DOI:
10.1039/C8SC04943B
(Edge Article)
Chem. Sci., 2019,
10, 1000-1007
Monitoring metal–amyloid-β complexation by a FRET-based probe: design, detection, and inhibitor screening†
Received
6th November 2018
, Accepted 5th December 2018
First published on 6th December 2018
Abstract
Aggregation of amyloidogenic peptides could cause the onset and progression of neurodegenerative diseases, such as Alzheimer's disease and Parkinson's disease. These amyloidogenic peptides can coordinate to metal ions, including Zn(II), which can subsequently affect the peptides' aggregation and toxicity, leading to neurodegeneration. Unfortunately, the detection of metal–amyloidogenic peptide complexation has been very challenging. Herein, we report the development and utilization of a probe (A-1) capable of monitoring metal–amyloid-β (Aβ) complexation based on Förster resonance energy transfer (FRET). Our probe, A-1, is composed of Aβ1–21 grafted with a pair of FRET donor and acceptor capable of providing a FRET signal upon Zn(II) binding even at nanomolar concentrations. The FRET intensity of A-1 increases upon Zn(II) binding and decreases when Zn(II)-bound A-1 aggregates. Moreover, as the FRET intensity of Zn(II)-added A-1 is drastically changed when their interaction is disrupted, A-1 can be used for screening a chemical library to determine effective inhibitors against metal–Aβ interaction. Eight natural products (out of 145 compounds; >80% inhibition) were identified as such inhibitors in vitro, and six of them could reduce Zn(II)–Aβ-induced toxicity in living cells, suggesting structural moieties useful for inhibitor design. Overall, we demonstrate the design of a FRET-based probe for investigating metal–amyloidogenic peptide complexation as well as the feasibility of screening inhibitors against metal-bound amyloidogenic peptides, providing effective and efficient methods for understanding their pathology and finding therapeutic candidates against neurodegenerative disorders.
Introduction
The number of aged people affected by neurodegenerative diseases has been increasing; however, the development of treatments for the diseases has not been successful due to the lack of understanding about their pathogenesis.1,2 The proposed risk factors of neurodegenerative diseases include metal ions [e.g., Zn(II)] and amyloidogenic peptides [e.g., amyloid-β (Aβ) and tau for Alzheimer's disease, α-synuclein for Parkinson's disease, and huntingtin for Huntington's disease].3–12 Toxic aggregates are formed upon aggregation of these amyloidogenic peptides, particularly in the presence of metal ions.2,13–15 The aggregation and conformational changes of such amyloidogenic peptides have been previously studied by luminescence, including Förster resonance energy transfer (FRET).16–21 In addition, the interactions between amyloidogenic peptides and metal ions (e.g., binding affinity and coordination geometry) have been investigated through multiple physical methods.8,9,22–26 Such approaches, however, require high concentrations of peptides and metal ions (e.g., high μM) presenting significant challenge in performing the experiments due to the aggregation-prone properties of amyloidogenic peptides. Unfortunately, detecting the formation of metal-bound amyloidogenic peptides with a straightforward and efficient method (e.g., monitoring a turn-on signal) at a low concentration (ca. nM) has not been reported. Herein, we report a FRET-based probe (A-1; Fig. 1 and Scheme 1), composed of Aβ1–21 grafted with a pair of FRET donor and acceptor, for monitoring metal–Aβ complexation at a nanomolar range with a turn-on FRET signal. The FRET intensity of A-1 was observed to increase upon binding to Zn(II) (green; Fig. S1†). Note that although other metal ions [particularly, Cu(II)] are reported to interact with Aβ,10,24 the use of our probe, A-1, is limited for paramagnetic metal ions, such as Cu(II), because its fluorescence is quenched (Fig. S1†). Additionally, the FRET signal of A-1 was changed when (i) Zn(II) binding of A-1 was interfered by the metal chelator, EDTA (ethylenediamine tetraacetic acid),29 or the compound, L2-b [N1N1-dimethyl-N4-(pyridin-2-ylmethyl)benzene-1,4-diamine],30,31 capable of forming a ternary complex with Zn(II)–Aβ; (ii) the probe was aggregated. Moreover, a library of natural products as inhibitors against metal–Aβ interaction was screened based on the change in the FRET responses of Zn(II)-treated A-1. 8 out of 145 natural products were identified as effective inhibitors (>80% inhibition) in vitro. Among the 8 molecules, 6 compounds were shown to lower the toxicity associated with Zn(II)–Aβ in living cells. Our studies demonstrate the feasibility of developing an efficient tactic to probe metal–amyloidogenic peptide complexation, along with its potential as a screening tool for drug discovery against neurodegenerative diseases.
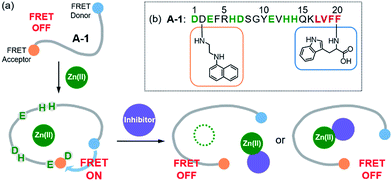 |
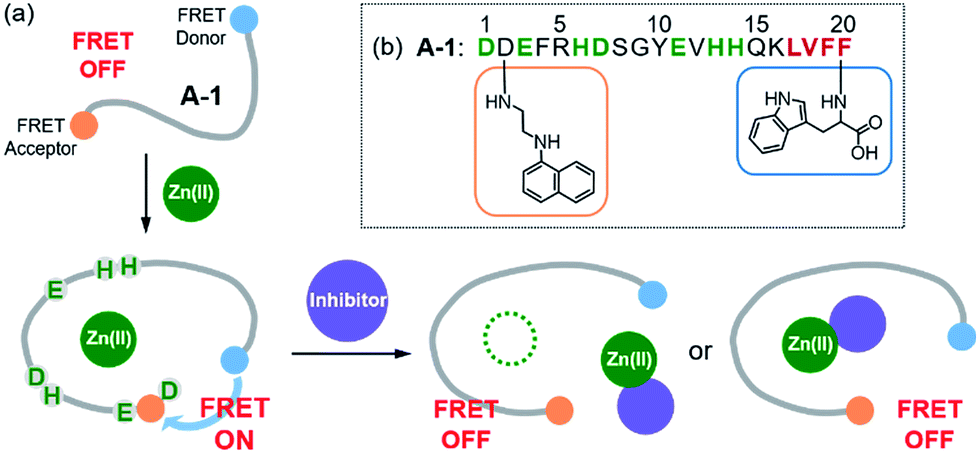
| | Fig. 1 Design principle and sequence of the FRET-based probe, A-1. (a) FRET responses of A-1 in the absence and presence of Zn(II) with and without inhibitors. (b) Amino acid sequence of A-1. A-1 is composed of Trp (blue box) at the C-terminus as a FRET donor and 1-naphthylethylenediamine conjugated to the side chain of the Asp (orange box) at the N-terminus as a FRET acceptor. Proposed amino acid residues for metal binding and a portion of the self-recognition site are indicated in green and red, respectively. | |
 |
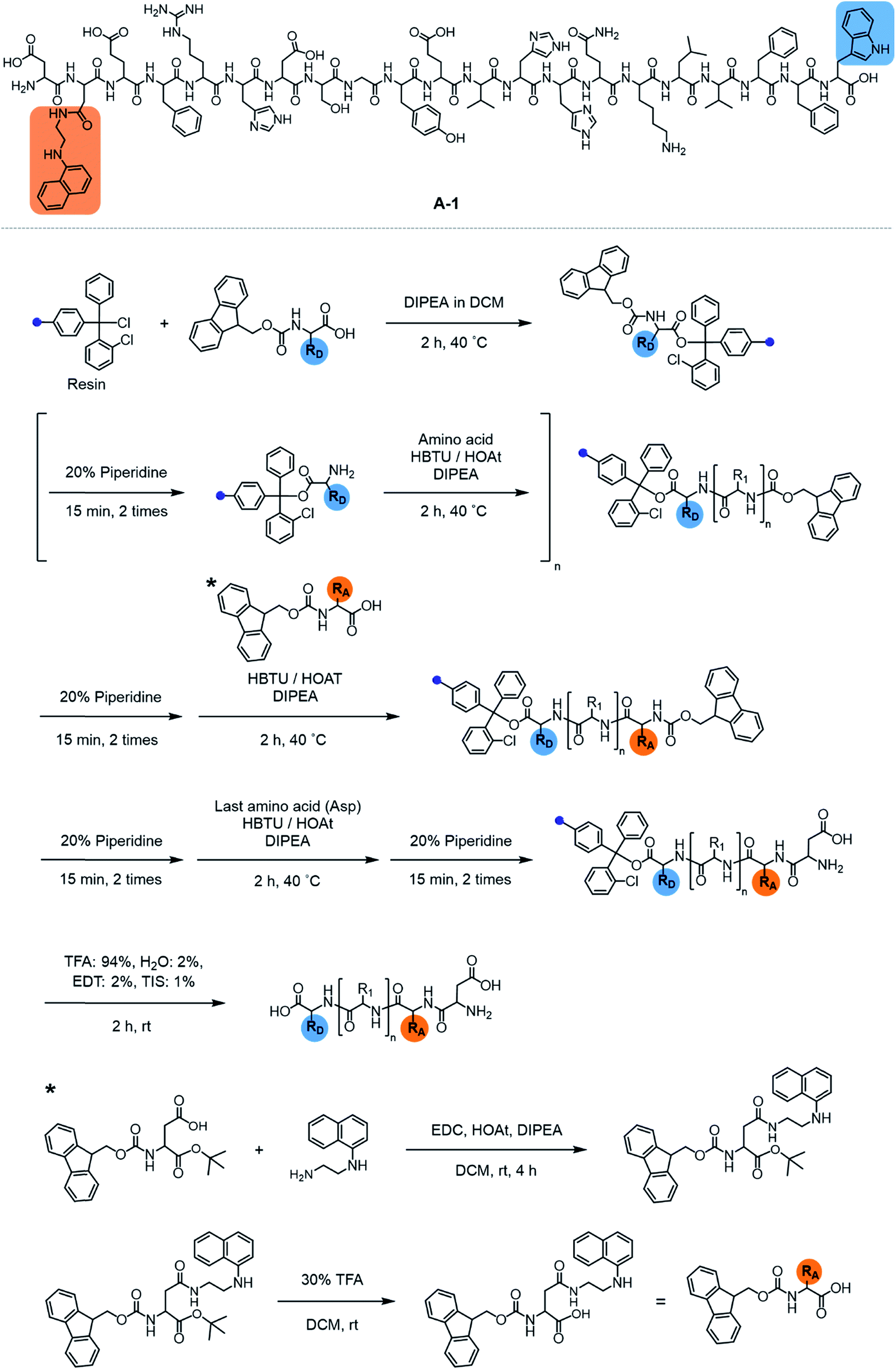
| | Scheme 1 Synthetic routes to A-1. | |
Results and discussion
Design and preparation of A-1
Our probe, A-1, was designed to have a FRET donor (Trp; λex = 280 nm, λem = 350 nm) and an acceptor (1-naphthylethylenediamine conjugated to the side chain of an Asp; λex = 350 nm, λem = 420 nm) for FRET at the C- and N-termini of the Aβ1–21 sequence, respectively (Fig. 1). Aβ1–21 was selected as the main framework of A-1 to include the metal binding site of Aβ (Fig. 1b; proposed metal binding residues highlighted in green, e.g., Asp1, Glu3, His6, Asp7, Glu11, His13, and His14).10,26,32–35 Thus, A-1 itself can interact with metal ions like Aβ. When A-1 was treated with Zn(II), the Zn(II)–A-1 complex was formed which was confirmed by mass spectrometry (MS) (Fig. S2†). Additionally, the binding affinity [Kd = 5.6 (±0.9) μM] of A-1 (5 μM) for Zn(II) was measured by a fluorescence measurement (Fig. S3a†), similar to the Kd values of Zn(II)–Aβ obtained using the same method from previous studies.36–38 Moreover, the progression of peptide aggregation could be observed because A-1 contains a portion of Aβ′s self-recognition site (Fig. 1b; red, Leu17–Phe20).10,33,39A-1 was synthesized through solid phase peptide synthesis. The detailed synthetic routes are described in Scheme 1 and Experimental section.†
FRET signal of A-1 upon binding to Zn(II)
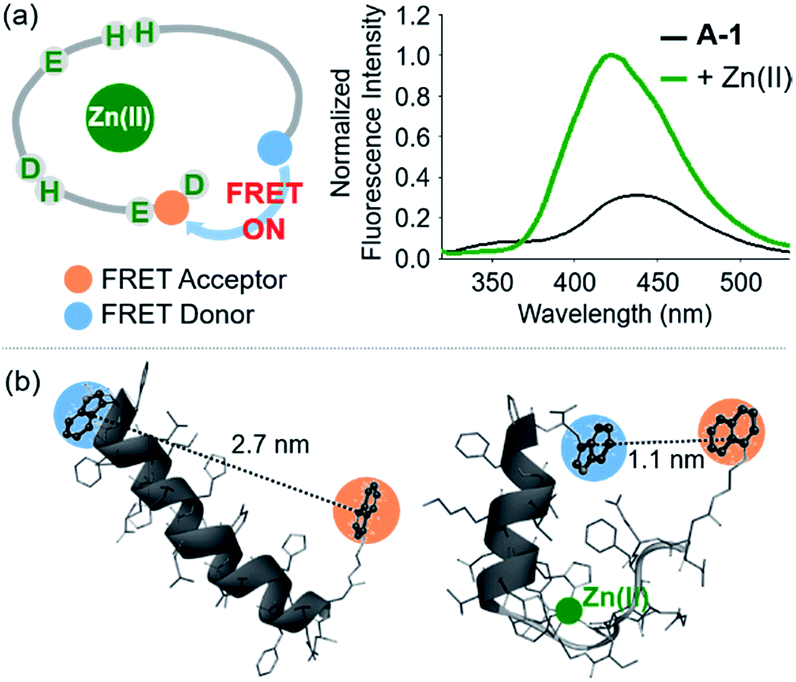
The presence of Zn(II) induced a significant turn-on FRET signal of A-1 by >2 fold compared to Zn(II)-free environment (Fig. 2a). In order to minimize the aggregation of Zn(II)–A-1 (vide infra; Fig. 3), along with consideration of our probe's Zn(II) binding property, 250–500 nM of the probe and 100 μM of Zn(II) were used for this study. As shown in Fig. S3b,† the fluorescence intensity of A-1 (500 nM) at 420 nm was enhanced upon titration and was saturated at ca. 100 μM of Zn(II). Since FRET occurs when a suitable donor and acceptor pair is in close proximity (1–10 nm) with the parallel orientation of the transition dipoles of the FRET donor and acceptor,40,41 an increase in the FRET intensity is indicative of A-1's folding upon Zn(II) binding (Fig. 2a). The possible conformations of metal-free and Zn(II)-bound A-1 were visualized by modeling with modifications of the previously reported structures of metal-free Aβ and Zn(II)-bound Aβ (PDB: 1AMC27 and 1ZE9,28 respectively; Fig. 2b). Without Zn(II), although the indole ring of the FRET donor and the naphthalene ring of the FRET acceptor are close enough for energy transfer (ca. 2.7 nm), they are not facing each other and shown to be unfavorable to have a dipole–dipole interaction for FRET (Fig. 2b; left). Upon interacting with Zn(II), however, the indole and naphthalene rings become closer (ca. 1.1 nm) than those in metal-free A-1 and are facing each other which could be favorable for the dipole–dipole interaction necessary for energy transfer, suggesting that an efficient FRET signal could be observed upon Zn(II) binding to the probe (Fig. 2b; right). Additionally, the emission spectrum was blue shifted by ca. 25 nm possibly due to an environmental change of the FRET acceptor, naphthylamine, when A-1 was folded with Zn(II) treatment (Fig. 2a; right). Note that we cannot rule out that intermolecular interactions resulted from A-1's propensity to aggregate may induce the FRET.
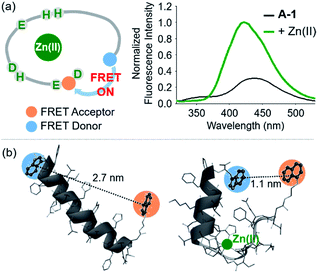 |
| | Fig. 2 FRET response of A-1 to Zn(II) and proposed structures of metal-free and Zn(II)-bound A-1. (a) Change in fluorescence upon incubation of A-1 (black) with Zn(II) (green). Conditions: [A-1] = 0.5 μM; [ZnCl2] = 100 μM; λex = 280 nm. (b) Proposed structures of metal-free A-1 (left) and Zn(II)-bound A-1 (right). The structures were generated by modifications of the previously reported structures of metal-free Aβ (PDB: 1AMC)27 and Zn(II)-bound Aβ (PDB:1ZE9).28 The approximate distances between the FRET donor and acceptor were indicated with dashed lines. | |
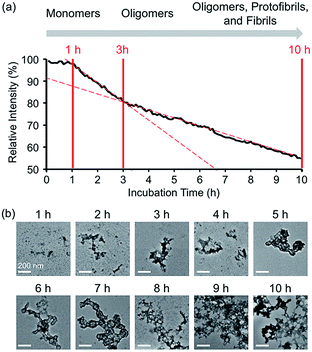 |
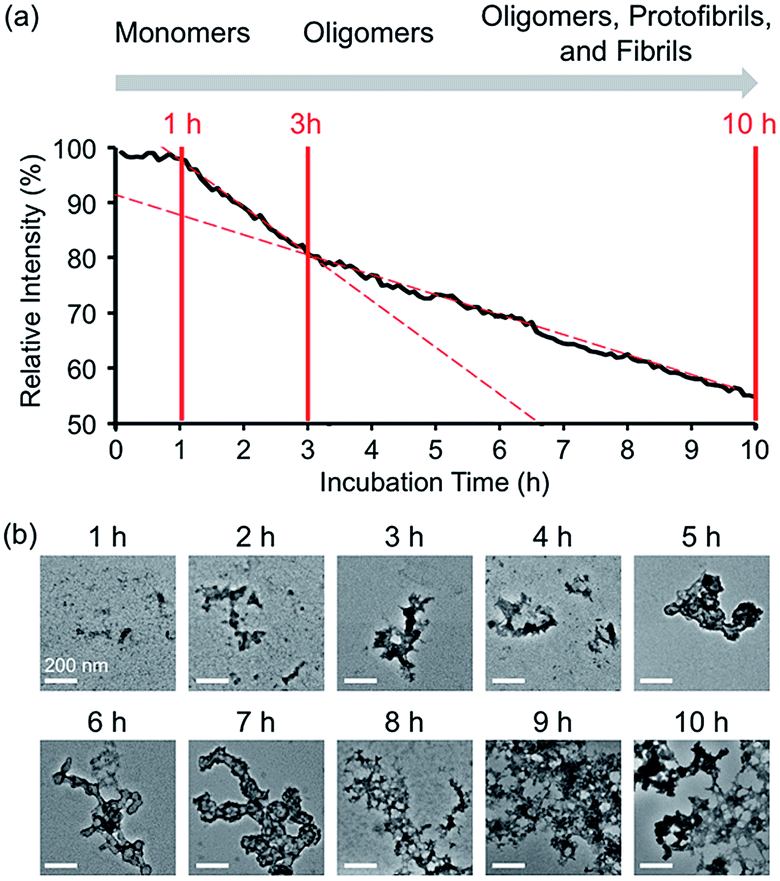
| | Fig. 3 Time-dependent fluorescent response and aggregation progression of Zn(II)-treated A-1. (a) Change in the FRET signal of A-1 with Zn(II) as a function of incubation time. (b) TEM images of Zn(II)-added A-1 aggregates generated at various incubation time points (scale bar = 200 nm). Conditions: [A-1] = 0.25 μM (for FRET) and 2.5 μM (for TEM); [ZnCl2] = 100 μM (for FRET) and 1 mM (for TEM); λex = 280 mm; λem = 420 nm; incubation up to 10 h; room temperature. | |
Aggregation of Zn(II)-bound A-1
In the absence of Zn(II), the FRET signal of A-1 reduced as a function of incubation time (ca. 70% and ca. 85% decrease after 1 and 3 h incubation, respectively; Fig. S4†). This lowered signal may be triggered by the aggregation of A-1 since the probe contains a portion of the self-recognition region of Aβ.10,33,39 In contrast, following incubation time, the FRET signal of Zn(II)-treated A-1 decreased (ca. 2% and ca. 18% decrease after 1 and 3 h incubation, respectively; Fig. 3a) at a slower rate compared to that of Zn(II)-free A-1 (Fig. S4†). This indicates that the aggregation of A-1 could be delayed by the presence of Zn(II), as observed with full-length Aβ40 (Fig. S5†). This difference could stem from the disparate conformations of Aβ aggregates generated upon the aggregation of metal–Aβ, distinct from those of metal-free Aβ aggregates.28,42 Thus, we analyzed the morphologies of Zn(II)–A-1 aggregates upon incubation by transmission electron microscopy (TEM). As depicted in Fig. 3b, small and amorphous aggregates were observed after 1 h incubation of Zn(II)-added A-1 followed by the detection of larger and more structured aggregates with longer incubation. Based on the variation of the FRET intensity as the probe aggregated, the aggregation process of Zn(II)–A-1 could be divided into three stages: (i) 0–1 h; (ii) 1–3 h; (iii) 3–10 h (Fig. 3a). Up to 1 h incubation, the FRET signal of Zn(II)–A-1 did not significantly decrease from the initial measurement. From 1 to 3 h, the FRET intensity of Zn(II)–A-1 dropped drastically and after 3 h incubation, the FRET responses of Zn(II)–A-1 were shown to be distinguishably reduced slower than those during the 1–3 h incubation period. This could be because Zn(II)–A-1 formed large-sized aggregates, including protofibrils and fibrils, which might restrict its rotation to limit the distance between the FRET donor and acceptor, along with its solubility in aqueous media. Thus, our FRET-based probe, A-1, could monitor the progression of Zn(II)–Aβ aggregation, distinct from metal-free Aβ aggregation.
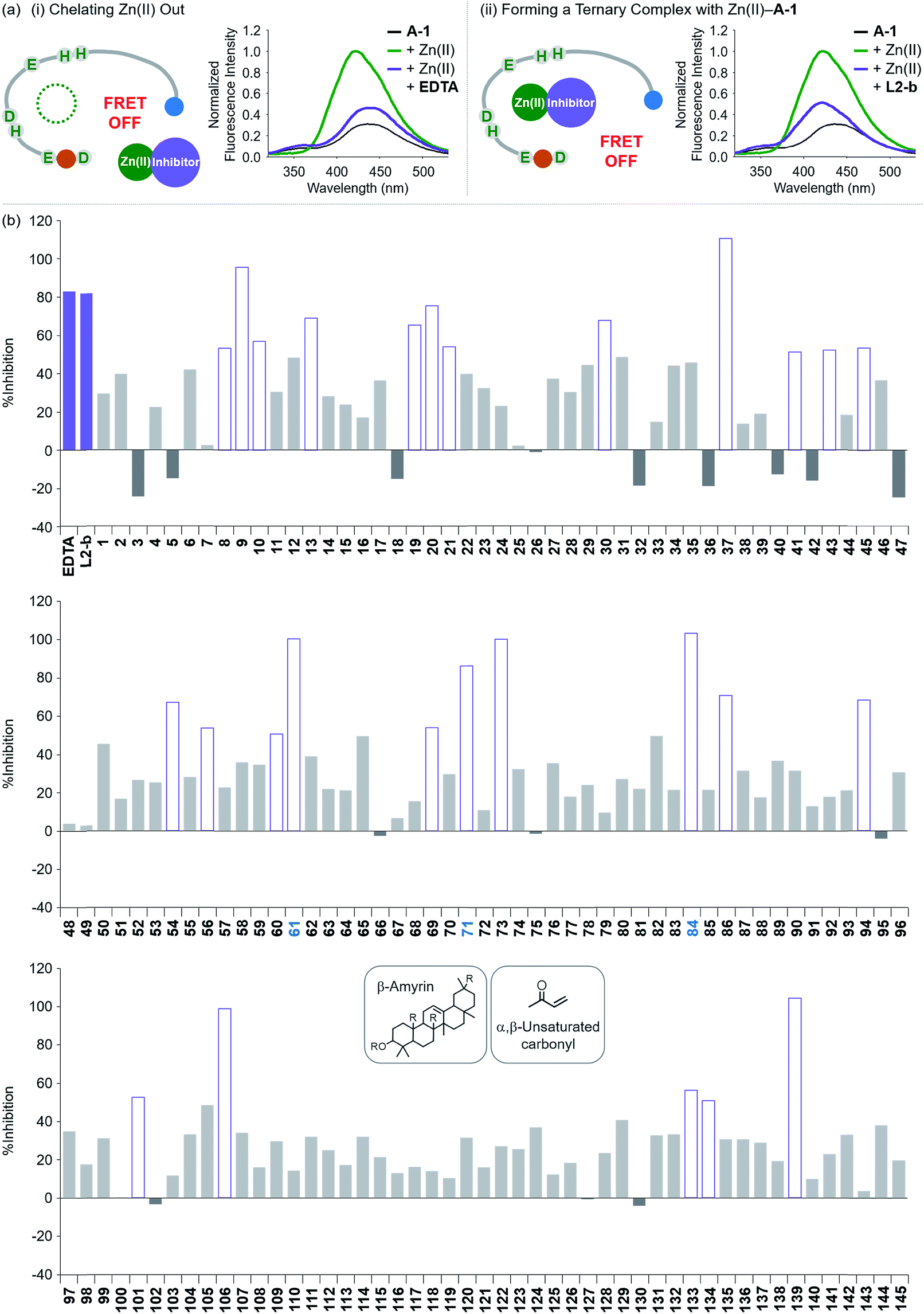
Screening inhibitors against Zn(II)–Aβ interaction
To evaluate whether Zn(II)-bound A-1 is an effective identification tool for inhibitors against Zn(II)–Aβ interaction, alteration of the FRET signal of Zn(II)–A-1 was monitored upon addition of the metal chelator (i.e., EDTA) or the molecule capable of forming a ternary complex with Zn(II)–Aβ (i.e., L2-b) (Fig. 4a and Table S1†).29–31 When EDTA was introduced to Zn(II)–A-1, the FRET intensity was reduced by 83%, compared to the FRET signal of Zn(II)–A-1, and the emission spectrum was red shifted back to that observed under Zn(II)-free conditions (Fig. 4a, i). This suggests that Zn(II) was chelated out from A-1 by EDTA, causing the probe to be unfolded. Furthermore, the treatment of L2-b to Zn(II)–A-1 exhibited a noticeably weaker FRET signal than Zn(II)–A-1 by 82%, but did not present the same emission spectrum as that of Zn(II)-free A-1 (Fig. 4a, ii). The fluorescence behavior of L2-b-added Zn(II)–A-1 implies that a ternary complex [e.g., L2-b–Zn(II)–A-1] could be formed and thus Zn(II) still interacts with A-1, but the distance between the FRET donor and acceptor may not be in close proximity. Note that the emission of Trp was not significantly changed at ca. 350 nm which was not absorbed by the FRET acceptor upon addition of EDTA and L2-b (Fig. 4a), indicating that the compounds did not affect the absorption and emission of the FRET donor. Together, our probe, A-1, demonstrates the ability to identify molecules with inhibitory activity towards metal–Aβ interaction.
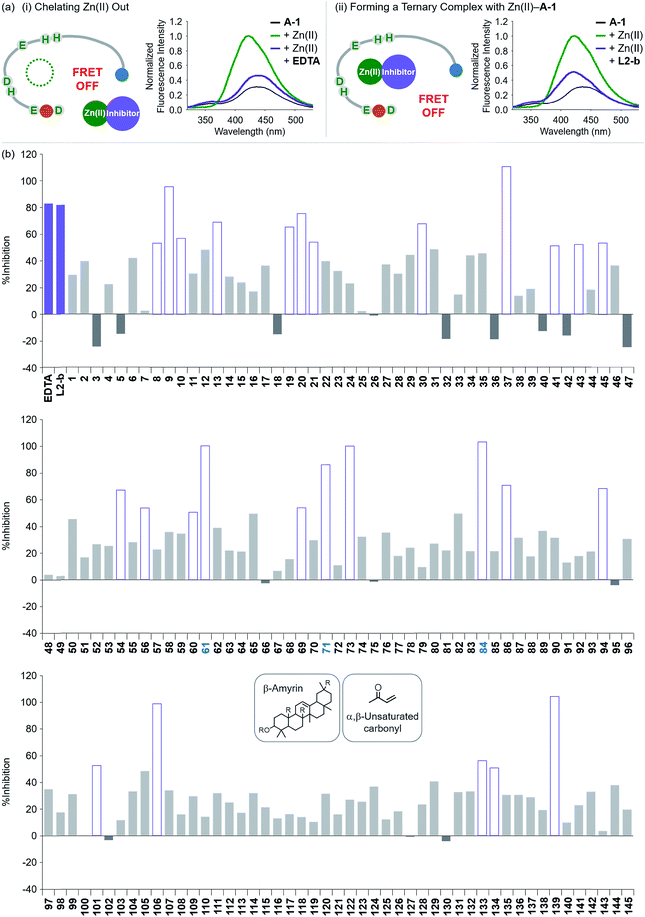 |
| | Fig. 4 Change in the FRET signal of Zn(II)-bound A-1 upon treatment with inhibitors against Zn(II)–Aβ interaction. (a) Fluorescent responses of A-1 in the presence of both Zn(II) and compounds: [(i) EDTA and (ii) L2-b]. (b) Inhibition (%) of Zn(II)–A-1 interaction by incubation with the natural products. Full data sets regarding the inhibition (%) of 145 natural products are summarized in Table S1.†61, 71, and 84 that contain both β-amyrin and α,β-unsaturated carbonyl groups and show >80% inhibition against Zn(II)–A-1 interaction are labeled in blue. Conditions: [A-1] = 0.3 μM; [ZnCl2] = 100 μM; [inhibitor] = 100 μM; incubation for 10 min; room temperature; λex = 280 nm; λem = 420 nm. | |
Moving forward, to confirm the screening capability of our FRET-based method to verify molecules as potential inhibitors against metal–Aβ interaction, we built up a chemical library containing 145 natural products that do not absorb the FRET signal of Zn(II)–A-1 at ca. 420 nm, similar to EDTA and L2-b (Fig. S6 and Table S1†). The FRET signal of Zn(II)–A-1 upon addition of natural products was compared to that of Zn(II)–A-1 to calculate % inhibition (Fig. 4b, S7, and Table S1†). In our library, (i) 15 molecules could not affect Zn(II) binding to A-1; (ii) 103 compounds showed 0 to 50% inhibition; (iii) 27 natural products induced a significant decrease in the fluorescence of Zn(II)–A-1 by >ca. 50% (Fig. 4b and Table S1†). Furthermore, among the 27 natural products (>ca. 50% inhibition), 8 compounds (i.e., 9, 37, 61, 71, 73, 84, 106, and 139) demonstrated >80% inhibitory activity against Zn(II)–A-1 interaction. Three compounds (i.e., 61, 71, and 84 out of 8 potent inhibitors; Fig. 4b and Table S1†) contain both β-amyrin moiety and α,β-unsaturated carbonyl groups, previously reported for controlling metal–Aβ aggregation.43 Note that the compounds containing an α,β-unsaturated carbonyl moiety could form a covalent adduct with Aβ possibly by reacting with Lys or His.44,45 To verify the covalent bond formation between one of the effective inhibitors, 61, and an Aβ fragment (Aβ28), the sample containing 61 and Aβ28 was monitored by MS. The MS measurement presented a covalent Aβ28–61 adduct at 1244 m/z (blue peak; Fig. S8a†). In addition, the tandem MS analysis of the peak at 1244 m/z indicated Aβ28 (at 1088 m/z) and 61 (at 471 m/z) confirming the formation of the covalent Aβ28–61 adduct (Fig. S8b†). Thus, our inhibitors containing an α,β-unsaturated carbonyl moiety have the potential to bind A-1. Overall, inhibitors against Zn(II)–Aβ interaction could be screened and identified by our probe, A-1, showing a variation in its FRET signal in the presence of Zn(II).
Influence of inhibitors on toxicity associated with Zn(II) and Zn(II)–Aβ
The effect of the 8 natural products that showed >80% inhibition against Zn(II)–A-1 interaction on the toxicity triggered by metal-free and Zn(II)-treated Aβ40 and Aβ42 (two major isoforms of Aβ)6,10 was determined in living cells. We first examined the toxicity of 10 natural products (i.e., 8 effective natural products: 9, 37, 61, 71, 73, 84, 106, and 139; 2 compounds which may not be able to disrupt Zn(II)–A-1 interaction: 7 and 48) in human neuroblastoma SH-SY5Y (5Y) cells. The tested compounds, except for 37 and 71, were not relatively toxic (>ca. 80% of cell viability at more than 10 μM) in the absence and presence of Zn(II) (Fig. 5a and S9†). Employing the relatively less toxic natural products (i.e., 7, 9, 48, 61, 73, 84, 106, and 139) with and without Zn(II), their impact on the toxicity induced by pre-incubated Aβ40 and Aβ42 with and without Zn(II) for 1 h at room temperature was analyzed. The natural products could not ameliorate the toxicity induced by metal-free Aβ (Fig. S10†). On the other hand, as depicted in Fig. 5b (purple), cell survival was improved by 6 natural products, determined as effective inhibitors against metal–A-1 interaction, even with the species of Zn(II)–Aβ. As expected, the compounds, 7 and 48, shown to hinder Zn(II) binding to Aβ by less than ca. 5% (Fig. 4b and Table S1†), were not able to mitigate the toxicity induced by both metal-free and Zn(II)-associated Aβ (Fig. 5b and S10;† gray). Thus, our FRET-based method employing A-1 demonstrates its practical utility to determine molecules that can affect metal–Aβ interaction and, as a result, alleviate metal–Aβ-linked cytotoxicity.
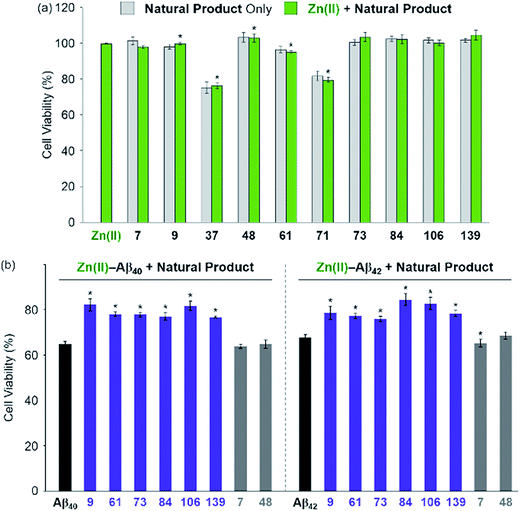 |
| | Fig. 5 Effect of the selected natural products on the cytotoxicity triggered by Zn(II) and Zn(II)–Aβ. (a) Toxicity of the selected natural products with and without Zn(II) in 5Y cells. Cells were treated with compounds (10 μM) in the absence (light gray) and presence (light green) of Zn(II) (same equivalent to compounds; 10 μM) for 24 h at 37 °C. (b) Aβ40 (left) or Aβ42 (right; 10 μM) with Zn(II) (10 μM) was pre-incubated at room temperature for 1 h and then treated to 5Y cells with compounds (10 μM) for 24 h. Cell viability (%) was determined by the MTT assay compared to that obtained upon treatement with a volume of H2O (1% v/v DMSO) equal to the samples added. Error bars represent the standard error of the mean from three independent experiments. *P < 0.05. | |
Conclusions
Since metal ions and amyloidogenic peptides (e.g., Aβ) can interact with each other and induce neurotoxicity, our understanding of such complexation is important to reveal their effects in the pathogenesis of neurodegenerative diseases. In order to verify the feasibility of monitoring metal–amyloidogenic peptide interactions, we employed Aβ as an example of amyloidogenic peptides to develop a FRET-based probe, A-1, to detect the metal binding of Aβ and the progression of metal–Aβ aggregation effectively and efficiently. Upon addition of Zn(II), the FRET signal of A-1 was significantly increased due to the folding of our probe. In addition, when the probe aggregated with Zn(II), its fluorescent response was altered in a distinct manner from that of metal-free case. Furthermore, by utilizing our FRET-based probe to screen a chemical library (total 145 compounds), we identified 6 natural products capable of significantly modulating metal–Aβ interaction (>80% inhibition) in vitro and diminishing cytotoxicity associated with Zn(II)–Aβ in living cells. Our overall studies illustrate the development of a strategy to monitor metal–Aβ interaction and its applicability towards searching potent inhibitors against metal–Aβ interaction. In the near future, for biological applications, new and optimized probes will be developed to monitor the interaction between Aβ and Zn(II) or other metal ions, including Cu(II), showing more sensitive fluorescent responses with lower energy profiles for excitation and emission (e.g., near-infrared region). Applying our tactic to other amyloidogenic peptides, their interactions with metal ions could be, and the inhibitors against metal–amyloidogenic peptide interaction could be identified.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Bio-Synergy Research Project (NRF-2012M3A9C4048775) (to S. J. C.); the National Research Foundation of Korea [NRF-2017M3A9C8031995 (to S. J. C.); NRF-2017R1A2B3002585 and NRF-2016R1A5A1009405 (to M. H. L.)]; KAIST (to M. H. L.). J. K. acknowledges the Global PhD fellowship program through the NRF funded by the Ministry of Education (NRF-2015HIA2A1030823). A. B. T. G. thanks the Korea Research Fellowship Program (NRF-2016H1D3A1938231) through NRF funded by the Ministry of Science and ICT. We thank Geewoo Nam for valuable help on preparation of the manuscript.
Notes and references
- T. Wyss-Coray, Nature, 2016, 539, 180–186 CrossRef CAS PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.8b00138.
- S.-H. Li and X.-J. Li, Trends Genet., 2004, 20, 146–154 CrossRef CAS PubMed.
- R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernández, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4294–4299 CrossRef CAS PubMed.
- J. H. Fox, J. A. Kama, G. Lieberman, R. Chopra, K. Dorsey, V. Chopra, I. Volitakis, R. A. Cherny, A. I. Bush and S. Hersch, PLoS One, 2007, 2, e334 CrossRef PubMed.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed.
- C. A. Ross and I. Shoulson, Parkinsonism Relat. Disord., 2009, 15, S135–S138 CrossRef PubMed.
- A. A. Valiente-Gabioud, V. Torres-Monserrat, L. Molina-Rubino, A. Binolfi, C. Griesinger and C. O. Fernández, J. Inorg. Biochem., 2012, 117, 334–341 CrossRef CAS PubMed.
- A. Binolfi, L. Quintanar, C. W. Bertoncini, C. Griesinger and C. O. Fernández, Coord. Chem. Rev., 2012, 256, 2188–2201 CrossRef CAS.
- M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 2013, 8, 856–865 CrossRef CAS PubMed.
-
M. W. Beck, A. S. Pithadia, A. S. DeToma, K. J. Korshavn and M. H. Lim, in Ligand Design in Medicinal Inorganic Chemistry, ed. T. Storr, Wiley, Chichester, 2014, ch. 10, pp. 257–286 Search PubMed.
- L. V. Kalia and A. E. Lang, Lancet, 2015, 386, 896–912 CrossRef CAS.
-
G. Bartzokis, P. H. Lu, T. A. Tishler and S. Perlman, Neurodegenerative Diseases and Metal Ions, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Wiley, Chichester, 2006, ch. 7, pp. 151–177 Search PubMed.
- S. Bolognin, L. Messori and P. Zatta, NeuroMol. Med., 2009, 11, 223–238 CrossRef CAS PubMed.
- L. Breydo and V. N. Uversky, Metallomics, 2011, 3, 1163–1180 RSC.
- S. Elbaum-Garfinkle and E. Rhoades, J. Am. Chem. Soc., 2012, 134, 16607–16613 CrossRef CAS PubMed.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- H. Tong, K. Lou and W. Wang, Acta Pharm. Sin. B, 2015, 5, 25–33 CrossRef PubMed.
- M.-m. Xu, W.-m. Ren, X.-c. Tang, Y.-h. Hu and H.-y. Zhang, Acta Pharmacol. Sin., 2016, 37, 719–730 CrossRef CAS PubMed.
- J. B. Warner IV, K. M. Ruff, P. S. Tan, E. A. Lemke, R. V. Pappu and H. A. Lashuel, J. Am. Chem. Soc., 2017, 139, 14456–14469 CrossRef PubMed.
- J. J. Ferrie, C. M. Haney, J. Yoon, B. Pan, Y.-C. Lin, Z. Fakhraai, E. Rhoades, A. Nath and E. J. Petersson, Biophys. J., 2018, 114, 53–64 CrossRef CAS PubMed.
- J. Danielsson, R. Pierattelli, L. Banci and A. Gräslund, FEBS J., 2007, 274, 46–59 CrossRef CAS PubMed.
- C. Talmard, A. Bouzan and P. Faller, Biochemistry, 2007, 46, 13658–13666 CrossRef CAS PubMed.
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Angew. Chem., Int. Ed., 2009, 48, 9273–9276 CrossRef CAS PubMed.
- T. Branch, P. Girvan, M. Barahona and L. Ying, Angew. Chem., Int. Ed., 2015, 54, 1227–1230 CrossRef CAS PubMed.
- B. Alies, A. Conte-Daban, S. Sayen, F. Collin, I. Kieffer, E. Guillon, P. Faller and C. Hureau, Inorg. Chem., 2016, 55, 10499–10509 CrossRef CAS PubMed.
- J. Talafous, K. J. Marcinowski, G. Klopman and M. G. Zagorski, Biochemistry, 1994, 33, 7788–7796 CrossRef CAS PubMed.
- S. Zirah, S. A. Kozin, A. K. Mazur, A. Blond, M. Cheminant, I. Ségalas-Milazzo, P. Debey and S. Rebuffat, J. Biol. Chem., 2006, 281, 2151–2161 CrossRef CAS PubMed.
- A. Takeda, H. Tamano, M. Tempaku, M. Sasaki, C. Uematsu, S. Sato, H. Kanazawa, Z. L. Datki, P. A. Adlard and A. I. Bush, J. Neurosci., 2017, 37, 7253–7262 CrossRef CAS PubMed.
- J.-S. Choi, J. J. Braymer, R. P. R. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21990–21995 CrossRef CAS PubMed.
- M. W. Beck, S. B. Oh, R. A. Kerr, H. J. Lee, S. H. Kim, S. Kim, M. Jang, B. T. Ruotolo, J.-Y. Lee and M. H. Lim, Chem. Sci., 2015, 6, 1879–1886 RSC.
- P. Zatta, D. Drago, S. Bolognin and S. L. Sensi, Trends Pharmacol. Sci., 2009, 30, 346–355 CrossRef CAS PubMed.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- K. J. Barnham and A. I. Bush, Chem. Soc. Rev., 2014, 43, 6727–6749 RSC.
- P. Faller, C. Hureau and G. La Penna, Acc. Chem. Res., 2014, 47, 2252–2259 CrossRef CAS PubMed.
- W. Garzon-Rodriguez, A. K. Yatsimirsky and C. G. Glabe, Bioorg. Med. Chem. Lett., 1999, 9, 2243–2248 CrossRef CAS PubMed.
- V. Tõugu, A. Karafin and P. Palumaa, J. Neurochem., 2008, 104, 1249–1259 CrossRef PubMed.
- S. L. Leong, T. R. Young, K. J. Barnham, A. G. Wedd, M. G. Hinds, Z. Xiao and R. Cappai, Metallomics, 2014, 6, 105–116 RSC.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
-
J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 1999 Search PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef CAS PubMed.
- J. T. Pedersen, J. Østergaard, N. Rozlosnik, B. Gammelgaard and N. H. H. Heegaard, J. Biol. Chem., 2011, 286, 26952–26963 CrossRef CAS PubMed.
- Y. Liu, A. Kochi, A. S. Pithadia, S. Lee, Y. Nam, M. W. Beck, X. He, D. Lee and M. H. Lim, Inorg. Chem., 2013, 52, 8121–8130 CrossRef CAS PubMed.
- K. Uchida and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 4544–4548 CrossRef CAS.
- V. Resch, C. Seidler, B.-S. Chen, I. Degeling and U. Hanefeld, Eur. J. Org. Chem., 2013, 2013, 7697–7704 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Table S1, and Fig. S1–S10. See DOI: 10.1039/c8sc04943b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Sang J.
Chung
*a and
Sang J.
Chung