
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled M12L24 nanospheres as a reaction vessel to facilitate a dinuclear Cu(I) catalyzed cyclization reaction†
Sergio
Gonell
 a,
Xavier
Caumes
a,
Nicole
Orth
b,
Ivana
Ivanović-Burmazović
b and
Joost. N. H.
Reek
*a
a,
Xavier
Caumes
a,
Nicole
Orth
b,
Ivana
Ivanović-Burmazović
b and
Joost. N. H.
Reek
*a
aHomogeneous, Supramolecular and Bio-Inspired Catalysis, Van 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, Amsterdam 1098XH, The Netherlands. E-mail: j.n.h.reek@uva.nl
bLehrstuhl für Bioanorganische Chemie, Department Chemie und Pharmazie Friedrich-Alexander-Universität Erlangen, Egerlandstrasse 3, Erlangen 91058, Germany
First published on 13th November 2018
Abstract
The application of large M12L24 nanospheres allows the pre-concentration of catalysts to reach high local concentrations, facilitating reactions that proceed through dinuclear mechanisms. The mechanism of the copper(I)-catalyzed cyclization of 4-pentynoic acid has been elucidated by means of a detailed mechanistic study. The kinetics of the reaction show a higher order in copper, indicating the formation of a bis-Cu intermediate as the key rate determining step of the reaction. This intermediate was further identified during catalysis by CIS-HRMS analysis of the reaction mixture. Based on the mechanistic findings, an M12L24 nanosphere was applied that can bind up to 12 copper catalysts by hydrogen bonding. This pre-organization of copper catalysts in the nanosphere results in a high local concentration of copper leading to higher reaction rates and turnover numbers as the dinuclear pathway is favored.
Introduction
Development in transition metal catalysis traditionally relies to a large extent on ligand modification as this greatly affects catalyst properties. Inspired by the working principles of enzymes, there has been increasing interest in controlling catalyst properties via the second coordination sphere by putting catalysts in well-defined cages.1 Control of both the activity and selectivity of various organic transformations2 has been reported, including Diels–Alder reactions with unusual product selectivity,2a–c and hydrolysis reactions that are greatly accelerated by stabilization of the intermediates.2d,e In addition, several metal-catalysed transformations3–6 have been carried out in molecular cages, also showing that the second coordination sphere impacts both the activity and selectivity of the catalyst. What these cages have in common is that they are relatively small, and as such, they accommodate one catalyst and perform a single transformation. Interestingly, Fujita has developed chemistry that enables the formation of much larger cages. Typical M12L24 nanospheres are around 5 nm in diameter and can contain up to 24 functional groups when appropriate building blocks are used.7 The Fujita group has utilized this by performing one-pot tandem reactions using two different spheres with catalysts that are usually not compatible.8 We used M12L24 nanospheres as reaction vessels in which extremely high local concentration of gold chloride complexes (1 M) resulted in increased reactivity.9 In subsequent work we generated guanidinium functionalized M12L24 nanospheres in which both the sulfonated catalyst and carboxylate containing substrates could be pre-concentrated, hence coined the nano-concentrator, leading to rate acceleration.10 We also demonstrated that pre-concentrating ruthenium complexes for electrochemical water oxidation greatly enhanced the reaction rate as this reaction proceeds via a dinuclear mechanism.11 We were interested if such a pre-concentration effect would generally work for reactions that operate via a dinuclear mechanism, and as such, wanted to extend this chemistry to other reactions. Transformations such as the Copper-catalysed Azide Alkyne Cyclisation (CuAAC)12 and the Kinugasa reaction13 are known to proceed through dinuclear mechanisms; therefore, we hypothesized that copper catalyzed cyclization of alkynoic acids may also proceed via a dinuclear pathway. There are only a few papers reported so far on this reaction, and the current reported protocols show that in, general, high catalyst loadings are required to get significant conversion.14 This reaction is mostly studied using complexes based on noble metals such as Ru,15 Ir,16 Rh,17 Pd,18 Pt,19 Ag20 or Au,11,21 and in these examples the mechanism is mononuclear, if reported at all. For the copper catalysed cyclization no mechanistic studies have been developed. Here we report a bench stable Xantphos based Cu(I) catalyst for the cyclization of 4-pentynoic acid. A detailed mechanistic study of this reaction indicates that the reaction follows a dinuclear mechanism. The second order in copper catalyst explains that the reported protocols require high catalyst loadings to get substantial conversion. In order to accelerate this reaction, the application of the guanidinium based M12L24 nanosphere has been applied, allowing the pre-organization of copper complexes (based on sulfonated Xantphos) to increase the local concentration of the catalyst. An increase of the reaction rate by a factor of 50 is observed in going from 1 catalyst to 12 catalysts per sphere. This strategy allows the reaction to proceed at relatively low overall catalyst loading, leading to high yields of the product.Results and discussion
Inspired by recent examples of Cu(I) catalysts based on the Xantphos ligand, we were interested in its possible application in the cyclization of alkynoic acids. The wide bite-angle of this ligand has proven to provide good catalytic properties to Cu(I) complexes for different transformations.22 A simple reaction of the Xantphos ligand with copper(I) or (II) acetate in THF leads to clean formation of the desired complexes (XantphosCu(I) and XantphosCu(II) respectively, Fig. 1) in quantitative yield. Both complexes are bench stable and were fully characterized by a combination of techniques (see the ESI† for details). | ||
| Fig. 1 Structure of XantphosCu(I) and XantphosCu(II). | ||
To evaluate the catalytic activity of the copper Xantphos complexes in cyclization reactions, 4-pentynoic acid (1) was chosen as a benchmark substrate. The reactions were carried out at room temperature, in deuterated acetonitrile and monitored by NMR spectroscopy for 24 hours (Table 1; see Fig. S41 in the ESI† for more details). In all cases the 5-member ring lactone (2) was obtained as the only product. In the absence of a base the reaction is slow and only low yields are observed after 24 hours (entry 1 to 4). However, in the presence of triethylamine (Et3N) as a base, an increased yield of the reaction from low to moderate was achieved when XantphosCu(I) was applied (entry 5). In all experiments the copper(I) based catalyst provided higher yields than its copper(II) analogue, underlining the importance of the oxidation state of the metal in the reactivity of the catalyst. It is important to notice that for both oxidation states, Xantphos complexes were more active than the copper acetate in the absence of any ligand.
|
|
||
|---|---|---|
| Entry | Conditionsa | Yieldb (%) |
| a Reaction conditions: [1] = 10 mM, [Cu] = 0.5 mM, CD3CN, RT, 24 h. b Yield determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. c [Et3N] = 1.5 mM. | ||
| 1 | XantphosCu(I) | 12 |
| 2 | XantphosCu(II) | 4 |
| 3 | CuOAc | 2 |
| 4 | Cu(OAc)2 | 0 |
| 5c | XantphosCu(I) + Et3N | 39 |
| 6c | XantphosCu(II) + Et3N | 17 |
| 7c | CuOAc + Et3N | 11 |
| 8c | Cu(OAc)2 + Et3N | 0 |

After having established that XantphosCu(I) under basic conditions gave the highest conversion we decided to investigate more thoroughly the kinetic parameters of the reaction through monitoring the conversion over time for reactions with various starting concentrations of the base and catalyst (see ESI Fig. S42–S44†).
In all experiments the conversion increased linearly with time in the first phase of the reaction. At higher conversion, the reaction rate increased even further, indicating a small negative order in substrate 1. From these data, the observed rate constants for the various experiments were calculated (−kobs = slope of the representation; see Fig. 2). The following empirical rate equation for the reaction was established, which notably shows a higher order in the copper catalyst; r = k[1]−y[Et3N]x[XantphosCu(I)]2.
 | ||
| Fig. 2 Dependence of the kobs of the cyclization of 4-pentynoic acid with the concentration of XantphosCu(I) (kobs obtained from Fig. S44 in the ESI†). | ||
The second order kinetics in catalyst concentration suggests that a dinuclear species is involved in the rate-determining step of the reaction, ergo two copper complexes are necessary for the reaction to proceed efficiently. Dinuclear activation has also been reported for other copper catalyzed reactions. For example, the copper-catalyzed azide–alkyne cycloaddition (CuAAC) has been reported to proceed via a dinuclear mechanism in which the alkyne is σ–π-activated by two different copper metals.12 It is interesting to note that in the few reported examples of Cu(I) catalyzed cyclization of acetylenic acids, the catalyst loading necessary to achieve good yields is high (10 mol%).14a–c The second order dependency on copper catalyst explains why high concentration of catalysts is required in these protocols.
To further study the reaction mechanism, a catalytic reaction was performed with a deuterated alkyne as the substrate (Fig. 3b, and S45 in the ESI†). If the reaction would proceed exclusively via π-activation, only one deuterated product would be expected to form. However, from the experiment it is clear that deuterium ends in both cis and trans positions with respect to the oxygen of the product (Fig. 3b), thus indicating that the reaction proceeds via a σ-copper complex. This implies that with XanthposCu(I) only terminal alkynes can be activated, and indeed, a reaction with an internal alkyne (4-hexynoic acid) as the substrate did not lead to any conversion. In order to obtain information on the resting state of the catalyst, the catalytic reaction was monitored by 31P NMR for 24 hours. Only one peak was observed, which is attributed to the copper acetate complex, suggesting that the resting state is a XantphosCu(pentynoate) species (ESI, Fig. S46†).
 | ||
| Fig. 3 (a) Proposed mechanism for the cyclization of 1 by XantphosCu(I) through dinuclear intermediates, (b) deuterium labeling experiment and 1H NMR spectrum after 24 h of reaction, showing the mixture of products (for reaction conditions see ESI Fig. S45†), (c) CSI-HRMS of the reaction mixture showing the peaks belonging to intermediates [XantphosCu]+ and B, together with the isotopic pattern of the peak corresponding to B (up experimental spectrum, down simulated spectrum; for detailed spectra and reaction conditions see ESI Fig. S47–S49†). | ||
A plausible mechanism based on these data is displayed in Fig. 3a. The reaction starts with XantphosCu(pentynoate) A, identified as the resting state, that with a second copper complex forms intermediate B, in which the alkyne is π-activated. A base is required to convert this species into a σ-activated complex that we tentatively have attributed to C (not detected in this study), which is the rate determining step of the reaction with the formation of the di-copper complex at pre-equilibrium. These steps of the mechanism are in agreement with the positive order in base, and the second order in XantphosCu(I) determined in the kinetic study. After intramolecular attack, intermediate D is formed, which yields the product and intermediate A, after protodemetallation with the acidic substrate. Further evidence for the mechanism of the reaction was achieved by in situ CSI-HRMS23 (Coldspray Ionization High Resolution Mass Spectrometry, Fig. 3c and S47–S49 in the ESI†) experiments to identify possible intermediates. Intermediates A and C are neutral and therefore not visible in the MS spectra; however, [XantphosCu]+ and intermediate B are both observed. The major peak in the mass spectra corresponds to [XantphosCu]+ which may partly stem from intermediate A after substrate loss. Importantly, the detection of intermediate B (Fig. 3c and S49 in the ESI†) strongly supports the proposed mechanism displayed in Fig. 3a.
As the reaction mechanism proceeds via a dinuclear intermediate, explaining the second order in the copper catalyst, we anticipated that this reaction could be facilitated by using a supramolecular strategy that pre-organizes the copper catalysts. Therefore we hypothesized that our previously reported nano-concentrator10,11 could be used to achieve high local concentrations of copper catalyst to accelerate the rate of the cyclization reaction. This type of assembly strongly binds sulfonated guests; thus a sulfonated analogue of XantphosCu(I) (SXantphosCu(I), Scheme 1b) was synthesized and fully characterized (see the ESI† for details).
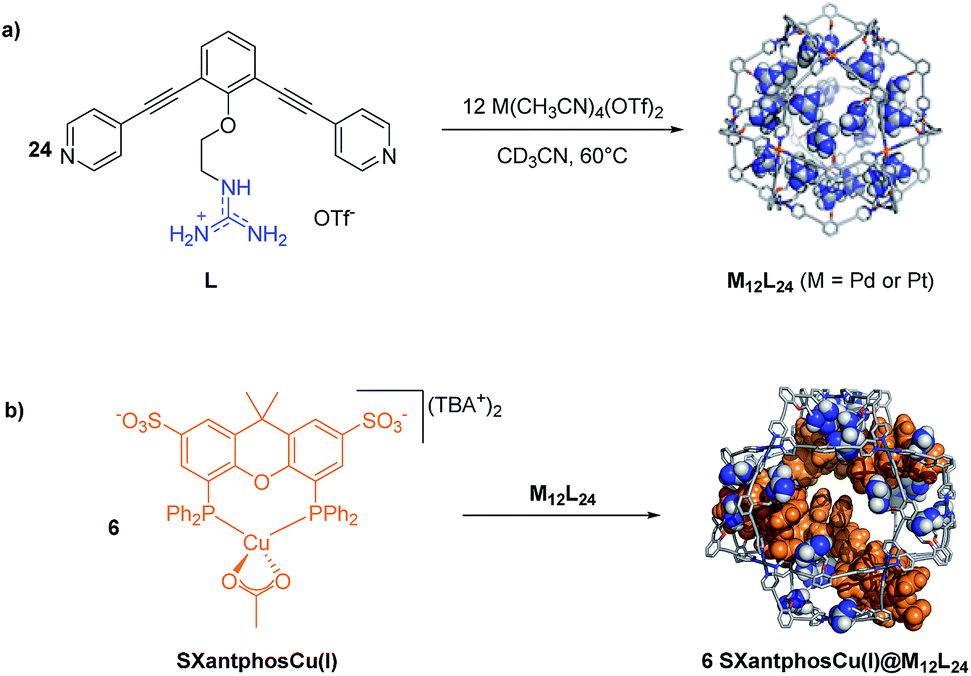 | ||
| Scheme 1 (a) Synthesis of nanospheres endohedrally functionalized with guanidinium groups. (b) Encapsulation of SXantphosCu(I) (TBA = tetrabutyl ammonium) in the nano-concentrator. | ||
The ability of the guanidinium spheres to encapsulate the SXantphosCu(I) catalyst was studied by diffusion-ordered NMR spectroscopy (DOSY). DOSY spectra were recorded for solutions containing the sphere and different amounts of SXantphosCu(I). In all the cases the spectra showed that the signals corresponding to the catalysts had the same D value as those belonging to the sphere (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = −9.6 m2 s−1), indicating strong binding of the guest copper complex to the guanidinium moieties in the sphere (see Fig. S1–S2 in the ESI†). When more than 12 equivalents of SXantphosCu(I) were added to the solution containing sphere, precipitation was observed, in agreement with our previous observations with mono-sulfonated guests.10 The binding between the spheres and SXantphosCu(I) was further confirmed by CSI-HRMS. The spectra corresponding to mixtures of the Pd12L24 sphere with different amounts of guest (4 and 12) show the presence of different species of type [Pd12L24 – xOTf + ySXantphosCu(I)]x−y (Fig. S3–S40 in the ESI,†y = 3–6 when 4 equivalents of guest are present and y = 8–13 for 12 equivalents).
D = −9.6 m2 s−1), indicating strong binding of the guest copper complex to the guanidinium moieties in the sphere (see Fig. S1–S2 in the ESI†). When more than 12 equivalents of SXantphosCu(I) were added to the solution containing sphere, precipitation was observed, in agreement with our previous observations with mono-sulfonated guests.10 The binding between the spheres and SXantphosCu(I) was further confirmed by CSI-HRMS. The spectra corresponding to mixtures of the Pd12L24 sphere with different amounts of guest (4 and 12) show the presence of different species of type [Pd12L24 – xOTf + ySXantphosCu(I)]x−y (Fig. S3–S40 in the ESI,†y = 3–6 when 4 equivalents of guest are present and y = 8–13 for 12 equivalents).
After having established that copper complexes can be pre-concentrated in nanospheres endohedrally functionalized with guanidinium groups, we explored the effect of changing the local concentration of copper catalysts in catalysis, while keeping the overall copper catalyst concentration constant. In line with a dinuclear mechanism, the lowest reaction rate (and yield) is observed when the lowest local concentration of SXantphosCu(I) within the nanosphere is used (Fig. 4 and in ESI Table S3 and Fig. S50†). When the copper/sphere ratio is one, the yield is lower than in the control experiment in the absence of the sphere (8% vs. 14%, Table S3 entry 1:Table S4, entry 2 in the ESI†). If the amount of SXantphosCu(I) per sphere is increased, both the yield and the reaction rate are much higher. A maximum of 80% yield of product 2 after 24 hours was achieved when the highest number of catalysts per sphere was used. Several control experiments confirmed that the role of the sphere is just to increase the local concentration of the catalyst and that other effects such as substrate pre-organization or the presence of guanidinium functional groups do not contribute to the increase in reaction rate (see Fig S41 and Table S4 in the ESI†).
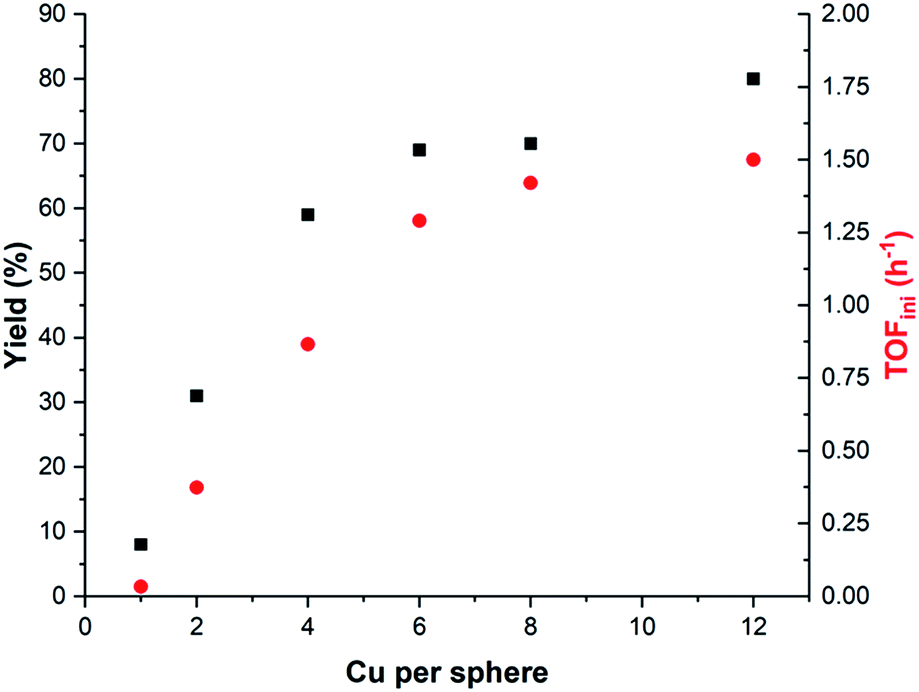 | ||
| Fig. 4 The yield and TOFini as a function of the number of SXantphosCu(I) catalysts within the sphere, while keeping the total copper concentration constant. For reaction conditions see Table S3.† | ||
The plot of both the TOFini and the yield against the number of catalysts within the sphere shows two different regimes. When the number of catalysts per sphere (at an overall constant copper concentration) is on average between 1 and 6, the TOFini increases almost linearly from 0.033 to 1.29 (Fig. 4). Increasing the number of Cu complexes further from 6 to 12 only slightly raises the TOFini to 1.50. Intrigued by those two kinetic regimes, we sought to probe the influence of the substrate and base concentration on the reaction rate. The influence of the base on the kinetics depends on the copper catalyst loading in the nanospheres (Fig. S51–S52 in the ESI†) and a partial order in the base was observed for 6 and 12 Cu per sphere. We also compared the TOFini as a function of the initial substrate concentration for the free complex XantphosCu(I) and the system at the highest local concentration (SXantphosCu(I)/sphere = 12) (Fig. 5). For XantphosCu(I), the TOFini remains the same in the window of substrate concentrations studied, in line with the earlier observed kinetics. Interestingly, an increase of the TOFini is observed when the reaction is performed with 12 SXantphosCu(I) in the nano-concentrator until 15 mM of 1 is achieved. Further increase of substrate concentration resulted in a decrease of TOFini, most likely as a result of cage precipitation. Importantly, the increase in TOFini with substrate concentration demonstrates that the kinetics has changed, now following r = k[1][Et3N]x, as the di-nuclear step is occurring rapidly when the copper catalysts are pre-organized in the sphere.
 | ||
| Fig. 5 Dependency of the TOFini on the substrate concentration at constant ratio [1]/[Et3N], when the XantphosCu(I) (red dots) is used or when 12 SXantphosCu(I) in the nanosphere are applied as the catalyst (black square). For reaction conditions see ESI Fig. S53† | ||
At lower catalyst loading (1 mol%) higher TON can be reached when the reaction is carried out at 50 °C. The TON (mol product/mol Cu) of the reaction was 94 (24 h, 94% yield, see Table S5†) when the reaction is carried out in the presence of the nanoconcentrator, which is almost 2.5 times higher than the TON when XantphosCu(I) is used under the same reaction conditions (24 h, 37% yield, see Table S5†). This again clearly demonstrates the effect of catalyst pre-organization for reactions that proceed via a dinuclear pathway.
Conclusions
In this contribution, we unraveled the mechanism of the Cu(I) (Xantphos) catalyzed cyclization of 4-pentynoic acid to the corresponding enol lactone. The reaction involves a bimetallic activation of the substrate, explaining why this type of reaction needs relatively high catalyst loading. Interestingly, pre-organization of copper catalysts based on SXanthphos (a sulfonated analogue) in a guanidinium functionalized M12L24 nanosphere leads to high local concentrations of catalysts and as a result improved reaction rates even when the average catalyst concentration is low. Having demonstrated that this supramolecular strategy facilitates reactions that proceed via dinuclear Cu(I) complexes, we can now extend these catalyst design strategies to related reactions based on abundant transition metals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the University of Amsterdam and by the European Research Council (ERC Advanced Grant 339786-NAT_CAT).Notes and references
- For reviews see: (a) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (c) D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed; (d) Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890 RSC; (e) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC; (f) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (g) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433 RSC; (h) M. Otte, ACS Catal., 2016, 6, 6491 CrossRef CAS.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS PubMed; (b) T. Iwasawa, R. J. Hooley and J. Rebek, Science, 2007, 317, 493 CrossRef CAS PubMed; (c) C. J. Walter, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Chem. Commun., 1993, 458–460 RSC; (d) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85 CrossRef CAS PubMed; (e) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed. Engl., 2013, 52, 7958 CrossRef CAS PubMed; (f) Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2013, 135, 16213 CrossRef CAS PubMed.
- (a) D. M. Dalton, S. R. Ellis, E. M. Nichols, R. A. Mathies, F. Dean Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 10128 CrossRef CAS PubMed; (b) D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 9202 CrossRef CAS PubMed; (c) Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358 CrossRef CAS PubMed; (d) D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS PubMed; (e) D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 9781 CrossRef CAS PubMed.
- M. A. Sarmentero, H. Fernández-Pérez, E. Zuidema, C. Bo, A. Vidal-Ferran and P. Ballester, Angew. Chem., Int. Ed., 2010, 49, 7489 CrossRef CAS PubMed.
- (a) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2014, 20, 4880 CrossRef CAS PubMed; (b) P. F. Kuijpers, M. Otte, M. Dürr, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, ACS Catal., 2016, 6, 3106 CrossRef CAS.
- (a) V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 113, 4401 CrossRef; (b) V. F. Slagt, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 1526 CrossRef CAS PubMed; (c) V. Bocokić, A. Kalkan, M. Lutz, A. L. Spek, D. T. Gryko and J. N. H. Reek, Nat. Commun., 2013, 4, 2670 CrossRef PubMed; (d) M. Kuil, T. Soltner, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2006, 128, 11344 CrossRef CAS PubMed; (e) T. Gadzikwa, R. Bellini, H. L. Dekker and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 2860 CrossRef CAS PubMed.
- For a review see: (a) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703 RSC . For endo-functionalisation see:; (b) S. Sota, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef PubMed; (c) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 1083–1085 CrossRef CAS PubMed; (d) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5133–5136 CrossRef CAS PubMed; (e) K. Suzuki, M. Kawano, S. Sato and M. Fujita, J. Am. Chem. Soc., 2007, 129, 10652–10653 CrossRef CAS PubMed; (f) T. Kikuchi, T. Murase, S. Sato and M. Fujita, Supramol. Chem., 2008, 20, 81–94 CrossRef CAS; (g) K. Suzuki, J. Iida, S. Sato, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 5780–5782 CrossRef CAS PubMed; (h) S. Sato, Y. Ishido and M. Fujita, J. Am. Chem. Soc., 2009, 131, 6064–6065 CrossRef CAS PubMed; (i) K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25–29 CrossRef CAS PubMed; (j) K. Suzuki, K. Takao, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2544–2545 CrossRef CAS PubMed; (k) K. Suzuki, K. Takao, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2011, 50, 4858–4861 CrossRef CAS PubMed; (l) K. Harris, Q. F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed; (m) C. J. Bruns, D. Fujita, M. Hoshino, S. Sato, J. F. Stoddart and M. Fujita, J. Am. Chem. Soc., 2014, 136, 12027–12034 CrossRef CAS PubMed . For exo-functionalisation see:; (n) N. Kamiya, M. Tominaga, S. Sato and M. Fujita, J. Am. Chem. Soc., 2007, 129, 3816–3817 CrossRef CAS PubMed; (o) M. Ikemi, T. Kikuchi, S. Matsumura, K. Shiba, S. Sato and M. Fujita, Chem. Sci., 2010, 1, 68–71 RSC; (p) T. Kikuchi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 15930–15932 CrossRef CAS PubMed; (q) K. Takao, K. Suzuki, T. Ichijo, S. Sato, H. Asakura, K. Teramura, K. Kato, T. Ohba, T. Morita and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 5893–5896 CrossRef CAS PubMed; (r) Q.-F. Sun, S. Sato and M. Fujita, Nat. Chem., 2012, 4, 330–333 CrossRef CAS PubMed; (s) F. Jiang, N. Wang, Z. Du, J. Wang, Z. Lan and R. Yang, Chem.–Asian J., 2012, 7, 2230–2234 CrossRef CAS PubMed.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS PubMed.
- R. Gramage-Doria, J. Hessels, S. H. A. M. Leenders, O. Tröppner, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2014, 53, 13380–13384 CrossRef CAS PubMed.
- Q. Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanovic-Burmazovic and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- F. Yu, D. Poole, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2018, 57, 11247–11251 CrossRef CAS PubMed.
- (a) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (b) B. T. Worrell, J. A. Malik and V. V. Fokin, Science (80-), 2013, 340, 457–460 CrossRef CAS PubMed.
- S. Santoro, R. Z. Liao, T. Marcelli, P. Hammar and F. Himo, J. Org. Chem., 2015, 80, 2649–2660 CrossRef CAS PubMed.
- (a) T. L. Mindt and R. Schibli, J. Org. Chem., 2007, 72, 10247–10250 CrossRef CAS PubMed; (b) C. Sun, Y. Fang, S. Li, Y. Zhang, Q. Zhao, S. Zhu and C. Li, Org. Lett., 2009, 11, 4084–4087 CrossRef CAS PubMed; (c) T. A. Fernandes, A. M. Galvão, A. M. Botelho do Rego and M. F. N. N. Carvalho, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3316–3323 CrossRef CAS; (d) S. Naidu and S. R. Reddy, ChemistrySelect, 2017, 2, 1196–1201 CrossRef CAS.
- M. Jiménez-Tenorio, M. Carmen Puerta, P. Valerga, F. Javier Moreno-Dorado, F. M. Guerra and G. M. Massanet, Chem. Commun., 2001, 259, 2324 RSC.
- M. J. Geier, C. M. Vogels, A. Decken and S. A. Westcott, Eur. J. Inorg. Chem., 2010, 2010, 4602–4610 CrossRef.
- (a) D. M. T. Chan, T. B. Marder, D. Milstein and N. J. Taylor, J. Am. Chem. Soc., 1987, 109, 6385–6388 CrossRef CAS; (b) S. Elgafi, L. D. Field and B. A. Messerle, J. Organomet. Chem., 2000, 607, 97–104 CrossRef CAS; (c) M. Viciano, E. Mas-Marzá, M. Sanaú and E. Peris, Organometallics, 2006, 25, 3063–3069 CrossRef CAS.
- (a) N. Nebra, J. Monot, R. Shaw, B. Martin-Vaca and D. Bourissou, ACS Catal., 2013, 3, 2930–2934 CrossRef CAS; (b) N. Á. Espinosa-Jalapa, D. Ke, N. Nebra, L. Le Goanvic, S. Mallet-Ladeira, J. Monot, B. Martin-Vaca and D. Bourissou, ACS Catal., 2014, 4, 3605–3611 CrossRef CAS; (c) J. Monot, P. Brunel, C. E. Kefalidis, N. Á. Espinosa-Jalapa, L. Maron, B. Martin-Vaca and D. Bourissou, Chem. Sci., 2016, 7, 2179–2187 RSC; (d) D. Ke, N. Á. Espinosa, S. Mallet-Ladeira, J. Monot, B. Martin-Vaca and D. Bourissou, Adv. Synth. Catal., 2016, 358, 2324–2331 CrossRef CAS.
- J. Alemán, V. del Solar and C. Navarro-Ranninger, Chem. Commun., 2010, 46, 454–456 RSC.
- V. Dalla and P. Pale, New J. Chem., 1999, 23, 803–805 RSC.
- (a) E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J.-P. Genêt and V. Michelet, J. Am. Chem. Soc., 2006, 128, 3112–3113 CrossRef CAS PubMed; (b) H. Harkat, J.-M. Weibel and P. Pale, Tetrahedron Lett., 2006, 47, 6273–6276 CrossRef CAS; (c) H. Harkat, A. Y. Dembelé, J.-M. Weibel, A. Blanc and P. Pale, Tetrahedron, 2009, 65, 1871–1879 CrossRef CAS.
- (a) H. Ito, A. Watanabe and M. Sawamura, Org. Lett., 2005, 7, 1869–1871 CrossRef CAS PubMed; (b) H. Ito, C. Kawakami and M. Sawamura, J. Am. Chem. Soc., 2005, 127, 16034–16035 CrossRef CAS PubMed; (c) J. Huang, J. Chan, Y. Chen, C. J. Borths, K. D. Baucom, R. D. Larsen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 3674 CrossRef CAS PubMed; (d) H. Ito, Y. Sasaki and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 15774–15775 CrossRef CAS PubMed; (e) C. Zhong, Y. Sasaki, H. Ito and M. Sawamura, Chem. Commun., 2009, 0, 5850 RSC; (f) H. Ohmiya, T. Moriya and M. Sawamura, Org. Lett., 2009, 11, 2145–2147 CrossRef CAS PubMed; (g) R. Alfaro, A. Parra, J. Alemán, J. L. García Ruano and M. Tortosa, J. Am. Chem. Soc., 2012, 134, 15165–15168 CrossRef CAS PubMed; (h) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, Angew. Chem., Int. Ed., 2012, 51, 3953–3956 CrossRef CAS PubMed; (i) S. Lee, D. Li and J. Yun, Chem.–Asian J., 2014, 9, 2440–2443 CrossRef CAS PubMed; (j) A. Parra, A. López, S. Díaz-Tendero, L. Amenós, J. Ruano and M. Tortosa, Synlett, 2015, 26, 494–500 CrossRef CAS; (k) H. Murayama, K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 2039–2041 CrossRef CAS PubMed.
- (a) S. Sakamoto, M. Fujita, K. Kim and K. Yamaguchi, Tetrahedron, 2000, 56, 955–964 CrossRef CAS; (b) K. Yamaguchi, J. Mass Spectrom., 2003, 38, 473–490 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03767a |
| This journal is © The Royal Society of Chemistry 2019 |