef CAS PubMed.
- N. Nakata, S. Fukazawa, N. Kato and A. Ishii, Organometallics, 2011, 30, 4490–4493 CrossRef CAS.
- R. A. Stockland, G. K. Anderson and N. P. Rath, J. Am. Chem. Soc., 1999, 121, 7945–7946 CrossRef CAS.
- D. E. Herbert and O. V. Ozerov, Organometallics, 2011, 30, 6641–6654 CrossRef CAS.
- E. Goto, R. A. Begum, S. Zhan, T. Tanase, K. Tanigaki and K. Sakai, Angew. Chem., Int. Ed., 2004, 43, 5029–5032 CrossRef CAS PubMed.
- Data on 6 are from single point calculations on the solid-state structure. See ESI† for further details.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- R. Gilbert-Wilson, L. D. Field and M. Bhadbhade, Inorg. Chem., 2014, 53, 12469–12479 CrossRef CAS PubMed.
- M. Plois, T. Wiegand and R. Wolf, Organometallics, 2012, 31, 8469–8477 CrossRef CAS.
- T. Cadenbach, T. Bollermann, C. Gemel and R. A. Fischer, Dalton Trans., 2009, 322–329 RSC.
- T. Steinke, M. Cokoja, C. Gemel, A. Kempter, A. Krapp, G. Frenking, U. Zenneck and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 2943–2946 CrossRef CAS PubMed.
- Y. Ishikawa, S. Kimura, K. Yamamoto and T. Murahashi, Chem.–Eur. J., 2017, 23, 14149–14152 CrossRef CAS PubMed.
- Y. Ishikawa, S. Kimura, K. Takase, K. Yamamoto, Y. Kurashige, T. Yanai and T. Murahashi, Angew. Chem., Int. Ed., 2015, 54, 2482–2486 CrossRef CAS PubMed.
- Y. Ishikawa, K. Yamamoto and T. Murahashi, Angew. Chem., Int. Ed., 2017, 129, 1366–1370 CrossRef.
- T. Sugawa, K. Yamamoto and T. Murahashi, Chem. Commun., 2018, 54, 5875–5878 RSC.
- T. Murahashi, N. Kato, T. Uemura and H. Kurosawa, Angew. Chem., Int. Ed., 2007, 46, 3509–3512 CrossRef CAS PubMed.
- T. Yamada, A. Mawatari, M. Tanabe, K. Osakada and T. Tanase, Angew. Chem., Int. Ed., 2009, 48, 568–571 CrossRef CAS PubMed.
- M. Tanabe, M. Kamono, K. Tanaka and K. Osakada, Organometallics, 2017, 36, 1929–1935 CrossRef CAS.
- M. Tanabe, N. Ishikawa, M. Chiba, T. Ide, K. Osakada and T. Tanase, J. Am. Chem. Soc., 2011, 133, 18598–18601 CrossRef CAS PubMed.
- M. Tanabe, A. Takahashi, T. Yamada and K. Osakada, Organometallics, 2013, 32, 1815–1820 CrossRef CAS.
- M. Tanabe, N. Ishikawa and K. Osakada, Organometallics, 2006, 25, 796–798 CrossRef CAS.
- T. Yamada, M. Tanabe, K. Osakada and Y.-J. Kim, Organometallics, 2004, 23, 4771–4777 CrossRef CAS.
- M. Tanabe, T. Yamada and K. Osakada, Organometallics, 2003, 22, 2190–2192 CrossRef CAS.
- Y.-J. Kim, S.-C. Lee, J.-I. Park, K. Osakada, J.-C. Choi and T. Yamamoto, Organometallics, 1998, 17, 4929–4931 CrossRef CAS.
- J. Braddock-Wilking, J. Y. Corey, K. A. Trankler, K. M. Dill, L. M. French and N. P. Rath, Organometallics, 2004, 23, 4576–4584 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- D. M. Dawson, J. M. Griffin, V. R. Seymour, P. S. Wheatley, M. Amri, T. Kurkiewicz, N. Guillou, S. Wimperis, R. I. Walton and S. E. Ashbrook, J. Phys. Chem. C, 2017, 121, 1781–1793 CrossRef CAS.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- S. Singh, H.-J. Ahn, A. Stasch, V. Jancik, H. W. Roesky, A. Pal, M. Biadene, R. Herbst-Irmer, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 2006, 45, 1853–1860 CrossRef CAS PubMed.
- C. Cui, S. Köpke, R. Herbst-Irmer, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and B. Wrackmeyer, J. Am. Chem. Soc., 2001, 123, 9091–9098 CrossRef CAS PubMed.
- J. Gauss, U. Schneider, R. Ahlrichs, C. Dohmeier and H. Schnöckel, J. Am. Chem. Soc., 1993, 115, 2402–2408 CrossRef CAS.
- A. B. Altman, C. D. Pemmaraju, C. Camp, J. Arnold, S. G. Minasian, D. Prendergast, D. K. Shuh and T. Tyliszczak, J. Am. Chem. Soc., 2015, 137, 10304–10316 CrossRef CAS PubMed.
- O. Ekkert, A. J. P. White, H. Toms and M. R. Crimmin, Chem. Sci., 2015, 6, 5617–5622 RSC.
- I. M. Riddlestone, S. Edmonds, P. A. Kaufman, J. Urbano, J. I. Bates, M. J. Kelly, A. L. Thompson, R. Taylor and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 2551–2554 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1908297, 1916497, 1916498, 1908298 and 1919750. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02750e |
| ‡ Current address: GSK Medical Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK. |
| This journal is © The Royal Society of Chemistry 2019 |