
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile approach to 2-alkoxyindolin-3-one and its application to the synthesis of N-benzyl matemone†
Makoto Shimizu *ab,
Hayao Imazatob,
Isao Mizotab and
Yusong Zhua
*ab,
Hayao Imazatob,
Isao Mizotab and
Yusong Zhua
aSchool of Energy Science and Engineering, Nanjing Tech University, Nanjing 211816, Jiangsu Province, China
bDepartment of Chemistry for Materials, Graduate School of Engineering, Mie University, Tsu, Mie 514-8507, Japan. E-mail: mshimizu@chem.mie-u.ac.jp
First published on 3rd June 2019
Abstract
2-Alkoxycarbonylindolin-3-one is synthesized from a methoxyglycine derivative via a 1,2-aza-Brook rearrangement followed by cyclization with bis(trimethylsilyl)aluminum chloride. A short-step synthesis of N-benzyl matemone is successfully carried out using the present indolin-3-one synthesis.
Introduction
Heterocyclic compounds possessing an oxindole1 skeleton have received considerable attention due to the widespread existence of naturally occurring bioactive materials containing this particular heterocycle. Among them bromine-containing and/or 2-alkoxy indolin-3-one and indole alkaloids such as matemone 3,2 cephalinone 4,3 and bromoaplysinopisin 6 (ref. 4) show intriguing bioactivities. Regarding matemone, it was isolated from the Indian Ocean sponge Iotrochota purpurea and its structure was elucidated in 2000. Matemone shows mild cytotoxicity against three cancer cell lines and marginal antibacterial activity against Staphylococcus aureus. We have been interested in the reactivity of α-iminoesters in umpolung reactions,5 and a facile indolin-3-one synthesis via aza-Brook rearrangement has been developed (Scheme 1).6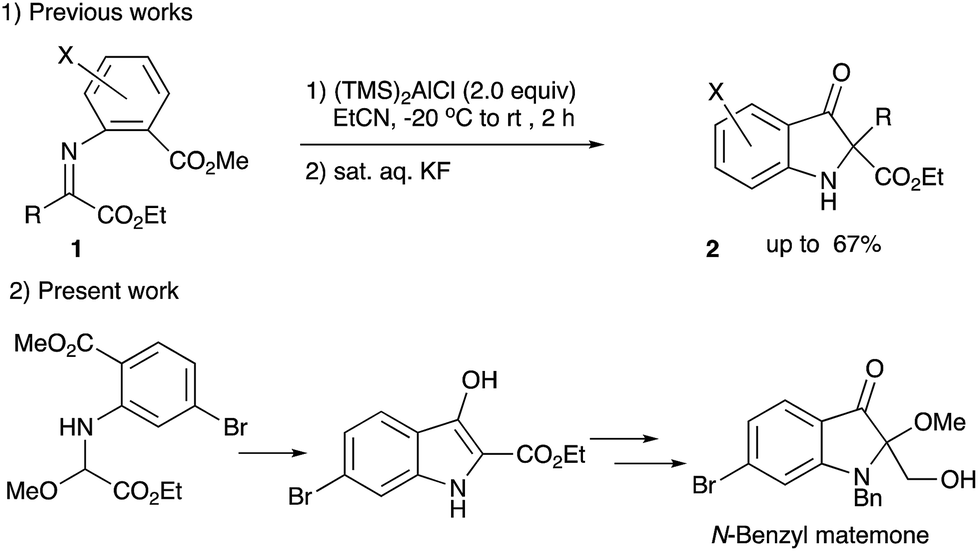 | ||
| Scheme 1 A new approach to indolin-3-ones and the present N-benzyl matemone synthesis. | ||
However, difficulties have been encountered regarding the substituents at the 2-position, i.e., only 2,2-disubstituted derivatives 2 could be synthesized by our previously reported procedure (compound 2, R = Ar or CO2R′).
For the construction of matemone and related structures, a procedure using the aldimine of type 1 (R = H) is needed; in particular, a facile approach to 2-mono-substituted indolin-3-one, a key intermediate is needed. We have now found that methoxyglycine derivative 11 serves as a good precursor to the aldimine 10, and 2-alkoxycarbonylindolin-3-one has been successfully synthesized using this particular imine precursor 11 (Scheme 2).
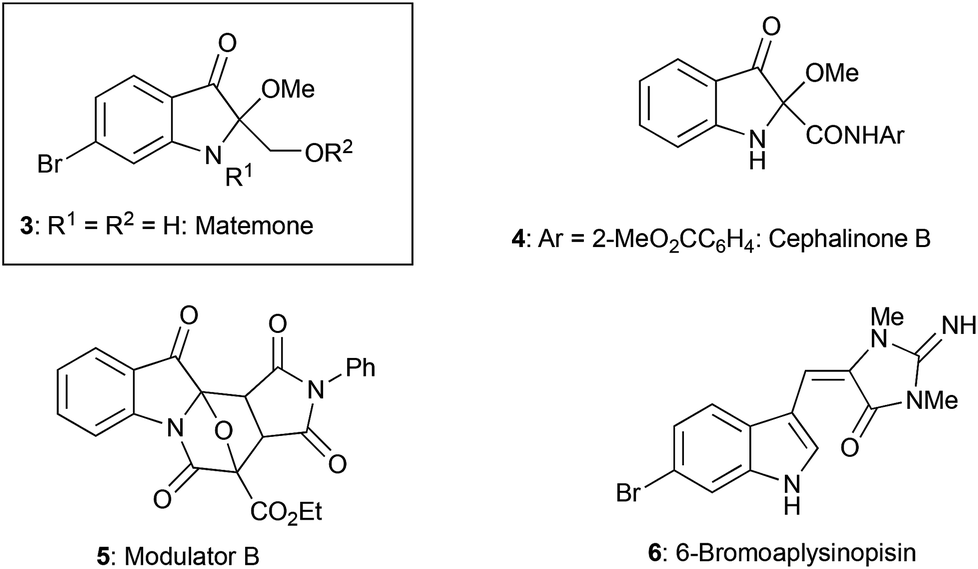 | ||
| Scheme 2 Bioactive compounds possessing 2-alkoxyindolin-3-one and a related structure. | ||
Results and discussion
For the synthesis of this particular aldimine 10, we examined several approaches, such as direct imination of glyoxylate through dehydration and oxidation of glycine derivatives 9 (MnO2, DDQ, NBS, etc.).7 However, none of the attempted procedures worked, and only complex mixtures were obtained (Scheme 3).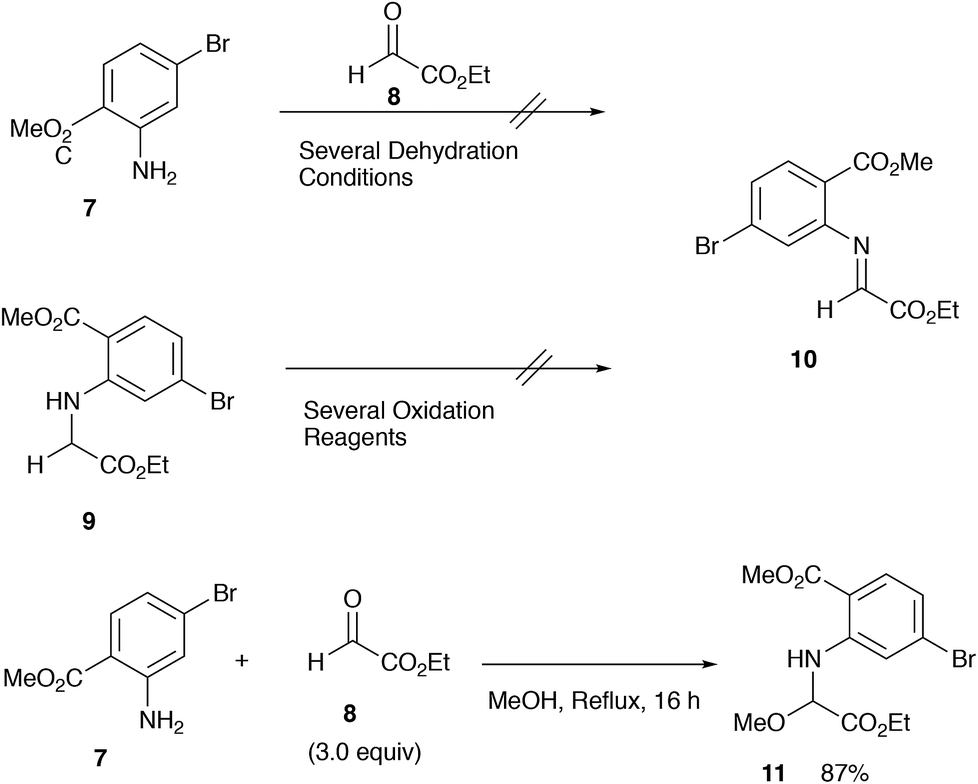 | ||
| Scheme 3 Attempted synthesis and a precursor 11 to the aldimine 10. | ||
We finally found that the methoxyglycine derivative 11 could be isolated in good yield and served as a stable imine precursor.8 Cyclization reaction of this methoxy amino diester 11 was carried out with (TMS)2AlCl,9 and the results are summarized in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Temperature | (TMS)2AlCl (equiv.) | Solvent | Yield of 12a (%) |
| a Isolated yield. | ||||
| 1 | −78 °C to rt | 2.0 | EtCN | 15 |
| 2 | −78 °C to rt | 4.0 | EtCN | 56 |
| 3 | −78 °C to rt | 6.0 | EtCN | 38 |
| 4 | −40 °C to rt | 4.0 | EtCN | 37 |
| 5 | −78 to 0 °C | 4.0 | EtCN | 36 |
| 6 | −78 to 50 °C | 4.0 | EtCN | 46 |
| 7 | −78 °C to rt | 4.0 | CH2Cl2 | 6 |
| 8 | −78 °C to rt | 4.0 | EtCN/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
50 |
| 9 | −78 °C to rt | 4.0 | Et2O | 26 |
| 10 | −78 °C to rt | 4.0 | THF | 38 |
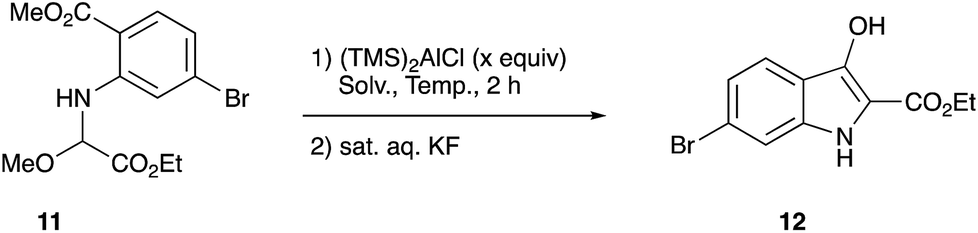
An initial examination using 2.0 equiv. of (TMS)2AlCl in EtCN as a solvent led to the formation of the desired indolin-3-one 12 in only 15% yield (entry 1). Increasing the amount of (TMS)2AlCl to 4.0 equiv. improved the yield to 56% (entry 2). However, the use of a large excess of the reagent decreased the yield (entry 3). Use of other solvents such as CH2Cl2, Et2O, and THF was unsuccessful (entries 7, 9 and 10). Regarding the reaction temperature, the treatment of the starting material 11 with (TMS)2AlCl at −78 °C, followed by warming the whole mixture to room temperature recorded the best result (entry 2). The following Scheme 4 shows a possible reaction pathway.
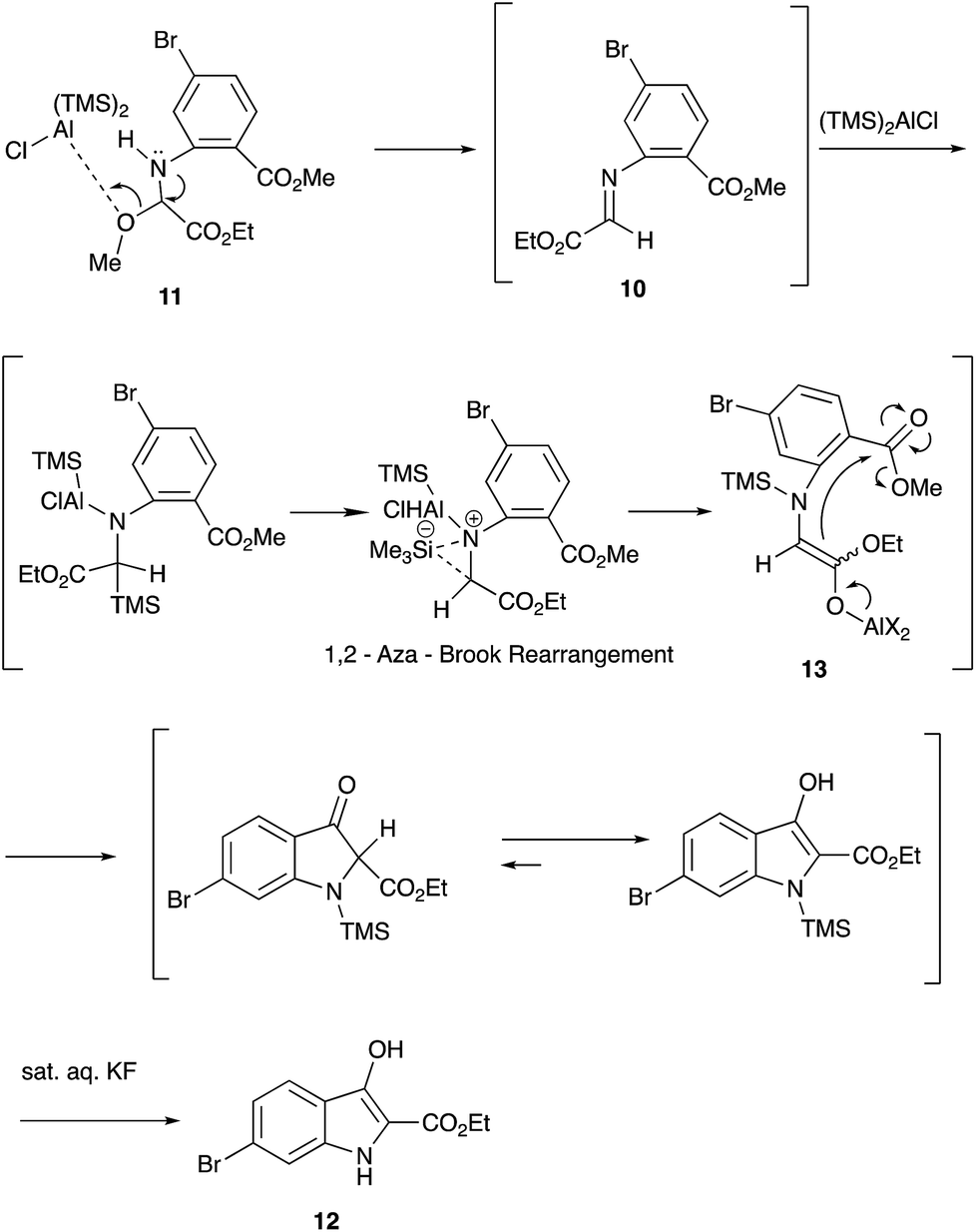 | ||
| Scheme 4 A proposed reaction mechanism of the indolin-3-one 12 synthesis. | ||
First, the aldimine 10 is formed in situ by the treatment of the methoxyglycine derivative 11 with bis(trimethylsilyl)aluminum chloride. The formation of the imine 10 was detected by a direct injection EI-MS (m/z 313). This imine 10 would be attacked by the second equivalent of bis(trimethylsilyl)aluminum chloride to form the aluminum enolate 13 via an aza-Brook rearrangement.10 A subsequent Dieckmann cyclization followed by hydrolysis gives the indolin-3-one 12 (Scheme 5).
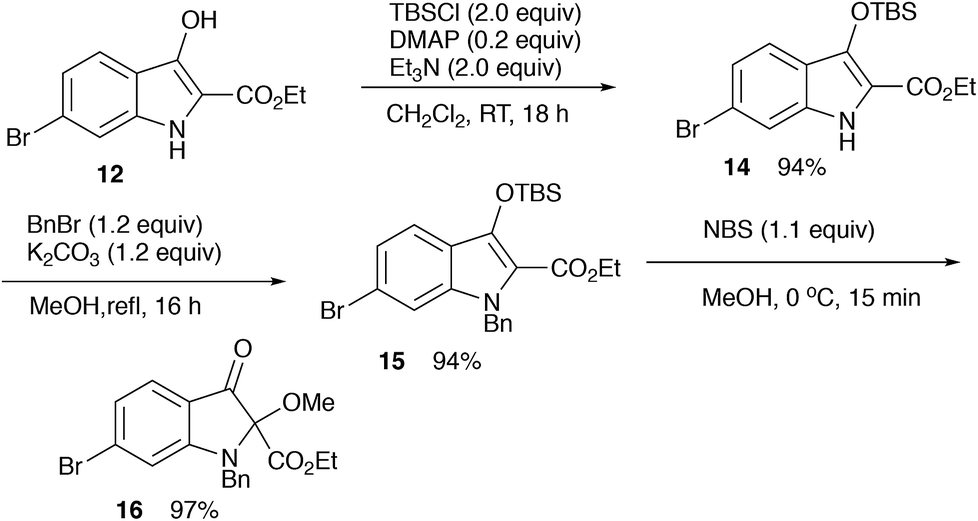 | ||
| Scheme 5 Introduction of 2-methoxy group. | ||
For the synthesis of matemone 3, the introduction of the methoxy group at the C-2 position is needed. After several attempts using a series of oxidation reagents, we found that the oxidation of the silyl enol ether 15 with NBS in methanol gave satisfactory results.11,12 However, selective reduction at the ester moiety was not successful.13 Bis-reduction at the ketone and the ester moieties followed by oxidation at the benzylic alcohol was also failed to give only complex mixtures. We then changed the order of the functional group transformations, i.e., reduction of the ester moiety, followed by the introduction of the methoxy group. This procedure worked well to give N-benzyl matemone 17 in high yield (Scheme 6).
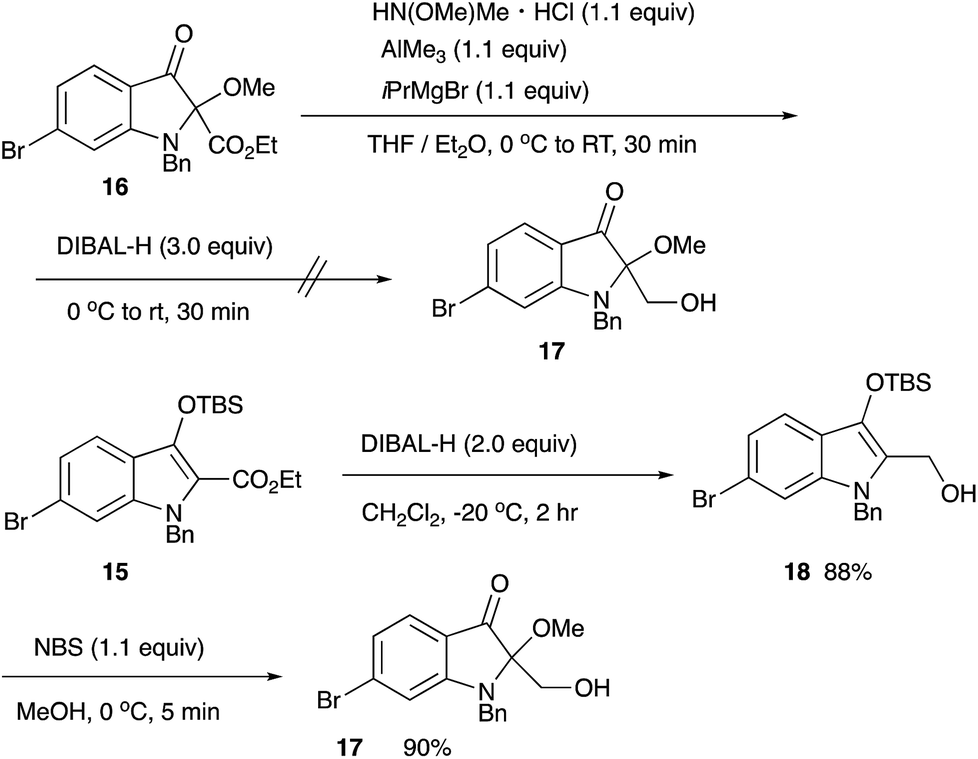 | ||
| Scheme 6 Synthesis of N-benzyl matemone. | ||
This intriguing oxidation into the methoxy derivative 17 is explicable in terms of the formation of the iminium species 19, which is attacked by methanol (Scheme 7).
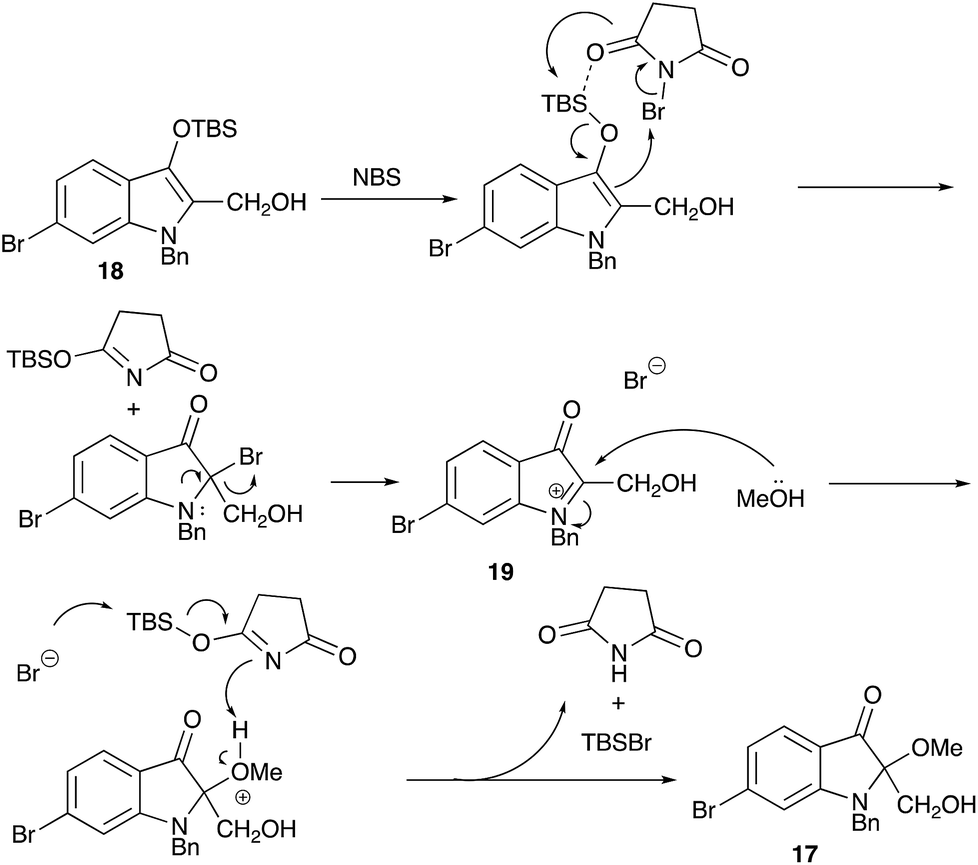 | ||
| Scheme 7 A proposed pathway for the introduction of a methoxy group. | ||
We next attempted removal of the benzyl group under a series of conditions (RSH/base, TMSI, Ca or Na/liq. NH3, H2/Pd or Pt, etc.). Although a small amount of matemone was detected by the mass spectra of the crude reaction mixtures, attempted isolation by silica gel chromatography was not successful. We also attempted the isolation as an acetate form by treatment of the whole reaction mixtures with an excess AcCl/base. However, the acetate was not isolated in sufficient quantity. Studies indicated that unprotected matemone was unstable due to a solvent-induced polymerization process.2 Therefore, matemone was immediately converted to the stable acetate derivative 3 (R2 = Ac), and detailed spectroscopic analyses were carried out with the acetate derivative. We found that N-protected matemone 18 was also reasonably stable and would be subject to further functional group interconversions.14
Conclusions
We have found that the methoxyglycine derivative 11 is a good precursor to the aldimine 10 derived from glyoxylate, and the subsequent treatment of this particular methoxyglycine 11 with bis(trimethylsilyl)aluminum chloride provides 2-alkoxycarbonylindolin-3-ones. Further oxidation of the silyl enol ether prepared from the 2-alkoxycarbonylindolin-3-one undergoes a facile oxidation reaction with NBS in methanol to give the 2-methoxy derivatives in high yields. This procedure has proved to be effective for the synthesis of N-benzyl matemone as a reasonably stable derivative. Although we have not examined the bioactivity of the N-benzyl matemone 17 yet, we will submit it and other derivatives to bioassay in due course.Experimental
General aspects
Infrared spectra were determined on a JASCO FT/IR-460 plus spectrometer. 1H NMR and 13C NMR spectra were recorded with a JEOL ECX-400P, or a JEOL A-500 spectrometer using tetramethylsilane as an internal standard. Mass spectra were recorded on a JEOL MS-700D spectrometer. Propionitrile (EtCN) and acetonitrile (MeCN) were distilled from phosphorus pentoxide and then from calcium hydride and stored over Molecular Sieves 4 Å. Dichloromethane (CH2Cl2) was distilled from calcium hydride and stored over Molecular Sieves 4 Å. Toluene was dried over calcium chloride, distilled, and stored over Molecular Sieves 4 Å. Diethyl ether (Et2O) and tetrahydrofuran (THF) were distilled from benzophenone ketyl immediately before use or purified by Glass Contour Organic Solvent Purification System of Nikko Hansen & Co., Ltd. MeOH was heated at reflux over magnesium for 5 h, distilled, and stored over Molecular Sieves 3 Å. Purification of products was performed by column chromatography on silica gel (Kanto Silica Gel 60N) and/or preparative TLC on silica gel (Merck Kiesel Gel GF254 or Wako Gel B-5F).Methyl 4-bromo-2-[(2-ethoxy-1-methoxy-2-oxoethyl)amino]-benzoate (11)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum and an argon balloon were placed methyl 2-amino-4-bromobenzoate (460.0 mg, 2.00 mmol) prepared according to the reported procedures,15 ethyl glyoxylate (1.23 mL, 6.00 mmol, 50% in toluene), and methanol (10.0 mL), respectively. The mixture was stirred at reflux for 16 h. After cooling to room temperature, the mixture was concentrated in vacuo to give a crude oil, which was purified by silica gel chromatography (nhexane:ethyl acetate = 6:1) to give the title compound 11 (604.6 mg, 87%) as white crystals.
Yield 87% (604.6 mg); white crystals; mp 86–88 °C; Rf = 0.50 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 1.36 (t, J = 7.3 Hz, 3H), 3.30 (s, 3H), 3.89 (s, 3H), 4.34 (q, J = 7.3 Hz, 2H), 5.26 (d, J = 6.4, 1H), 6.88–6.91 (m, 1H), 7.13 (d, J = 1.8 Hz, 1H), 7.79 (d, J = 8.7 Hz, 1H), 8.98 (d, J = 6.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 51.6, 51.9, 62.2, 81.4, 110.9, 116.1, 120.7, 129.5, 132.7, 148.6, 167.9, 168.0; IR (neat) 3329, 2952, 1743, 1693, 1571, 1505, 1240, 1095, 1064, 769 cm−1; HRMS (EI) calcd for C13H16BrNO5 (M)+ 345.0212 found 345.0196.
General procedure: synthesis of ethyl 6-bromo-3-hydroxy-1H-indole-2-carboxylate (12) (Table 1)
Under an argon atmosphere, a solution of methyl 4-bromo-2-[(2-ethoxy-1-methoxy-2-oxoethyl)amino]-benzoate 11 (100.0 mg, 0.29 mmol) in EtCN (30.0 mL) was placed at −78 °C and to it was added a propionitrile solution (10 mL) of (TMS)2AlCl, which was prepared by mixing aluminum chloride (52.0 mg, 0.39 mmol) and (TMS)3Al·Et2O (0.62 mL, 0.77 mmol, 1.25 M in Et2O) at room temperature in another flask. After the mixture was stirred for 2 hours at room temperature, to it was added saturated aqueous potassium fluoride followed by a saturated aqueous Rochelle's salt to quench the reaction. The whole mixture was extracted with ethyl acetate (10 mL × 3). The combined organic phases were washed with brine, dried over Na2SO4, and concentrated in vacuo to give a crude product. Purification by silica gel column chromatography (nhexane:ethyl acetate = 4:1 as an eluent) gave ethyl 6-bromo-3-hydroxy-1H-indole-2-carboxylate 12 (44.8 mg, 56%) as yellow crystals.
Yield 56% (44.8 mg); mp 167–169 °C; yellow crystals; Rf = 0.32 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 6.9 Hz, 3H), 4.39 (q, J = 6.9 Hz, 2H), 7.04–7.06 (m, 1H), 7.48–7.49 (m, 1H), 7.58–7.60 (m, 1H), 8.79 (s, 1H), 10.70 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.2, 58.7, 108.1, 113.6, 115.5, 118.0, 119.9, 120.3, 134.2, 142.8, 160.9; IR (neat) 3341, 1672, 1608, 1583, 1308, 1240, 1141, 1104, 1018, 770 cm−1; HRMS (EI) calcd for C11H10BrNO3 (M)+ 282.9844 found 282.9842.
Ethyl 6-bromo-3-[(tert-butyldimethylsilyl)oxy]-1H-indole-2-carboxylate (14)
In a 50 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum and an argon balloon was placed ethyl 6-bromo-3-hydroxy-1H-indole-2-carboxylate (125.4 mg, 0.44 mmol), DMAP (0.09 mmol, 10.8 mg), triethylamine (0.12 mL, 0.88 mmol) and CH2Cl2 (10 mL), and to it was added a solution of TBDMSCl (0.88 mmol, 132.6 mg) in CH2Cl2 (4 mL). After the mixture was stirred for 16 h at room temperature, it was concentrated in vacuo to give a crude oil, which was purified by silica gel column chromatography (nhexane:ethyl acetate = 6:1) to give the title compound 14 (165.0 mg, 94%) as white crystals.
Yield 94% (165.0 mg); white crystals; mp 135–136 °C; Rf = 0.54 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 0.21 (s, 6H), 1.09 (s, 9H), 1.43 (t, J = 6.9, 3H), 4.44 (q, J = 7.3, 2H), 7.19–7.17 (m, 1H), 7.47–7.49 (m, 2H), 8.65 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −4.2, 14.7, 18.3, 25.7, 60.7, 114.4, 114.7, 120.0, 120.6, 121.5, 123.0, 133.8, 139.7, 161.7; IR (neat) 3313, 2952, 1675, 1568, 1472, 1316, 1240, 1145, 851, 783 cm−1; HRMS (EI) calcd for C17H24BrNO3Si (M)+ 397.0709 found 397.0707.
Ethyl 1-benzyl-6-bromo-3-[(tert-butyldimethylsilyl)oxy]-1H-indole-2-carboxylate (15)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum and an argon balloon was placed ethyl 6-bromo-3-[(tert-butyldimethylsilyl)oxy]-1H-indole-2-carboxylate (37.0 mg, 0.09 mmol), K2CO3 (15.2 mg, 0.11 mmol), benzyl bromide (0.01 mL) and MeCN (15 mL), and to it was added a solution of TBDMSCl (132.6 mg, 0.88 mmol) in MeCN (15 mL, 0.11 mmol). After the mixture was stirred for 16 h at reflux, it was filtered through a plug of cotton and concentrated in vacuo to give a crude oil, which was purified on silica gel TLC (nhexane:ethyl acetate = 5:1) to give the title compound 15 (165.0 mg, 94%) as a colourless oil.
Yield 94% (41.1 mg); colourless oil; Rf = 0.68 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 0.18 (s, 6H), 1.08 (s, 9H), 1.28 (t, J = 7.1, 3H), 4.31 (q, J = 7.3, 2H), 5.65 (s, 2H), 6.95–6.97 (m, 2H), 7.16–7.25 (m, 4H), 7.43–7.44 (m, 1H), 7.49–7.51 (m, 1H); 13C NMR (100 MHz, CDCl3) δ −4.2, 14.5, 18.4, 25.8, 48.1, 60.3, 133.3, 116.0, 119.5, 120.1, 121.7, 123.1, 126.0, 127.1, 128.6, 137.4, 138.1, 141.1, 161.6; IR (neat) 2930, 2857, 1698, 1532, 1437, 1329, 1257, 1119, 830, 781 cm−1; HRMS (EI) calcd for C24H30BrNO3Si (M)+ 487.1178 found 487.1163.
Ethyl 1-benzyl-6-bromo-2-methoxy-3-oxoindoline-2-carboxylate (16)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum and an argon balloon was placed NBS (59.1 mg, 0.33 mmol) and MeOH (6.0 mL), and to it was added a solution of ethyl 1-benzyl-6-bromo-3-[(tert-butyldimethylsilyl)oxy]-1H-indole-2-carboxylate (59.1 mg, 0.30 mmol) in MeOH (4 mL) at 0 °C. After the mixture was stirred for 15 min at 0 °C, to it was added saturated aqueous K2CO3 to quench the reaction. The whole mixture was extracted with ethyl acetate (50 mL × 3). The combined organic phases were washed with brine, dried over Na2SO4, and concentrated in vacuo to give a crude product, which was purified by silica gel column chromatography (nhexane:ethyl acetate = 4:1) to give the title compound 16 (117.6 mg, 97%) as a yellow oil.
Yield 97% (117.6 mg); yellow oil; Rf = 0.42 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 1.09–1.13 (m, 3H), 3.27 (s, 3H), 3.96–4.10 (m, 2H), 4.47–4.60 (m, 2H), 6.90–7.05 (m, 2H), 7.29–7.37 (m, 5H), 7.44–7.48 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 13.8, 46.6, 52.1, 32.5, 112.7, 118.0, 122.7, 126.0, 127.1, 127.7, 128.8, 134.2, 135.9, 161.4, 165.1, 193.5; IR (neat) 2930, 1721, 1605, 1465, 1311, 1260, 1148, 1098, 906, 699 cm−1; HRMS (EI) calcd for C17H16BrNO3 (M)+ 403.0419 found 403.0414.
1-Benzyl-6-bromo-3-[(tert-butyldimethylsilyl)oxy-1H-indol-2-yl]methanol (18)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum and an argon balloon was placed a solution of ethyl 1-benzyl-6-bromo-3-[(tert-butyldimethylsilyl)oxy]-1H-indole-2-carboxylate (23.0 mg, 0.05 mmol) in CH2Cl2 (5.0 mL), and to it was added dropwise DIBAL-H (0.09 mL, 0.09 mmol, 10% in n-hexane) at −20 °C. After the mixture was stirred for 30 min at 0 °C, to it was added saturated aqueous Rochelle's salt to quench the reaction. The whole mixture was filtered through a Celite pad, and was extracted with CH2Cl2 (10 mL × 3). The combined organic phases were dried over Na2SO4, and concentrated in vacuo to give a crude product, which was purified on silica gel TLC (nhexane:ethyl acetate = 4:1) to give the title compound 18 (19.6 mg, 88%) as a yellow green oil.
Yield 88% (19.6 mg); yellow green oil; Rf = 0.31 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 0.18 (s, 6H), 1.08 (s, 9H), 4.65 (d, J = 5.5 Hz, 2H), 5.38 (s, 2H), 6.92–6.94 (m, 2H), 7.14–7.42 (m, 6H); 13C NMR (100 MHz, CDCl3) δ −4.4, 18.2, 25.8, 46.8, 53.5, 112.4, 116.4, 119.8, 120.1, 122.2, 125.0, 125.7, 127.4, 128.8, 132.6, 135.3, 137.8; IR (neat) 3413, 2931, 2858, 1584, 1468, 1364, 1253, 1189, 1008, 829, 781 cm−1; HRMS (EI) calcd for C22H28BrNO2Si (M)+ 445.1073 found 445.1073.
1-Benzyl-6-bromo-2-(hydroxymethyl)-2-methoxyindolin-3-one (17)
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum and an argon balloon was placed NBS (89.4 mg, 0.50 mmol) and MeOH (25.0 mL), and to it was added a solution of 1-benzyl-6-bromo-3-[(tert-butyldimethylsilyl)oxy-1H-indol-2-yl]methanol (203.8 mg, 0.46 mmol) in MeOH (4 mL) at 0 °C. After the mixture was stirred for 5 min at 0 °C, to it was added saturated aqueous K2CO3 to quench the reaction. The whole mixture was extracted with ethyl acetate (50 mL × 3). The combined organic phases were dried over Na2SO4, and concentrated in vacuo to give a crude product, which was purified on silica gel TLC (nhexane:ethyl acetate = 3:1) to give the title compound 17 (150.4 mg, 90%) as a yellow green oil.
Yield 90% (150.4 mg); Rf = 0.19 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3) δ 3.12 (s, 3H), 3.65 (d, J = 0.0 Hz, 1H), 3.85–3.91 (m, 1H), 4.59 (s, 2H), 6.90–6.96 (m, 2H), 7.36–7.40 (m, 5H), 7.42–7.44 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 45.2, 52.0, 63.9, 112.0, 114.5, 121.9, 122.7, 125.5, 126.7, 127.8, 129.4, 149.8, 213.3; IR (neat) 3462, 2929, 1716, 1606, 1472, 1312, 1092, 1053, 937, 755 cm−1; HRMS (EI) calcd for C17H16BrNO3 (M)+ 361.0314 found 361.0297.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) and on Innovative Areas “Organic Synthesis Based on Reaction Integration. Development of New Methods and Creation of New Substances” from JSPS and MEXT.Notes and references
- (a) T. Kawasaki, A. Ogawa, Y. Takashima and M. Sakamoto, Tetrahedron Lett., 2003, 44, 1591 CrossRef CAS; (b) P. N. Wyrembak and A. D. Hamilton, J. Am. Chem. Soc., 2009, 131, 4566 CrossRef CAS PubMed; (c) K. Okuma, N. Matsunaga, N. Nagahora, K. Shioji and Y. Yokomori, Chem. Commun., 2010, 5822 Search PubMed; (d) Y. Sun and R. Fan, Chem. Commun., 2010, 6834 RSC; (e) W. Sun, L. Hong and R. Wang, Chem.–Eur. J., 2011, 17, 6030 CrossRef CAS PubMed; (f) A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354 CrossRef CAS PubMed.
- (a) I. Carletti, B. Banaigs and P. Amade, J. Nat. Prod., 2000, 63, 981 CrossRef CAS PubMed; (b) S.-S. Wen, Z.-F. Zhou, J.-A. Xiao, J. Li, H. Xiang and H. Yang, New J. Chem., 2017, 41, 11503 RSC; (c) For a review see, G. W. Gribble in Progress in Heterocyclic Chemistry, ed by G. W. Gribble and J. A. Joule, Pergamon, Kidlington, vol. 15, 2003, pp. 58–74 Search PubMed.
- (a) P. S. Baran and E. J. Corey, J. Am. Chem. Soc., 2002, 124, 7904 CrossRef CAS PubMed; (b) P.-L. Wu, Y.-L. Hsu and C.-W. Jao, J. Nat. Prod., 2006, 69, 1467 CrossRef CAS PubMed; (c) L. A. Adams, M. W. N. Valente and R. M. Williams, Tetrahedron, 2006, 62, 5195 CrossRef CAS; (d) D. D. O'Rell, F. G. H. Lee and V. Boekelheide, J. Am. Chem. Soc., 1972, 94, 3205 CrossRef; (e) S. Tsukamoto, H. Umaoka, K. Yoshikawa, T. Ikeda and H. Hirota, J. Nat. Prod., 2010, 73, 1438 CrossRef CAS PubMed; (f) A. Karadeolian and M. A. Kerr, Angew. Chem., Int. Ed., 2010, 49, 1133 CrossRef CAS PubMed; (g) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2010, 75, 6830 CrossRef CAS PubMed.
- P. H. B. França, D. P. Barbosa, D. L. da Silva, Ê. A. N. Ribeiro, A. E. G. Santana, B. V. O. Santos, J. M. Barbosa-Filho, J. S. S. Quintans, R. S. S. Barreto, L. J. Quintans-Júnior and J. X. de Araújo-Júnior, BioMed Res. Int., 2014, 1 Search PubMed.
- For N-alkylation to α-imino esters in our laboratory, see, (a) M. Shimizu and Y. Niwa, Tetrahedron Lett., 2001, 42, 2829 CrossRef CAS; (b) Y. Niwa, K. Takayama and M. Shimizu, Tetrahedron Lett., 2001, 42, 5473 CrossRef CAS; (c) Y. Niwa, K. Takayama and M. Shimizu, Bull. Chem. Soc. Jpn., 2002, 75, 1819 CrossRef CAS; (d) Y. Niwa and M. Shimizu, J. Am. Chem. Soc., 2003, 125, 3720 CrossRef CAS PubMed; (e) M. Shimizu, H. Itou and M. Miura, J. Am. Chem. Soc., 2005, 127, 3296 CrossRef PubMed; (f) M. Shimizu, Pure Appl. Chem., 2006, 78, 1867 CAS; (g) M. Shimizu, I. Hachiya and I. Mizota, Chem. Commun., 2009, 874 RSC; (h) I. Mizota, K. Tanaka and M. Shimizu, Tetrahedron Lett., 2012, 53, 1847 CrossRef CAS; (i) T. Nishi, I. Mizota and M. Shimizu, Pure Appl. Chem., 2012, 84, 2609 CAS; (j) S. Hata, T. Maeda and M. Shimizu, Bull. Chem. Soc. Jpn., 2012, 85, 1203 CrossRef CAS; (k) M. Shimizu, D. Kurita and I. Mizota, Asian J. Org. Chem., 2013, 2, 208 CrossRef CAS; (l) T. Sano, I. Mizota and M. Shimizu, Chem. Lett., 2013, 42, 995 CrossRef CAS; (m) I. Mizota, Y. Matsuda, S. Kamimura, H. Tanaka and M. Shimizu, Org. Lett., 2013, 15, 4206 CrossRef CAS PubMed; (n) H. Tanaka, I. Mizota and M. Shimizu, Org. Lett., 2014, 16, 2276 CrossRef CAS PubMed; (o) K. Koyama, I. Mizota and M. Shimizu, Pure Appl. Chem., 2014, 86, 755 CAS; (p) M. Shimizu, M. Tateishi and I. Mizota, Chem. Lett., 2014, 43, 1752 CrossRef; (q) I. Mizota, T. Maeda and M. Shimizu, Tetrahedron, 2015, 71, 5793 CrossRef CAS; (r) I. Mizota and M. Shimizu, Chem. Rec., 2016, 16, 688 CrossRef CAS PubMed; (s) T. Tanaka, I. Mizota, K. Umezu, A. Ito and M. Shimizu, Heterocycles, 2017, 95, 830 CrossRef CAS; (t) M. Kawanishi, I. Mizota, K. Aratake, H. Tanaka, K. Nakahama and M. Shimizu, Bull. Chem. Soc. Jpn., 2017, 90, 395 CrossRef CAS; (u) I. Mizota, Y. Nakajima, A. Higashino and M. Shimizu, Arabian J. Sci. Eng., 2017, 42, 4249 CrossRef CAS; (v) I. Mizota, C. Ueda, Y. Tesong, Y. Tsujimoto and M. Shimizu, Org. Lett., 2018, 20, 2291 CrossRef CAS PubMed; (w) K. Nakahama, M. Suzuki, M. Ozako, I. Mizota and M. Shimizu, Asian J. Org. Chem., 2018, 7, 910 CrossRef CAS.
- M. Shimizu, Y. Takao, H. Katsurayama and I. Mizota, Asian J. Org. Chem., 2013, 2, 130 CrossRef CAS.
- (a) L. Blackburn and R. J. K. Taylor, Org. Lett., 2001, 3, 1637 CrossRef CAS PubMed; (b) T. Mukaiyama, A. Kawana, Y. Fukuda and J. Matsuo, Chem. Lett., 2001, 390 CrossRef CAS; (c) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, Angew. Chem., 2003, 115, 4211 CrossRef; (d) M. S. Kwon, S. Kim, S. Park, W. Bosco, R. K. Chidrala and J. Park, J. Org. Chem., 2009, 74, 2877 CrossRef CAS; (e) E. Zhang, H. Tian, S. Xu, X. Yu and Q. Xu, Org. Lett., 2013, 15, 2704 CrossRef CAS; (f) R. Kumar, E. H. Gleißner, E. G. V. Tiu and Y. Yamakoshi, Org. Lett., 2016, 18, 184 CrossRef CAS.
- For the formation of hemiaminal ethers, see, (a) G. Li, F. R. Fronczek and J. C. Antilla, J. Am. Chem. Soc., 2008, 130, 12216 CrossRef CAS; (b) K. Xu, Z. Wang, J. Zhang, L. Yu and J. Tan, Org. Lett., 2015, 17, 4476 CrossRef CAS PubMed; (c) A. Beltran, E. Alvarez, M. M. Diaz-Requejo and P. J. Pereza, Adv. Synth. Catal., 2015, 357, 2821 CrossRef CAS; (d) M. Li, B. Luo, Q. Liu, Y. Hu, A. Ganesan, P. Huang and S. Wen, Org. Lett., 2014, 16, 10 CrossRef CAS PubMed; (e) H. Yu and J. Shen, Org. Lett., 2014, 16, 3204 CrossRef CAS; (f) For a related work, see, T. Kano, T. Yurino, D. Asakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 5532 CrossRef CAS PubMed.
- (a) M. A. Avery, W. K. M. Chong and C. Jennings-White, J. Am. Chem. Soc., 1992, 114, 974 CrossRef CAS; (b) L. Rçsch, G. Altnau and W. H. Otto, Angew. Chem., Int. Ed., 1981, 20, 581 CrossRef.
- (a) A. G. Brook and J. M. Duff, J. Am. Chem. Soc., 1974, 96, 4692 CrossRef CAS; (b) T. Honda and M. Mori, J. Org. Chem., 1996, 61, 1196 CrossRef CAS; (c) M. Suginome, T. Fukuda and Y. Ito, J. Organomet. Chem., 2002, 643, 508 CrossRef.
- For the bromination of silyl enol ethers, see, (a) R. H. Reuss and A. Hassner, J. Org. Chem., 1974, 39, 1785 CrossRef CAS; (b) L. Blanco, P. Amice and J. M. Conia, Synthesis, 1976, 194 CrossRef CAS; (c) G. F. Hambly and T. H. Chan, Tetrahedron Lett., 1986, 27, 2563 CrossRef CAS.
- For the oxidative formation of iminium salts from amino ketene silyl acetals, see, S. Hata, H. Koyama and M. Shimizu, J. Org. Chem., 2011, 76, 9670 CrossRef CAS PubMed.
- (a) J. L. Luche and A. L. Gemal, J. Am. Chem. Soc., 1979, 101, 5848 CrossRef CAS; (b) M. T. Reetz, B. Wenderoth and R. Peter, J. Chem. Soc., Chem. Commun., 1983, 406 RSC; (c) K. Maruoka, S. Saito, A. B. Concepcion and H. Yamamoto, J. Am. Chem. Soc., 1993, 115, 1183 CrossRef CAS; (d) G. Bastug, S. Dierick, F. Lebreux and I. E. Mark, Org. Lett., 2012, 14, 1306 CrossRef CAS PubMed; (e) H. Fujioka, K. Yahata, O. Kubo, Y. Sawama, T. Hamada and T. Maegawa, Angew. Chem., Int. Ed., 2011, 50, 12232 CrossRef CAS PubMed; (f) F. J. Barrios, B. C. Springer and D. A. Colby, Org. Lett., 2013, 15, 3082 CrossRef CAS PubMed.
- Although we attempted to synthesize N,O-bis-TBS derivative of the compound 12 or its TIPS counterpart as a trialkylsilyl-protected derivative, upon treatment with an excess TBS-Cl, only the mono-O-TBS derivative 14 was isolated after work-up, whereas an excess TIPS-Cl treatment led to the recovery of the starting material 12..
- (a) P. Imming, I. Imhof and M. Zentgraf, Synth. Commun., 2001, 31, 3721 CrossRef CAS; (b) B. D. Allison, V. K. Phuong, L. C. McAtee, M. Rosen, M. Morton, C. Prendergast, T. Barrett, G. Lagaud, J. Freedman, L. Li, X. Wu, H. Venkatesan, M. Pippel, C. Woods, M. C. Rizzolio, M. Hack, K. Hoey, X. Deng, C. King, N. P. Shankley and M. H. Rabinowitz, J. Med. Chem., 2006, 49, 6371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02204j |
| This journal is © The Royal Society of Chemistry 2019 |