
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative steps during the biosynthesis of squalestatin S1†
Karen E.
Lebe
and
Russell J.
Cox
 *
*
Institute for Organic Chemistry, BMWZ, Leibniz Universität Hannover, Schneiderberg 38, 30167 Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
First published on 15th November 2018
Abstract
The squalestatins are a class of highly complex fungal metabolites which are potent inhibitors of squalene synthase with potential use in the control of cholesterol biosynthesis. Little is known of the chemical steps involved in the construction of the 4,8-dioxa-bicyclo[3.2.1]octane core. Here, using a combination of directed gene knockout and heterologous expression experiments, we show that two putative non-heme-iron-dependent enzymes appear to catalyse a remarkable series of six consecutive oxidations which set up the bioactive core of the squalestatins. This is followed by the action of an unusual copper-dependent oxygenase which introduces a hydroxyl required for later acetylation.
Introduction
Squalestatin S1 1 (also known as zaragozic acid)1 is a picomolar inhibitor of squalene synthase (SS) and is produced by various fungi.2 Isotopic feeding experiments using 13C-labelled acetates show that 1 consists of two polyketides: a benzoate-primed hexaketide, which forms the core moiety; and a dimethylated tetraketide ester side chain. More sophisticated experiments using [1-13C]-acetate in combination with 18O2 showed that the carbon skeleton of 1 is heavily oxidised during biosynthesis and that oxygens at positions 1, 3, 5, 6, 7 and 12 are derived from atmospheric oxygen (Scheme 1) to form the 4,8-dioxa-bicyclo[3.2.1]octane core.3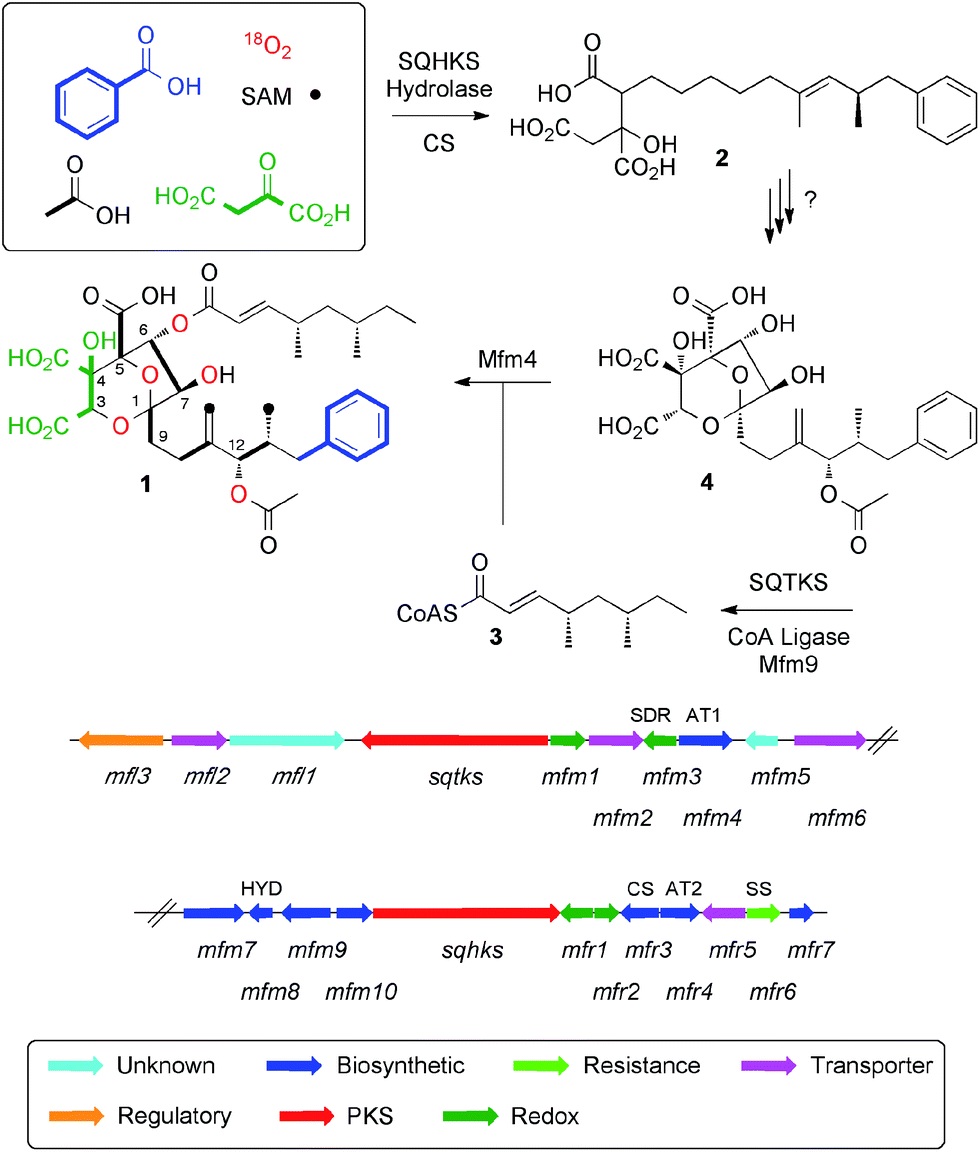 | ||
| Scheme 1 Squalestatin S1 1, biosynthetic origin of heavy atoms and the biosynthetic gene cluster in Phoma MF5453. Abbreviations: SAM, S-adenosyl methionine; SQTKS, squalestatin tetraketide synthase; SQHKS, squalestatin hexaketide synthase; SDR, short-chain dehydrogenase; AT, acyl transferase; HYD, hydrolase; CS, citrate synthase; SS, squalene synthase. | ||
We have reported the biosynthetic gene cluster (BGC) which encodes the construction of 1 in Phoma species,4 (Fig. 1) and more recently Tang and coworkers have described a homologous cluster in the fungus Curvularia lunata.5 These BGC contain two PKS genes, one of which encodes squalestatin tetraketide synthase (SQTKS)6 and the other encodes squalestatin hexaketide synthase (SQHKS). The clusters also each contain a gene for a citrate synthase-like (CS) protein which is also present in maleidride biosynthetic gene clusters7 and the CS is known to attach oxaloacetate groups to the α-carbons of polyketides. Tang's group showed through heterologous expression that SQHKS and CS, combined with a hydrolase, form the hexaketide citrate 2 that appears to be the first enzyme-free intermediate in the pathway (Scheme 1).5 Our experiments have shown that the final step of the pathway involves addition of the squalestatin tetraketide 3 to O-6 of intermediate 4 catalysed by the acyl transferase Mfm4.4 This protein shows broad substrate selectivity and is probably responsible for the wide range of related compounds known.1
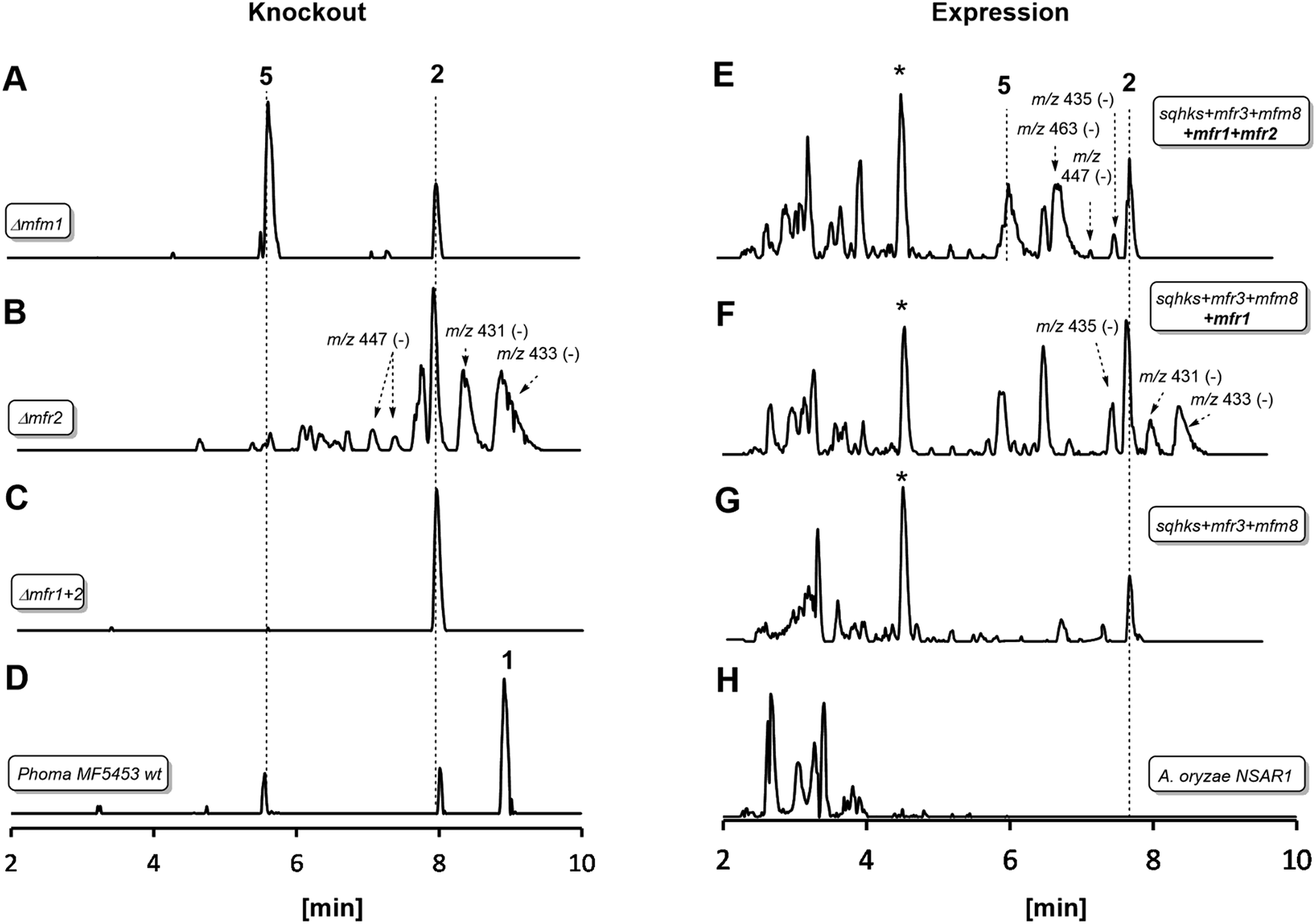 | ||
| Fig. 1 LCMS traces for key experiments (ES− total ion current). Left hand panel shows results of knockout experiments. Right hand panel shows results of heterologous expression experiments in Aspergillus oryzae. Indicated molecular ions in ES− mode. See text for HRMS data and ESI† for MSMS and partial NMR analysis. * Unrelated compound (see ESI†). A. oryzae in experiments (E), (F) and (G) also contained genes to create benzoyl CoA. All m/z values are for the observed [M − H]− species. See text for description of individual panels. | ||
However the steps leading to the construction of the highly functionalised core of the squalestatins responsible for the potent inhibition of SS remain enigmatic.8 Our previous work showed that the squalestatin S1 1 gene cluster contains few obvious oxygenase encoding genes despite the requirement for up to seven oxidative steps. We now report a series of experiments that elucidate the genes and biosynthetic steps leading to the construction of the 4,8-dioxabicyclo-[3.2.1]octane motif and oxygenation at C-12.
Results
The SQS1 1 BGC in Phoma MF5453 has been previously described.4 It consists of 22 contiguous genes spanning ca 50 Kb of the genome. Initial bioinformatic examination revealed few possible genes encoding potential oxygenases despite the fact that several oxidative steps must be required to form the core of 1. The gene mfm3 encodes an NAD(P)H-dependent short-chain dehydrogenase/reductase (SDR) which is unlikely to be able to oxygenate. Initial BLASTp analysis of mfr1 and mfr2 showed no significant homology to proteins of known function, but more detailed structural analysis using PHYRE-2 (ref. 9) (see ESI†) suggested that mfr1 might encode a non-heme iron protein as it shows some structural similarity (although less than 20% sequence identity) to CytC3 which is involved in halogenation of aminobutyric acid in soil Streptomycetes.10 Mfr2 showed even lower similarity to other known proteins, but since it is 41.1% identical to Mfr1 this pair of enzymes might be involved in oxygenation. The gene mfm1 encodes a protein which shows some structural similarity to copper-dependent oxygenases (e.g. peptidylglycine alpha-amidating monooxygenases, see ESI†).11 No other genes in the SQS1 1 BGC appeared to encode oxygenases and we therefore focussed our efforts on these three targets.Extensive attempts to obtain quantities of soluble Mfm1 using the expression hosts Escherichia coli, Saccharomyces cerevisae and Spodoptera frugiperda failed. Likewise, soluble Mfr1 and Mfr2 have not yet been obtained from E. coli or S. cerevisae. This ruled out the possibility of investigating the oxidation steps in vitro. We thus turned to a combination of heterologous expression and targetted knockout (KO) experiments.
In an initial gene KO experiment using Phoma MF5453, the neighbouring genes mfr1 and mfr2 were deleted together using the bipartite gene inactivation method reported by Nielsen and coworkers.12 This led to abolition of SQS1 1 biosynthesis (Fig. 1C), and formation of a compound with molecular formula C23H32O7 (calc. [M − H]− HRMS 419.2070, measured 419.2070). This compound was purified and methylated to 2A with TMS-CHN2 (ref. 13) prior to full NMR structure determination which proved it to be the hexaketide citrate 2 (ref. 14) (m/z [M − H]− 419) showing that Mfr1 and Mfr2 must act early in the pathway, and that the squalestatin hexaketide synthase (SQHKS) fully reduces at C-1 (1-numbering). MSMS analysis of this compound in ES− mode shows facile and distinctive losses of water and CO2.
Individual KO of mfr1 alone gave the same chemotype as the dual KO (see ESI†), but KO of mfr2 alone gave a mixture of 2 plus several compounds in very low titres with m/z values ([M − H]−) of 431.2, 433.2, 435.2 (very weak) and 447.2 (Fig. 1B). LC-HRMS analysis (see Scheme 2 and ESI† for details) confirmed these to be oxidised congeners of 2. MSMS analysis of the m/z 433, 431 and 447 compounds showed losses of CO2 and H2O again, but also facile loss of the oxaloacetate (m/z −132, see ESI†) moiety proving the oxidations to have occurred on the hexaketide backbone. Partial 1H NMR of isolated compounds supported the presence of structural features including the phenyl and dimethylated triketide, but further structural information could not be determined by NMR for these compounds (see ESI†) due to their very low titres and consequent difficulties of purification.
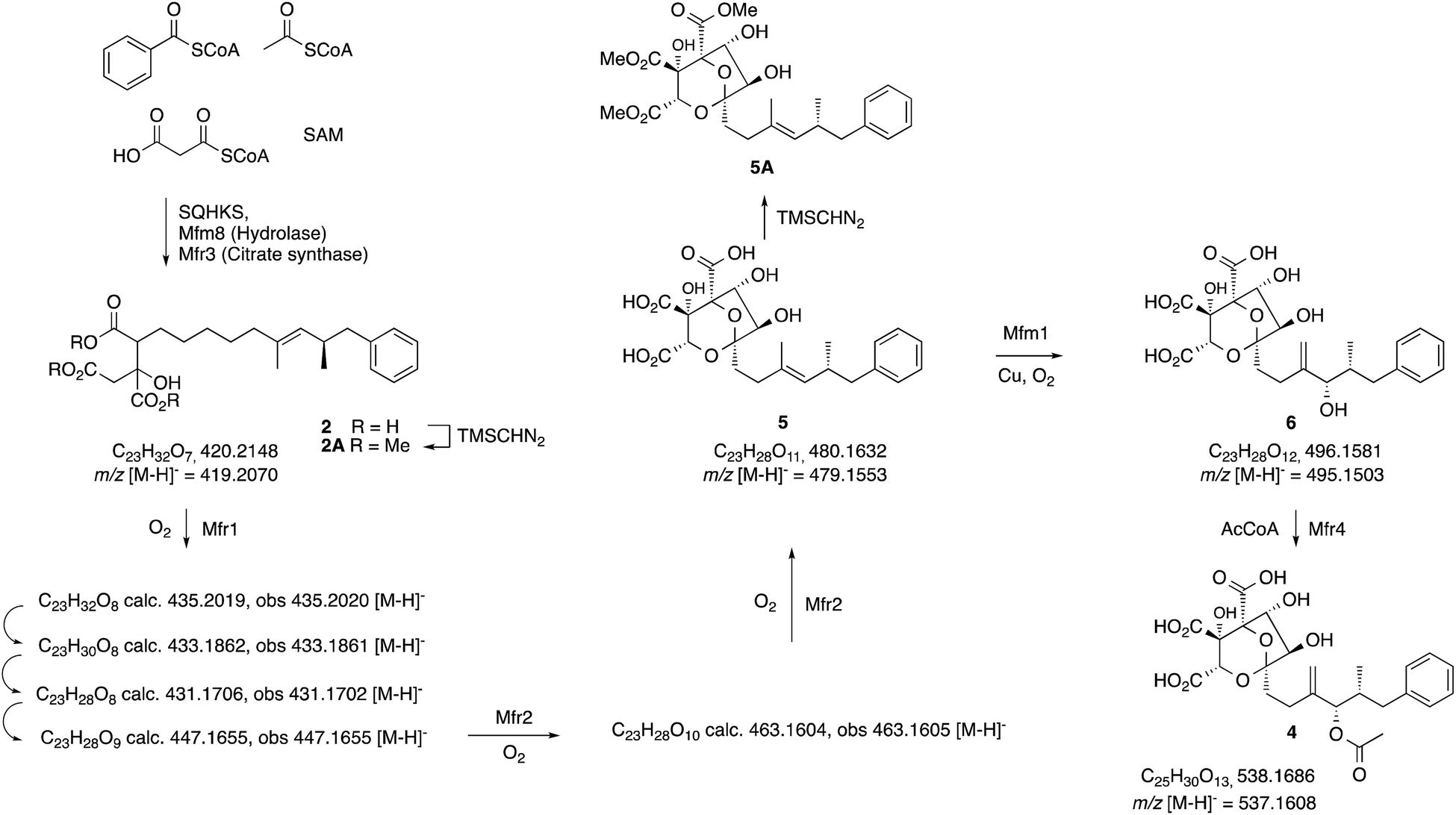 | ||
| Scheme 2 Observed intermediates from KO and expression experiments. All m/z values are for the observed [M − H]− species. | ||
Heterologous expression experiments were then deployed to examine these transformations in more detail using Aspergillus oryzae as the host, and the modular fungal expression system described by Lazarus and coworkers.15 Tang and coworkers have already shown that the C. lunata hexaketide synthase (clz14 = sqhks) must be co-expressed with a hydrolase (clz11 = mfm8) and the citrate synthase (clz17 = mfr3). Benzoyl CoA is the likely starter unit for SQHKS and this is presumably supplied endogenously in the Aspergillus nidulans host used by Tang et al. to produce the hexaketide citrate 2 in very low titre (ca 0.1 mg L−1).5A. oryzae does not appear to be able to synthesise benzoyl CoA.16 In initial experiments we therefore supplemented fermentations with benzoyl SNAC,17 which is a benzoyl CoA mimic, but with no success. We then used a different strategy and produced benzoyl CoA in situ by cotransformation of A. oryzae with benzoyl CoA biosynthesis-encoding genes from Strobilurus tenacellus17 with the MF5453 SQHKS, CS and hydrolase genes. In our hands this produced a better titre of the hexaketide citrate 2 (ca 1 mg L−1 after purification, Fig. 1G). Although 2 was produced in limiting amounts, we decided it was worth attempting to extend the pathway by adding the genes encoding the oxidations, despite the risk of reducing titres still further.
Absence of CS, the hydrolase or the benzoyl CoA-forming enzymes from the expression strains resulted in no observed product. Likewise, knockout of mfr3 encoding the CS in the wild-type (WT) Phoma strain abolished biosynthesis of 1 without formation of any observable intermediate (see ESI†). However, coexpression of the mfr1 oxidase with sqhks, the mfm8 hydrolase and the mfr3 citrate synthase then produced a very similar panel of oxidised congeners as observed in the mfr2 KO experiment in Phoma, albeit in very low titres (Fig. 1F). However, higher concentrations of the m/z 435 (m/z [M − H]−) compound were observed. Comparison of retention times, HRMS, MSMS and partial 1H NMR data, with the compound from the mfr2 KO experiment showed them to be the same.
Introduction of mfr2 to the A. oryzae expression system then gave clear production of a new compound 5 (m/z 479.2 [M − H]−, Fig. 1E), again in low titre, which was characterised by HRMS and MSMS. A less oxidised congener of 5 was also observed with an m/z value of 463.2 ([M − H]−, see ESI†). This was supported by LC-HRMS, MSMS and partial 1H NMR (ESI†), although full NMR structure elucidation was again prevented by low titre.
The gene mfm1 appears to encode a copper-dependent oxygenase. Its KO created a Phoma strain which produced 5 in high yield (20 mg L−1, Fig. 1A), together with smaller amounts of 2. This shows that the Mfm1 oxidation step follows the Mfr1 and Mfr2 oxygenations. Compound 5 was purified and converted to its trimethylester 5A (Scheme 2) which facilitated full characterisation by NMR.
The usual production media for 1 contains additional copper (2.9 μM). In order to probe the copper-dependency of the Mfm1-catalysed step we incubated WT Phoma MF5453 in media prepared without the addition of copper ions, and in these experiments we observed a significant reduction in the production of 1, and production of 5 instead (see ESI†).
Finally, the gene mfr4 is predicted to encode an acetyltransferase. Its KO in Phoma MF5453 led to abolition of 1 production and production of 2, 5 and 6 instead (see ESI†). The structure of 6 was confirmed by comparison to the previously described complete hydrolysis product of 1.4
Discussion
These results, combined with our earlier results and those of Tang and coworkers provide a full biosynthetic pathway for the production of 1 in which the order of the enzymes encoded by the squalestatin BGC is now elucidated (Scheme 3). The hexaketide synthase, primed by benzoyl CoA, produces the dimethylated tetraketide citrate 2 in cooperation with the hydrolase and CS proteins. Previous speculation about the oxygen at C-1 could not resolve whether this position was fully reduced by SQHKS, or left as a ketone or alcohol, because late formation of the C-1 acetal could exchange the oxygen. Our results show that 2 is the substrate for stepwise oxidation of this position by the putative non-heme iron oxygenase Mfr1, most likely to an alcohol (7, m/z 435.2 [M − H]−), a ketone (8, m/z 433.2 [M − H]−) and then an unsaturated ketone (9, m/z 431.2 [M − H]−). Further oxidation could give the 3,4-epoxide (10, m/z 447.2, [M − H]−, Scheme 3). Two peaks corresponding to the calculated mass of 10 (Fig. 1B) may correspond to keto and hemiacetal forms. These intermediates are present in very low titres in the Phoma MF5453 KO strain and proved extremely difficult to purify, probably because of facile shunts and degradations of 8–10 due to their electrophilic nature. In A. oryzae, production of 2 at 1 mg L−1, while higher than previous reports, considerably constrains the amounts of 7–10 which can be observed. However MSMS data convincingly shows that the oxidative modifications performed by Mfr1 leading to 7–10 occur on the hexaketide and not on the oxaloacetate moiety of 2.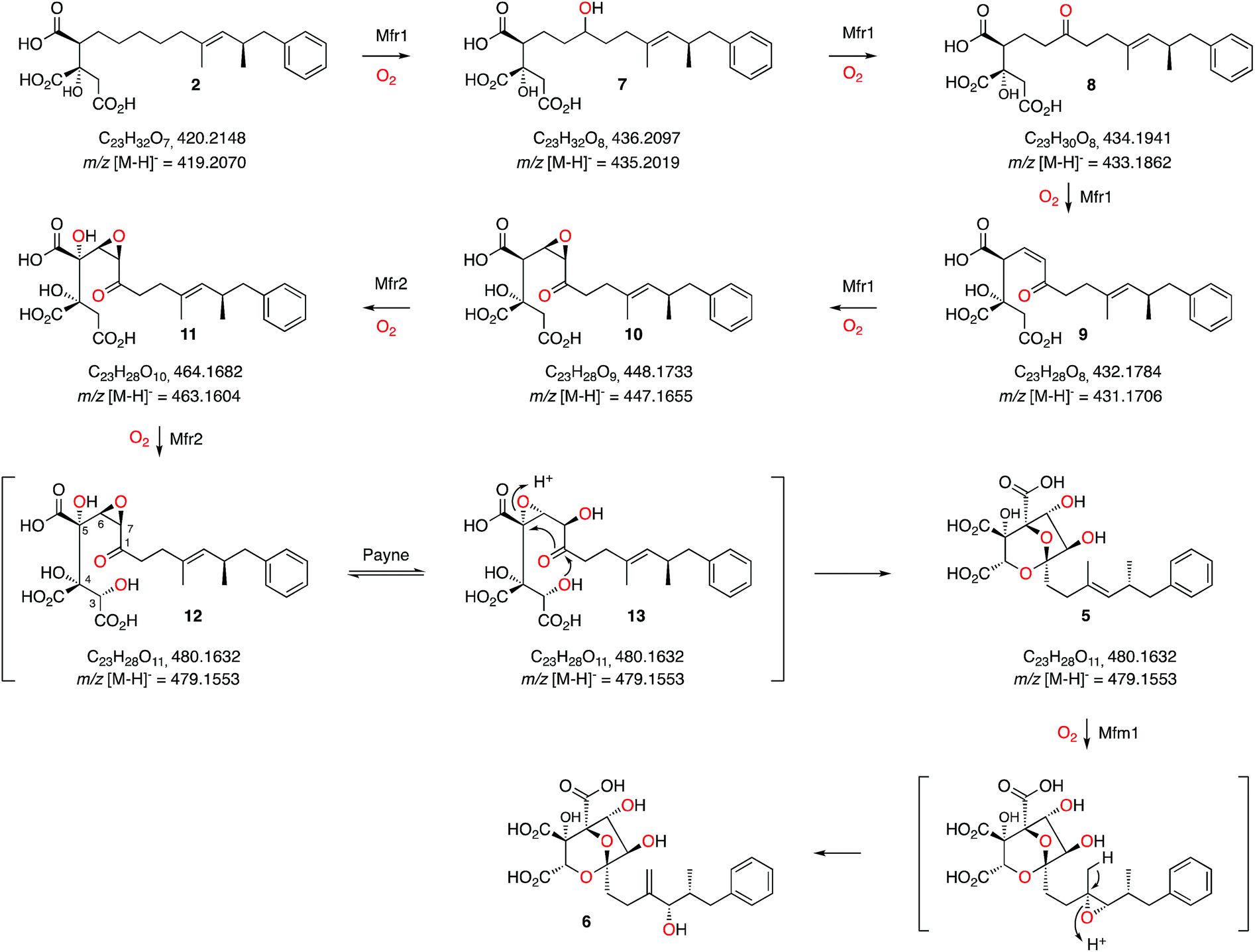 | ||
| Scheme 3 Proposed sequence of oxidative steps during the formation of 6. Masses are given for both the neutral and anionic species where observed. | ||
The MS analysis is consistent with Mfr2 hydroxylating at C-5 and C-3 to give 11 and 12, although low titres and instability of these compounds again precluded full structural elucidation. However MSMS analysis of 11 and earlier compounds 2 and 7–10 showed retro-aldol loss of unmodified oxaloacetate (m/z 132) which was not observed for 5 (see ESI†) indicating that hydroxylation at C-3 of 11 is probably the final oxidative step catalysed by Mfr2. Facile Payne rearrangement of 12 would furnish 13 which could undergo acetal formation and concomitant transannular epoxide opening to give the observed intermediate 5 (Scheme 3).
It is clear from our results that intermediates on the pathway are highly unstable and it is thus fascinating to consider how such a pathway could have evolved as the later oxidation products of Mfr1 alone appear to be transient species. Interestingly, intermediate 2 has been isolated from producing organisms as a minor cometabolite of 1 and it is known to be a micromolar inhibitor of SS.15 Gain by the pathway of Mfr1 and Mfr2 (possibly by duplication), however, significantly increases the potency of the pathway product. Recent efficient synthetic routes towards the squalestatins illustrate the mandatory requirement for protecting group strategies when nucleophilic and electrophilic species are in close proximity, emphasising the instability of intermediates 7 to 13.18 Nature, however, appears to solve this problem by using catalysts which can passage unstable intermediates rapidly from 2 towards stable products such as 5.
Oxidation of the 11,12-olefin of 5 probably involves epoxidation catalysed by the copper-dependent oxygenase Mfm1 and rearrangement to give the allylic alcohol 6 (Scheme 2). A similar epoxidation/rearrangement sequence is performed by the cytochrome P450 oxygenase LovA during the biosynthesis of lovastatin.19
The oxidative pathway that converts 2 to 6 determined here is fully consistent with the earlier 18O2 feeding experiments.3 Acetylation of 6 at O-9 (Mfr4) then gives 4 which is already known as the substrate for the final acylation reaction catalysed by Mfm4.4 Thus three enzymes, Mfr1, Mfr2 and Mfm1 appear to catalyse a remarkably efficient series of seven sequential oxidations to convert polyketide citrate 2 to the highly functionalised squalestatin precursor 6.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KEL thanks the Leibniz Universität Hannover for funding. DFG is thanked for the provision of LCMS equipment (INST 187/621-1). Simon Blazy, Annika Stein and Michelangelo Marasco are thanked for technical assistance. Dr Gerald Dräger is thanked for assistance with HRMS and MSMS.Notes and references
- J. D. Bergstrom, M. M. Kurtz, D. J. Rew, A. M. Amend, J. D. Karkas, R. G. Bostedor, V. S. Bansal, C. Dufresne, F. L. VanMiddlesworth and O. D. Hensens, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 80–84 CrossRef CAS.
- M. J. Dawson, J. E. Farthing, P. S. Marshall, R. F. Middleton, M. J. O'Neill, A. Shuttleworth, C. Stylli, R. M. Tait, P. M. Taylor and H. G. Wildman, J. Antibiot., 1992, 45, 639–647 CrossRef CAS PubMed.
- C. A. Jones, P. J. Sidebottom, R. J. Cannell, D. Noble and B. A. Rudd, J. Antibiot., 1992, 45, 1492–1498 CrossRef CAS PubMed.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
- N. Liu, Y.-S. Hung, S.-S. Gao, L. Hang, Y. Zou, Y.-H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS PubMed.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- A. J. Szwalbe, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 51, 17088–17091 RSC; K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2016, 55, 6784–6788 CrossRef CAS PubMed.
- P. A. Procopiou, B. Cox, E. J. Bailey, M. J. Bamford, A. P. Craven, M. G. Lester, B. W. Dymock, A. D. McCarthy, J. G. Houston, P. J. Sharratt, J. L. Hutson, M. A. Snowden, B. E. Kirk, S. J. Spooner, M. Sareen, N. S. Watson, J. J. Scicinski, J. Widdowson, P. J. Sharratt, M. A. Snowden, N. S. Watson and R. J. Williams, J. Med. Chem., 1996, 39, 1413–1422 CrossRef CAS PubMed.
- S. Mezulis, C. M. Yates, M. N. Wass, M. J. E. Sternberg and L. A. Kelley, Nat. Protoc., 2015, 10, 845–858 CrossRef PubMed.
- C. Wong, D. G. Fujimori, C. T. Walsh and C. L. Drennan, J. Am. Chem. Soc., 2009, 131, 4872–4879 CrossRef CAS PubMed.
- S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Nat. Struct. Biol., 1999, 6, 976–983 CrossRef CAS PubMed.
- M. L. Nielsen, L. Albertsen, G. Lettier, J. B. Nielsen and U. H. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed.
- E. Kuehnel, D. D. R. Laffan, G. C. Lloyd-Jones, T. M. del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075–7078 CrossRef CAS PubMed.
- G. Harris, C. Dufresne, H. Joshua, L. Koch, D. Zink, P. Salmon, K. Göklen, M. Kurtz, D. Rew, J. Bergstrom and K. Wilson, Bioorg. Med. Chem. Lett., 1995, 5, 2403–2408 CrossRef CAS.
- K. A. K. Pahirulzaman, K. Williams and C. M. Lazarus, Methods Enzymol., 2012, 517, 241–260 CAS.
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef PubMed.
- M. I. Kim, S. J. Kwon and J. S. Dordick, Org. Lett., 2009, 11, 3806–3809 CrossRef CAS PubMed.
- H. A. A. Almohseni, H. H. Al Mamari, A. Valade, H. O. Sintim and D. M. Hodgson, Chem. Commun., 2018, 1–3 Search PubMed.
- J. Barriuso, D. T. Nguyen, J. W. H. Li, J. N. Roberts, G. MacNevin, J. L. Chaytor, S. L. Marcus, J. C. Vederas and D.-K. Ro, J. Am. Chem. Soc., 2011, 133, 8078–8081 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures and analytical data. See DOI: 10.1039/c8sc02615g |
| This journal is © The Royal Society of Chemistry 2019 |