
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGram scale production of 1-azido-β-D-glucose via enzyme catalysis for the synthesis of 1,2,3-triazole-glucosides†
Jaggaiah N. Gorantlaa,
Salila Pengthaisonga,
Sunaree Choknuda,
Teadkait Kaewpuangb,
Tanaporn Manyuma,
Vinich Promarakb and
James R. Ketudat Cairns *a
*a
aSchool of Chemistry, Institute of Science, & Center for Biomolecular Structure, Function and Application, Suranaree University of Technology, Nakhon Ratchasima 30000, Thailand. E-mail: cairns@sut.ac.th; Fax: +66 44 224185; Tel: +66 44 224193
bDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
First published on 20th February 2019
Abstract
The production of analytical amounts of azido sugars is used as a means of verifying catalytic acid/base mutations of retaining glycosidase, but application of this process to preparative synthesis has not been reported. The catalytic acid/base mutant of Thermoanaerobacterium xylanolyticus GH116 β-glucosidase, TxGH116D593A, catalyzed the gram scale production of 1-azido-β-D-glucose (1) from p-nitropheyl-β-D-glucopyranoside (pNPGlc) and azide via a transglucosylation reaction. Overnight reaction of the enzyme with pNPGlc and NaN3 in aqueous MES buffer (pH 5.5) at 55 °C produced 1 (3.27 g), which was isolated as a white foamy solid in 96% yield. This 1 was successfully utilized for the synthesis of fifteen 1,2,3-triazole-β-D-glucosyl derivatives (2–16) containing a variety of functional groups, via click chemistry.
Introduction
Enzymes are well known for their unique ability to catalyze stereospecific reactions and produce stereoselective products.1–3 Retaining β-glycosidases are one group of enzymes displaying this property via generation of products with the same β-anomeric configuration as their substrates. The retaining β-glycosidase mechanism involves the displacement of the aglycone leaving group from the anomeric carbon of the sugar by the catalytic nucleophile with acid-assistance by the catalytic acid/base, which protonates the leaving group.4,5 The α-glycosyl enzyme intermediate, thus formed, is resolved by attack of a water molecule or other nucleophile on the anomeric carbon to displace the catalytic nucleophile residue and release a β-sugar or β-glycoside, respectively. This second step is generally facilitated by the deprotonation of the incoming nucleophile (water) molecule by the catalytic acid/base residue.Acid/base mutants of β-glycosidase enzymes, in which the catalytic carboxylate is replaced with a nonionizable amino acid sidechain, have been frequently reported to catalyze the formation of β-glucosides without significant hydrolysis.4,5 In an early study with the acid/base mutant of Agrobacterium β-glucosidase, the substrates 2,4-dinitrophenyl-β-D-glucopyranoside (DNPGlc) and 4-nitrophenyl-β-D-glucopyranoside (pNPGlc) served as donor substrates for the transglucosylation of various nucleophiles, including formate, azide, acetate, cyanide and benzoate.5 Similarly, the Thermoanaerobacterium xylanolyticum TxGH116 β-glucosidase acid/base mutant, TxGH116D593A, was previously produced and rescued by azide to verify that the D593 acts as a catalytic acid/base.6 Kinetic characterization of transglucosylation reactions catalyzed by β-glucosidase catalytic acid/base mutants to produce 1-azido-β-D-glucose (1) has also been reported,5–7 but exploration of this reaction as a preparative method has not been reported.
Synthesis of 1-azido-β-D-glucose (1) has also been reported from unprotected glucose using 1,3-dimethylimidazolinium salts, base and NaN3 in deuterium oxide,8–10 and other reagents.11,12 1-Azido-β-D-glucose (1) has been used in the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction for the construction of triazole-glucosides.13–16 Triazole-glucosides exhibit a wide range of glucosidase,17–19 glycogen phosphorylase,20–22 and carbonic anhydrase23,24 inhibitory activities (Fig. 1). The triazole-glucosides also exhibit binding affinity towards various proteins,25–28 and were found to be potential radiopharmaceuticals,29,30 pesticides,31 water soluble prodrugs, and inhibitors against cancer associated carbonic anhydrases,32,33 and to have antinociception34 activities (Fig. 1). Triazole-glucosides and their various activities have been reviewed.35,36
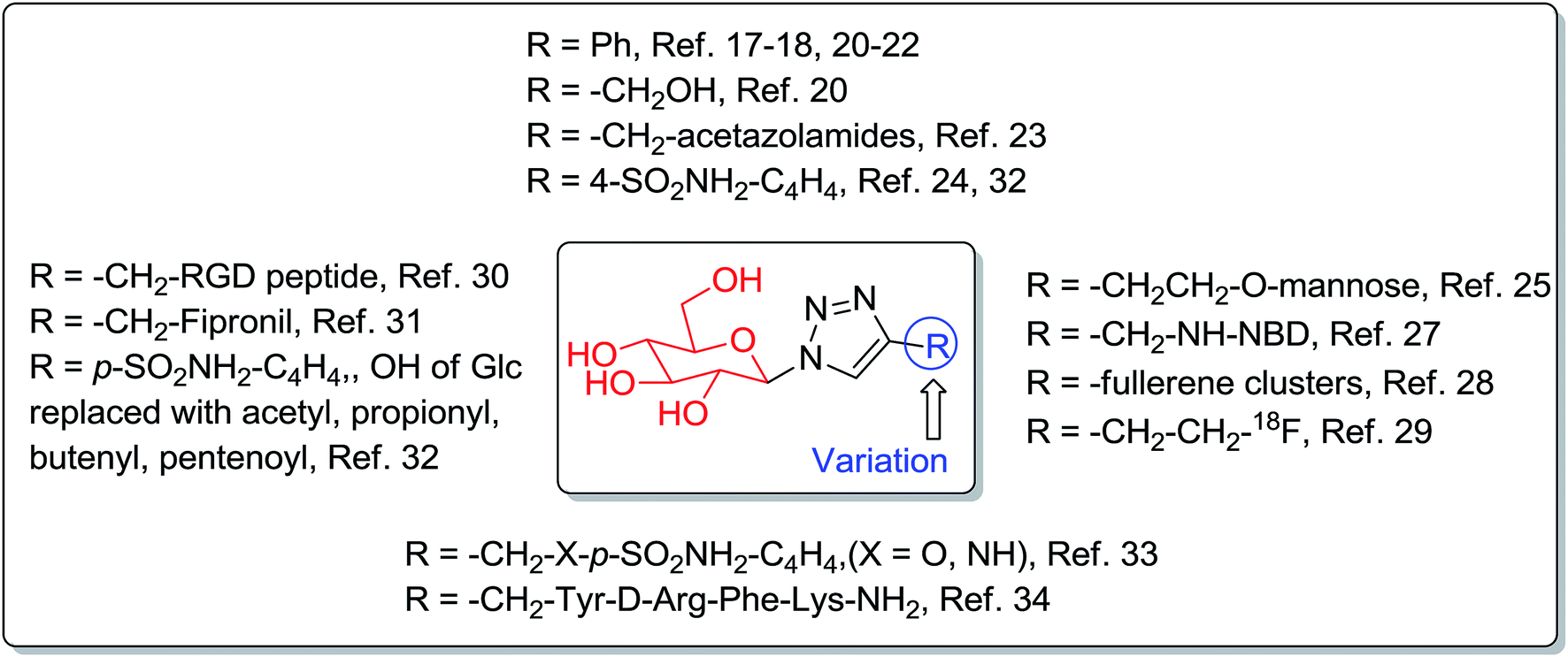 | ||
| Fig. 1 Reported bioactive 1,2,3-triazole-β-D-glucosides. | ||
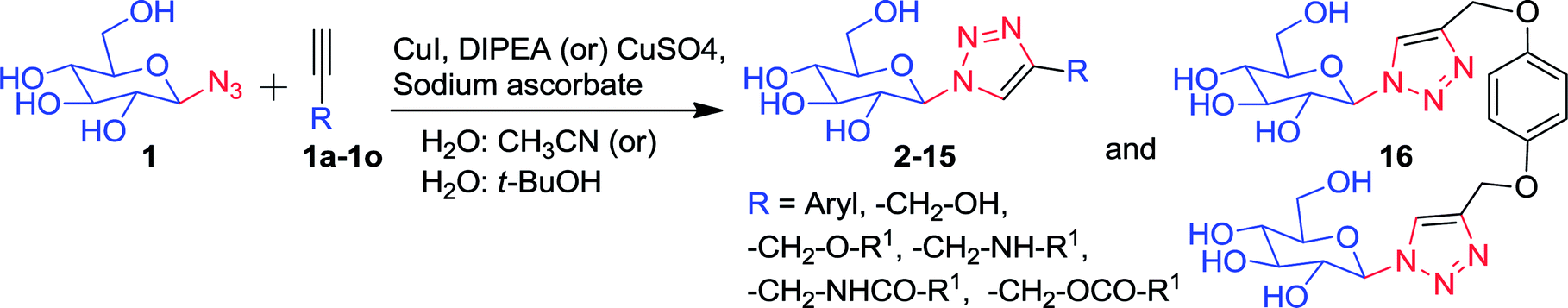
In the present study, a catalytic process for the gram scale production of the 1-azido-β-D-glucose (1) by an enzyme (TxGH116D593A) in water was developed. The purified enzyme catalyzed the production of gram scale amounts of 1 from pNPGlc and sodium azide in aqueous MES buffer (pH = 5.5), in an overnight reaction at 55 °C. After the overnight incubation, thin layer chromatography (TLC) indicated the nearly complete conversion of pNPGlc substrate into 1, by the enzyme via transglucosylation with azide as the acceptor nucleophile. The product (1) could be conveniently purified and reacted with various alkynes to form a variety of 1,2,3-triazole-β-D-glucosyl derivatives (2–16), without the need for further deprotection of the sugar.
Results and discussion
Initially, we optimized the catalytic transglucosylation reaction conditions by varying the concentration of enzyme, substrate, and sodium azide, while the MES buffer was maintained at pH 5.5, based on previous pH optimization. Following these initial experiments, the production was screened with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:6, 1:9, 1:12 and 1:15 (w/w) ratios of enzyme:pNPGlc substrate, 100 mM sodium azide and 50 mM MES buffer, pH 5.5, in 1 mL water, and yellow color was observed after the 24 h incubation at 55 °C. TLC (Fig. 2) showed the formation of a new polar compound, with the Rf of 1-azido-β-D-glucose and near complete conversion of substrate, except at the 1:15 ratio, where the substrate conversion was clearly not completed.
:3, 1:6, 1:9, 1:12 and 1:15 (w/w) ratios of enzyme:pNPGlc substrate, 100 mM sodium azide and 50 mM MES buffer, pH 5.5, in 1 mL water, and yellow color was observed after the 24 h incubation at 55 °C. TLC (Fig. 2) showed the formation of a new polar compound, with the Rf of 1-azido-β-D-glucose and near complete conversion of substrate, except at the 1:15 ratio, where the substrate conversion was clearly not completed.
 | ||
| Fig. 2 TLC profile of 1-azido-β-D-glucose (1) formation after 24 h from 3 mg/1 mg to 15 mg/1 mg and 5 g/370 mg of pNPGlc/enzyme (TxGH116D593A). In each case, detection under UV light is shown on the left and carbohydrate staining with 10% sulphuric acid in ethanol and charring is shown on the right. The numbers below the TLC lanes indicate ratios of pNPGlc to enzyme. | ||
Again, we ran the same experiment as explained above with 1:13 and 1:14 (w/w) ratios of enzyme:substrate, the substrate was almost completely converted into a product, as indicated by TLC (Fig. 2). Taking advantage of this optimization of reaction conditions, we used the 1:14 (w/w) ratio of enzyme:substrate for the large scale synthesis of 1-azido-β-D-glucose (1), from pNPGlc and azide catalyzed by TxGH116D593A in a 370 mL reaction volume (see ESI, Fig. S1†). After 24 hours of reaction at 55 °C, the reaction mixture was lyophilized to dryness, redissolved in methanol (MeOH) and purified by silica gel column chromatography. The pNP product eluted in 100% ethyl acetate (EtOAc, 300 mL), and then the unreacted pNPGlc was isolated as a mixture with 1-azido-β-D-glucose (1) (2.645 g) (see ESI Fig. S1†), with EtOAc/MeOH (95:5, v/v). The product 1 (800 mg) (see ESI Fig. S1†), was isolated in pure form by elution with EtOAc/MeOH (94:6, v/v). Finally, a trace of glucose byproduct (see ESI Fig. S1†) was eluted with EtOAc/MeOH (1:1, v/v). The pure 1-azido-β-D-glucose (1) was dried under reduced pressure to give a white foamy solid, Rf = 0.36 (CHCl3/MeOH/NH3, 7:2.8:0.2, v/v/v). Compound 1 was characterized with 1H, 13C NMR and HRMS analysis and the 1H NMR spectrum resembles that reported in the literature (see ESI†).6 In compound 1, the anomeric proton (H-1) shifted to downfield and merged with water solvent residual peak at δ 4.8 ppm in 600 MHz (see ESI page S4†), but the H-1 proton was observed at δ 4.55 ppm (H-1, d, J = 9 Hz, 1H) in the 400 MHz spectrum (see ESI page S3†).
To increase the yield, the obtained mixture (2.645 g) was redissolved in 6 mL of water/methanol (95:5, v/v) and separated over a Sephadex LH-20 column in the same solvent (ESI Fig. S2†). The product (1) eluted first in pure form, followed by a minor amount of pNPGlc. Drying this portion of product yielded an additional 2.470 g of compound 1 as a foamy white solid. Added with the 0.800 g from the first step, the 3.270 g of compound 1 represented 96% of the theoretical yield from the 5 g of pNPGlc used in the reaction. This was similar to the yield of 230 mg obtained from 330 mg of pNPGlc (98% yield) that we obtained in a preliminary small-scale reaction, suggesting reproducibly efficient conversion at milligram to gram scale. The purified compound (1) was thus ready for the synthesis of β-D-glucosyl-1,2,3-triazole derivatives via a click chemistry.
Compound 1 was subsequently joined with different compounds in which a propargyl group was attached to an alcohol, ethers, esters, amines, and amides (Fig. 3), as well as phenyl acetylenes in a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reactions, giving rise to the 1,2,3-triazole-β-D-glucosyl derivatives 2–16 (Fig. 4, Table 1).
 | ||
| Fig. 3 Structures of alkynes (1a–o) used in click chemistry. | ||
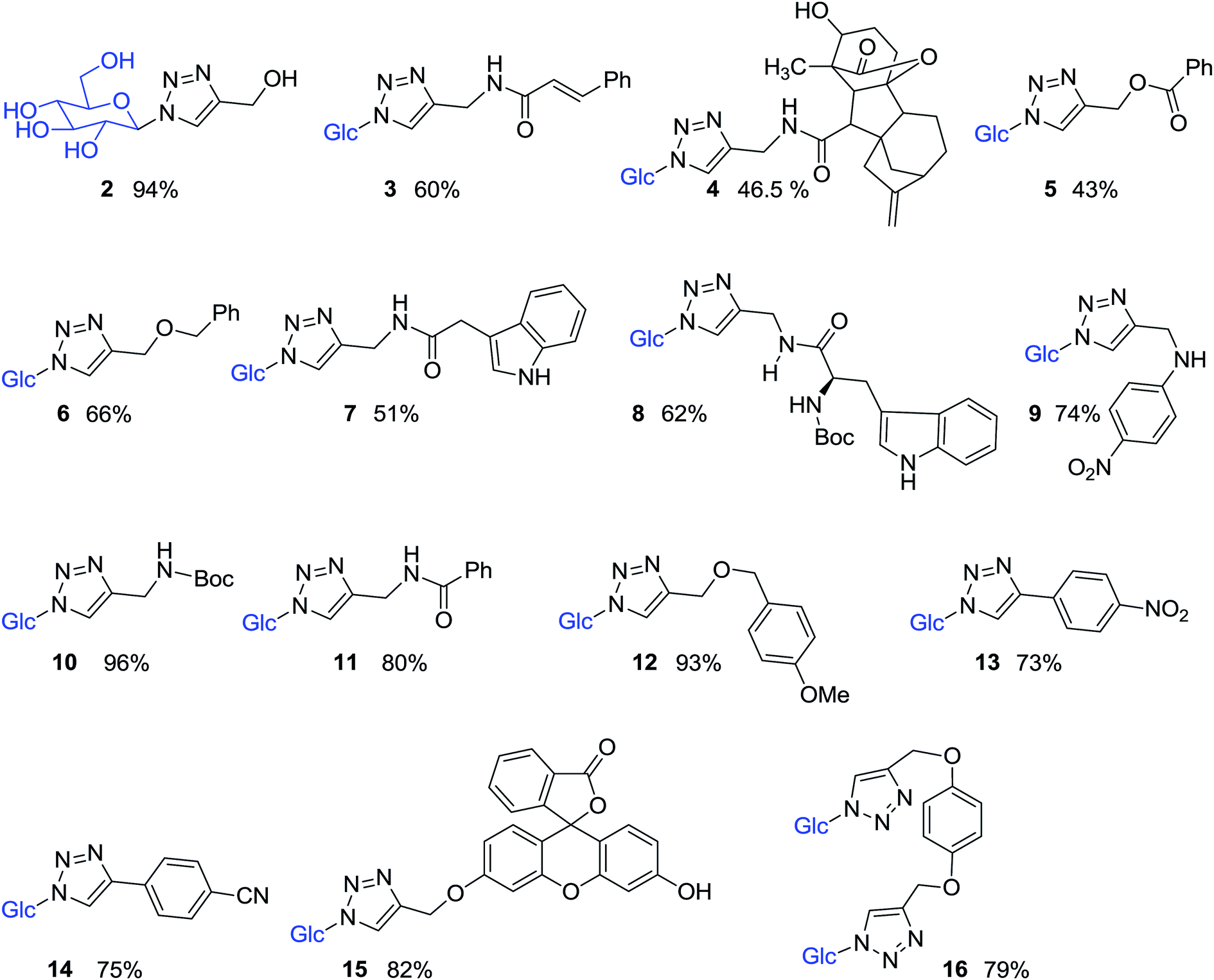 | ||
| Fig. 4 Structures of 1,2,3-triazole-β-D-glucosyl derivatives 2–16. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkyne | Cu salt (eq.) | Base (eq.) | Solvent system | Triazole-glucoside | Isolated yield |
| 1 | 1a | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
2 | 94% |
| 2 | 1b | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
3 | 60% |
| 3 | 1c | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
4 | 46.5% |
| 4 | 1d | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
5 | 43% |
| 5 | 1e | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
6 | 66% |
| 6 | 1f | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
7 | 51% |
| 7 | 1g | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
8 | 62% |
| 8 | 1h | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
9 | 78% |
| 9 | 1i | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
10 | 96% |
| 10 | 1j | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
11 | 80% |
| 11 | 1k | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
12 | 93% |
| 12 | 1l | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
No reaction | — |
| 13 | 1m | CuI (2) | DIPEA (2) | H2O:CH3CN (1:2) |
No reaction | — |
| 14 | 1l | CuSO4·5H2O (0.1) | NaASc (0.2) | H2O:t-BuOH (1:5) |
13 | 73% |
| 15 | 1m | CuSO4·5H2O (0.1) | NaASc (0.2) | H2O:t-BuOH (1:5) |
14 | 75% |
| 16 | 1n | CuSO4·5H2O (0.2) | NaASc (0.4) | H2O:t-BuOH (1:1) |
15 | 82% |
| 17 | 1o | CuSO4·5H2O (0.2) | NaASc (0.4) | H2O:t-BuOH (1:2) |
16 | 79% |
Initially, except for 1a, all the alkynes 1b–1k and 1n–o (Fig. 3) were freshly prepared from their respective starting materials by EDC coupling, reductive amination and alkylation reactions. The phenyl acetylenes (1l–m), were obtained from their respective aldehydes by the Seyferth–Gilbert homologation reaction.37
Pure 1-azido-β-D-glucose (1) was successfully coupled with propargyl groups attached to hydroxyl, ether, amine, amide and ester functional groups by copper-catalyzed azide–alkyne cycloaddition (CuAAC) reactions at room temperature. In all the reactions, two equivalents each of CuI and DIPEA were used for the coupling of 1 and alkynes 1a–k (Fig. 3) giving rise to most of the water soluble 1,2,3-triazole-β-D-glucosyl derivatives 2,20 3,38 4, 5, 6,39 7, 8, 9, 10,38 11,38 12, in good to excellent yields (Fig. 4, Table 1). When the phenyl acetylenes were reacted with compound 1 in the same solvent system, no triazole product was observed, prompting us to explore different click reaction conditions. Later, we found the phenyl acetylenes (1l–m) successfully reacted with compound 1 when catalytic amounts of CuSO4·5H2O (10%) and sodium ascorbate (20%) were used in water and tert-butanol (1:5, v/v), leading to the formation of triazole-glucoside derivatives 13 (ref. 40) and 14 (Fig. 4, Table 1). In the above reaction conditions in water:tert-butanol (1:1 and 1:2, v/v), the compounds 15 and 16 (ref. 41) (Fig. 4, Table 1), were obtained by joining compound 1 with mono-propargylated fluorescein and di-propargylated hydroquinone (1n and 1o), respectively.
It should be noted that compounds such as 4, 5 and 15 (Fig. 4), could be difficult to produce from click reactions with tetra-acetyl-1-azido-β-D-glucose, since the internal ester and lactone groups may be liable during deprotection reactions. For instance, the deprotection of the glucose in generation of the glucose ester of gibberellin 4 (GA4, the starting material for compound 4) led to relatively low yield.7 In fact, for click chemistry, the tetra-acetyl-1-azido-β-D-glucose could be deprotected before coupling and there are synthetic routes to 1-azido-β-D-glucose directly.8–12 However, the enzymatic generation of compound 1 reported here represents is a convenient method for its generation. Compound 1 can then be coupled to such labile groups without the need for deprotection.
Conclusions
The enzyme TxGH116D593A was successfully produced in a multi-milligram scale and subsequently utilized for the gram scale synthesis of 1-azido-β-D-glucose (1) in aqueous medium. The compound 1 was further conjugated with different alkynes via a 1,2,3-triazole as a linker. The complete process proceeds in a safe and convenient way for the generation of novel 1,2,3-triazole-β-D-glucosyl derivatives (2–16) in a short route by avoiding deprotection steps. In addition, compound 1 was produced from pNPGlc in an inexpensive way, when compared with the commercial price of the product. This is the foremost method developed for the gram scale synthesis of compound 1 using an enzyme (TxGH116D593A), and its utilization for the synthesis of glucoconjugates. Compound 1 is widely used in different fields of synthesis and 2–16 are under screening for their inhibitory activities.Methods and experimental section
Silica gel 60 F254 aluminum TLC plates were used to monitor the reactions with short-wavelength ultraviolet light and by charring the TLC plate after spraying with 10% sulfuric acid in ethanol to visualize the spots. Column chromatography was performed on silica gel 60–200 mesh and Sephadex LH-20 resin. 1H NMR and spectra were recorded at 400 MHz and 600 MHz and 13C NMR spectra at 100 MHz, 125 MHz and 150 MHz on a Bruker Avance III 600 MHz instrument with a Cryo probe Prodigy. Either deuterated water (D2O), or deuterated DMSO (CD6SO) was used as the solvent. Chemical shifts are given in parts per million and coupling constants in hertz. HR-ESI-MS analysis was performed on a Thermo Scientific Exactive Mass Spectrometer with ions denoted in m/z.Process for the gram scale production of 1-azido-β-D-glucose (1) from pNP-glucose using enzyme (TxGH116D593A) and purification of 1
The TxGH116D593A enzyme was produced and purified as previously described,6 but without cleavage and removal of the fusion tag. Compound 1 was produced in reactions with 100 mM NaN3 (since higher concentrations did not produce significantly more product, data not shown), in 50 mM 4-morpholineethanesulphonic acid (MES buffer), pH 5.5, and varying concentrations of pNPGlc (Henan Tianfu Chemical Co. Ltd., Zhengzhou, China) substrate and TxGH116D593A enzyme. For the large-scale reaction, 5 g pNPGlc (45 mM) was reacted with 370 mg TxGH115D593A (0.01 mM) as catalyst in a total volume of 370 mL in a 1 L Duran bottle (see ESI Fig. S1†). After 24 hours of incubation in a water bath at 55 °C, the yellow-colored reaction mixture was frozen and subjected to lyophilization (ESI Fig. S1†) for 48 h. The resulting yellow-colored dried mixture was redissolved in methanol, the insoluble material was removed by filtration through cotton and the soluble portion was adsorbed on silica gel and loaded onto a silica gel column.Purification of 1-azido-β-D-glucose (1) using silica gel column chromatography
The silica gel with the adsorbed yellow-colored crude mixture was loaded onto a silica gel (60–200 mesh) column (18.4 cm × 4 cm), in ethyl acetate (EtOAc, 100%). The column was washed with 100% EtOAc (300 mL) to elute the p-nitrophenol, and then with EtOAc/MeOH (95:5, v/v, 5 × 150 mL) to elute the unreacted pNPGlc in a mixture with 1-azido-β-D-glucose (1). Elution with EtOAc/MeOH (94:6, v/v, 5 × 150 mL) gave pure product (see ESI Fig. S1†). Finally, the column was eluted with EtOAc/MeOH (1:1, v/v) to elute the trace of glucose (see ESI Fig. S1†). The compound was monitored by charring the TLC plate after spraying with 10% (v/v) sulfuric acid in ethanol. The eluent fractions containing pure compound 1 and compound 1 mixed with pNPGlc were separately concentrated under reduced pressure. The pure 1-azido-β-D-glucose (1) dried to a white foamy solid (800 mg), Rf = 0.36 (CHCl3/MeOH/NH3, 7:2.8:0.2, v/v/v). The mixture of pNPGlc and compound 1 dried to yield white powder (2.645 g) (ESI Fig. S1†).
Purification of mixture of 1-azido-β-D-glucose (1) and pNP-β-D-glucoside by Sephadex LH-20 column chromatography
The obtained mixture (2.645 g, ESI Fig. S1†) was redissolved in 6 mL of water:methanol (95:5, v/v) and loaded onto an 18 cm × 3.5 cm Sephadex LH-20 column (see ESI Fig. S2†). The column was eluted with 2 × 100 mL of water/methanol (95:5, v/v). The product (1) eluted in pure form, after which the minor amount of pNPGlc eluted. The product purity in the selected fractions was shown by TLC (ESI Fig. S2†), and these fractions were pooled into a round bottom flask, and freeze-dried, resulting in 2.470 g of compound 1 as a white foamy solid (ESI Fig. S2†).
1-Azido-β-D-glucose (1)
1H NMR (D2O, 400 MHz): δ 4.55 (H-1, d, J = 9 Hz, 1H), 3.73 (H-6a, dd, J = 2, 12.4 Hz, 1H), 3.55 (H-6b, dd, J = 5.6, 12.8, Hz, 1H), 3.37–3.30 (H-4, H-5, m, 2H), 3.25 (H-2, appt, J = 9.4 Hz, 1H), and 3.072 (H-3, appt, J = 8.5 Hz, 1H); 1H NMR (D2O, 600 MHz). In the 600 MHz NMR analysis, the anomeric proton peak merged with the solvent peak. The chemical shifts of the observed peaks were δ 3.89 (H-6a, d, J = 12 Hz, 1H), 3.72 (H-6b, d, J = 12 Hz, 1H), 3.51–3.48 (H-4, H-5, m, 2H), 3.40 (H-2, appt, J = 9 Hz, 1H), and 3.24 (H-3, appt, J = 9 Hz, 1H); 13C NMR (D2O, 150 MHz) δ 90.1, 77.9, 75.7, 72.8, 69.2 and 60.6; HR-ESI-MS m/z [M + Na]+ C6H11N3O5Na calc'd for 228.05964, found 228.05975.Compound (2)
To a solution of 1 [26 mg, 0.126 mmol, 1 equivalent (eq.)] in water/acetonitrile (1:2, 2 mL) was added propargyl alcohol 1a (22 μL, 0.38 mmol, 3 eq.), followed by copper iodide (48 mg, 0.252 mmol, 2 eq.) and DIPEA (44 μL, 0.252 mmol, 2 eq.), and the resulting yellow color reaction mixture was stirred at room temperature overnight. TLC indicated the complete conversion of the starting material into a polar compound. The reaction mixture was diluted with 5 mL methanol and then passed through a celite pad by washing with 10 mL methanol; the filtrate was concentrated under reduced pressure. The resulting crude product was redissolved in 4 mL methanol and adsorbed on silica gel and subjected to purification by silica gel chromatography in solvent ethyl acetate/methanol (80:20, v/v), which gave rise to pure compound 2 (31 mg, 94% yield) as a white solid. Rf = 0.13 (EtOAc/MeOH, 9:1, v/v); 1H NMR (D2O, 600 MHz) δ 8.39 (s, 1H), 5.94 (d, J = 8 Hz, 1H), 4.94 (s, 2H), 4.24–4.20 (m, 1H), 4.09 (d, J = 11.7 Hz, 1H), 3.97–3.90 (m, 3H), and 3.82–3.81 (m, 1H); 13C NMR (D2O, 150 MHz) δ 147.2, 123.4, 87.5, 78.9, 76.0, 72.3, 69.0, 60.5 and 54.7; HR-ESI-MS m/z [M + Na]+ calc'd for C9H15N3O6Na 284.08585, found 284.08640.
Compound 3
To a solution of 1 (25 mg, 0.121 mmol, 1 eq.) and propargylcinnamide (1b) (45 mg, 0.243 mmol, 2 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (46 mg, 0.243 mmol, 2 eq.) and DIPEA (42 μL, 0.243 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature for 2.5 h. TLC indicated the formation of a polar compound. The reaction mixture was filtered through a celite pad, washed with methanol (15 mL), adsorbed on silica gel and purified by column chromatography using ethyl acetate/methanol (90:10, v/v), which afforded compound 3 (28 mg, 60%) as a white solid. Rf = 0.2 (EtOAc/MeOH, 8.5:1.5, v/v); 1H NMR (D2O, 600 MHz) δ 8.14 (s, 1H), 7.60 (bs, 2H), 7.51 (d, J = 15.7 Hz, 1H), 7.51 (bs, 3H), 6.61 (d, J = 15.7 Hz, 1H), 5.72 (d, J = 8.7 Hz, 1H), 4.59 (s, 2H), 3.97 (appt, J = 8.5 Hz, 1H), 3.88 (d, J = 12 Hz, 1H), 3.76–3.68 (m, 3H), and 3.60 (appt, J = 9 Hz, 1H); 13C NMR (D2O, 150 MHz) δ 168.8, 141.6, 134.3, 130.3, 129.0, 128.0, 123.1, 119.8, 87.4, 78.9, 75.9, 72.3, 68.9, 60.4, and 34.6; HR-ESI-MS m/z [M + Na]+ calc'd for C18H22N4NaO6 413.14370, found 413.14374.
Compound 4
To a solution of 1 (20 mg, 0.097 mmol, 1.2 eq.) and compound 1c (30 mg, 0.081 mmol, 1 eq.) in water/acetonitrile (1:2, v/v, 1.5 mL), were added copper iodide (31 mg, 0.162 mmol, 2 eq.) and DIPEA (28 μL, 0.162 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature 1 h. TLC indicated the almost complete conversion of the starting material into a polar compound. The reaction mixture was filtered through a celite pad, washed with methanol (15 mL), adsorbed on silica gel, and purified by chromatography using ethyl acetate/methanol (90:10, v/v) as solvent. Compound 4 (26 mg, 46.5% yield) was obtained as a pale brown solid. It had a silica gel TLC Rf = 0.30 (EtOAc/MeOH 9:1, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 8.12 (s, 1H), 5.71 (d, J = 8.7 Hz, 1H), 4.99 (s, 1H), 4.85 (s, 1H), 4.52–4.44 (m, 3H), 3.96 (appt, J = 8.5 Hz, 1H), 3.90 (d, J = 12 Hz, 1H), 3.80–3.75 (m, 2H), 3.71–3.68 (m, 2H), 3.62–3.59 (m, 1H), 3.07 (d, J = 10 Hz, 1H), 2.62–2.60 (m, 2H), 2.06–2.04 (m, 2H), 1.91–1.85 (m, 2H), 1.80–1.56 (m, 6H), 1.35–1.29 (m, 2H), and 1.03 (s, 3H); 13C NMR (D2O, 150 MHz): δ 181.9, 174.1, 158.3, 123.4, 106.5, 97.1, 87.4, 78.9, 75.9, 72.2, 69.6, 69.0, 60.5, 55.0, 53.0, 52.5, 51.3, 51.1, 43.6, 38.8, 36.6, 34.3, 30.8, 27.2, 26.5, 15.6, and 13.7; HR-ESI-MS m/z [M + Na]+ calc'd for C28H38N4NaO9 597.25365, found 597.25415.
Compound 5
To a solution of 1 (25 mg, 0.121 mmol, 1 eq.) and propargyl-benzoate 1d (35 mg, 0.304 mmol, 1.8 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (46 mg, 0.243 mmol, 2 eq.) and DIPEA (42 μL, 0.243 mmol, 2 eq.), and the resulting yellow color reaction mixture was stirred at room temperature overnight. TLC indicated the formation of a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (20 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (90:10, v/v) as solvent gave rise to compound 5 (19 mg, 43% yield) as a white solid. Silica gel TLC gave an Rf = 0.26 (EtOAc/MeOH, 9:1, v/v as solvent); 1H NMR: (D2O, 600 MHz) δ 8.37 (s, 1H), 8.01 (d, J = 6.6 Hz, 2H), 7.67 (s, 1H), 7.51 (d, J = 6.1 Hz, 2H), 5.78 (d, J = 8.8 Hz, 1H), 5.51 (s, 2H), 4.01 (appt, J = 8.6 Hz, 1H), 3.91 (d, J = 12 Hz, 1H), 3.79–3.71 (m, 3H), 3.63 (appt, J = 8.9 Hz, 1H); 13C NMR (D2O, 150 MHz) δ 168.2, 134.0, 129.5, 129.0, 128.8, 125.0, 87.6, 78.9, 75.9, 72.3, 69.0, 60.5, and 57.9; HR-ESI-MS m/z [M + Na]+ calc'd for C16H19N3NaO7 388.11207 amu, found 388.11189.
Compound 6
To a solution of 1 (18 mg, 0.087 mmol, 1 eq.) and propargyl-benzyl ether 1e (63 mg, 0.435 mmol, 5 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (33 mg, 0.174 mmol, 2 eq.) and DIPEA (30 μL, 0.174 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature overnight. TLC indicated formation of a polar compound, just below to the starting material. The reaction mixture was filtered through a celite pad, washed with methanol (15 mL), adsorbed on silica gel and purified by silica gel chromatography using ethyl acetate/methanol (90:10, v/v) as solvent to afford compound 6 (20 mg, 66% yield) as a pale yellow viscous solid. Silica gel TLC showed an Rf = 0.25 (EtOAc/MeOH, 9:1, v/v, as solvent); 1H NMR: (D2O, 600 MHz); δ 8.24 (s, 1H), 7.44–7.42 (m, 5H), 5.76 (d, J = 8.9 Hz, 1H), 4.75 (s, 2H, merged with solvent peak), 4.65 (s, 2H), 4.01 (appt, J = 8.9 Hz, 1H), 3.92 (d, J = 12 Hz, 1H), 3.80–3.71 (m, 3H), and 3.64 (appt, J = 9 Hz, 1H); 13C NMR: (D2O, 150 MHz): δ 136.9, 128.8, 128.7, 128.4, 124.5, 87.4, 78.9, 75.9, 72.3, 72.3, 69.0, 62.2, and 60.4; HR-ESI-MS m/z [M + Na]+ calc'd for C16H21N3NaO6 374.13281, found 374.13373.
Compound 7
To a solution of 1 (27 mg, 0.131 mmol, 1 eq.) and propargyl-indole-3-acetamide 1f (45 mg, 0.210 mmol, 1.6 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (50 mg, 0.263 mmol, 2 eq.) and DIPEA (46 μL, 0.263 mmol, 2 eq.), and the resulting yellow colored mixture was stirred at room temperature for 4 h. Silica gel TLC indicated the almost complete conversion of the starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (20 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (90:10, v/v) as solvent afforded compound 7 (27.3 mg, 51% yield) as a pale brown solid. Silica gel TLC showed an Rf = 0.30 (with EtOAc/MeOH, 9:1, v/v, as solvent); 1H NMR (D2O, 600 MHz):δ 7.68 (s, 1H), 7.50 (appt, J = 8.4 Hz, 2H), 7.28 (s, 1H), 7.23 (appt, J = 7.2 Hz, 1H), 7.12 (appt, J = 7.2 Hz, 1H), 5.58 (d, J = 9 Hz, 1H), 4.42 (s, 2H), 3.86 (d, J = 11.4 Hz, 1H), 3.82–3.79 (m, 4H), 3.66.3.62 (m, 2H), and 3.55 (appt, J = 9.6 Hz, 1H); 13C NMR (D2O, 150 MHz): δ 175.4, 145.3, 136.3, 126.5, 125.1, 122.4, 122.2, 119.5, 118.3, 112.0, 107.5, 87.3, 78.8, 75.8, 72.2, 68.9, 60.4, 34.6, and 32.5; HR-ESI-MS m/z [M + Na]+ calc'd for C19H23N5NaO6 440.15460, found 440.15543.
Compound 8
To a solution of 1 (22 mg, 0.107 mmol, 1 eq.) and propargyltryphtophanamide 1g (44 mg, 0.128 mmol, 1.2 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (41 mg, 0.214 mmol, 2 eq.) and DIPEA (37 μL, 0.214 mmol, 2 eq.), and the resulting reaction mixture was stirred at room temperature for 30 min. TLC indicated the complete conversion of starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (25 mL), and adsorbed on silica gel. Purification by chromatography using ethyl acetate/methanol (92:8, v/v) as solvent afforded compound 8 (35.5 mg, 62% yield) as a white solid. Silica gel TLC gave an Rf = 0.2 (EtOAc/MeOH, 8.5:1.5, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 7.60 (d, J = 7.8 Hz, 1H), 7.50 (s, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.22 (appt, J = 7.2 Hz, 1H), 7.13 (appt, J = 7.2 Hz, 1H), 7.05 (s, 1H), 5.64 (d, J = 9 Hz, 1H), 4.40–4.18 (m, 3H), 3.97–3.93 (m, 2H), 3.78 (dd, J = 5.4, 12.6 Hz, 1H), 3.74–3.73 (m, 2H), 3.69 (appt, J = 9 Hz, 1H), 3.61 (appt, J = 9 Hz, 1H), 3.14 (s, 2H), 1.34 (s, 6H) and 1.22 (s, 3H); 13C NMR (D2O, 150 MHz): δ 174.4, 157.2, 136.0, 126.7, 124.4, 121.8, 119.2, 118.3, 111.8, 108.7, 87.3, 78.8, 75.9, 72.1, 68.9, 60.4, 55.6, 34.1, 27.5, and 27.1; HR-ESI-MS m/z [M + Na]+ calc'd for C25H34N6NaO8 569.23358, found 569.23474.
Compound 9
To a solution of 1 (25 mg, 0.121 mmol, 1 eq.) and p-nitro-benzyl-propargylamine 1h (32 mg, 0.18 mmol, 1.5 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (46 mg, 0.242 mmol, 2 eq.) and DIPEA (42 μL, 0.242 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature for 6 h. TLC indicated the completion of starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (20 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (70:30, v/v) as solvent afforded compound 9 (35 mg, 74% yield) as white solid. Silica gel TLC gave an Rf = 0.29 (EtOAc/MeOH, 7:3, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 8.23 (d, J = 6.9 Hz, 1H), 8.1 (s, 1H), 7.56 (d, J = 6.7 Hz, 1H), 5.75 (d, J = 8.5 Hz, 1H), 4.94 (s, 2H), 4.06–4.02 (m, 3H), 3.93 (d, J = 12 Hz, 1H), 3.81–3.71 (m, 3H), and 3.64 (appt, J = 9 Hz, 1H); 13C NMR (D2O, 150 MHz): δ 147.2, 129.7, 123.9, 87.5, 79.0, 76.0, 72.3, 69.0, 60.5, 54.5, and 50.9; HR-ESI-MS m/z [M + Na]+ calc'd forC16H21N5NaO7 418.13387, found 418.13445.
Compound 10
To a solution of 1 (26 mg, 0.126 mmol, 1 eq.) and Boc-propargylamine 1i (59 mg, 0.378 mmol, 3 eq.) in water/acetonitrile (1:2, v/v, 2 mL) were added copper iodide (48 mg, 0.253 mmol, 2 eq.) and DIPEA (44 μL, 0.253 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature for 3 h. TLC indicated the complete conversion of the starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (15 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (96:4, v/v) as solvent afforded compound 10 (44 mg, 96% yield) as a white solid. Silica gel TLC demonstrated an Rf = 0.35 (with EtOAc/MeOH, 8.5:1.5, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 8.11 (s, 1H), 5.72 (d, J = 8.9 Hz, 1H), 4.36 (s, 2H), 3.99 (appt, J = 8.5 Hz, 1H), 3.91 (d, J = 12 Hz, 1H), 3.79–3.70 (m, 3H), 3.62 (appt, J = 9 Hz, 1H), and 1.47 (s, 9H); 13C NMR (D2O, 150 MHz): δ 158.0, 122.9, 87.4, 78.9, 75.9, 72.3, 69.0, 60.4, and 27.6; HR-ESI-MS m/z [M + Na]+ calc'd for C14H24N4NaO7 383.15427, found 383.15500.
Compound 11
To a solution of 1 (20 mg, 0.097 mmol, 1 eq.) and propargyl-benzamide 1j (31 mg, 0.195 mmol, 2 eq.) in water/acetonitrile (1:2, v/v, 2 mL), were added copper iodide (37 mg, 0.195 mmol, 2 eq.) and DIPEA (34 μL, 0.195 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature for 40 minutes. TLC indicated the complete conversion of the starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (15 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (90:10, v/v) as solvent afforded compound 11 (28 mg, 80% yield) as a white solid. Silica gel TLC demonstrated an Rf = 0.27 (with EtOAc/MeOH, 8.5:1.5, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 8.15 (s, 1H), 7.76 (d, J = 7.2 Hz, 2H), 7.59 (appt, J = 7.6 Hz, 1H), 7.50 (appt, J = 8.4 Hz, 2H), 5.71 (d, J = 9.6 Hz, 1H), 4.68 (s, 2H), 3.96 (appt, J = 9 Hz, 1H), 3.87 (d, J = 11.4 Hz, 1H), 3.76–3.66 (m, 3H), and 3.59 (appt, J = 9.6 Hz, 1H); 13C NMR (D2O, 150 MHz): δ 170.9, 145.1, 133.2, 132.3, 128.8, 127.1, 123.1, 87.4, 78.8, 75.9, 72.2, 68.9, 60.4, and 34.9; HR-ESI-MS m/z [M + Na]+ calc'd for C16H20N4NaO6 387.12805, found 387.12903.
Compound 12
To a solution of 1 (50 mg, 0.243 mmol, 1 eq.) and PMB-propargylether 1k (129 mg, 0.731 mmol, 3 eq.) in water/acetonitrile (1:2, v/v, 4 mL) were added copper iodide (93 mg, 0.486 mmol, 2 eq.) and DIPEA (85 μL, 0.486 mmol, 2 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature overnight. TLC indicated the complete conversion of the starting material into a polar spot. The reaction mixture was filtered through a celite pad, washed with methanol (25 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (90:10, v/v) as solvent afforded compound 12 (87 mg, 93% yield) as a pale yellow solid. Silica gel TLC demonstrated an Rf = 0.27 (with EtOAc/MeOH, 8.5:10, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 8.17 (s, 1H), 7.32 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 7.8 Hz, 2H), 5.71 (d, J = 9.6 Hz, 1H), 4.68 (s, 2H), 4.54 (s, 2H), 3.96 (appt, J = 9 Hz, 1H), 3.88 (d, J = 12.4 Hz, 1H), 3.80 (s, 3H), 3.76–3.66 (m, 3H), and 3.59 (appt, J = 9 Hz, 1H); 13C NMR (D2O, 150 MHz): δ 158.9, 144.4, 130.4, 129.5, 124.4, 87.4, 78.9, 75.9, 72.2, 71.9, 68.9, 61.9, 60.4, and 55.4; HR-ESI-MS m/z [M + Na]+ calc'd for C17H23N3NaO7 is 404.14337, found 404.14513.
Compound 13
A solution of 1 (25 mg, 0.0121 mmol, 1 eq.) and 4-nitro-phenylacetylene 1l (27 mg, 0.182 mmol, 1.5 eq.) in water/tert-butanol (1:5, v/v, 3.5 mL) was heated in a 50 °C water bath until the reaction mixture became homogeneous and then CuSO4·5H2O (3 mg, 0.0121 mmol, 0.1 eq.) and sodium ascorbate (5 mg, 0.0242 mmol, 0.2 eq.), were added and the resulting colorless reaction mixture was stirred at room temperature overnight. TLC indicated the complete conversion of the starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (15 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (94:6, v/v) as solvent afforded compound 13 (31 mg, 73% yield) as a white solid. Silica gel TLC (ran twice), demonstrated an Rf = 0.20 (with EtOAc/MeOH, 9:1, v/v, as solvent); 1H NMR (DMSO-d6, 600 MHz): δ 9.13 (s, 1H), 8.34 (d, J = 8.4 Hz, 2H), 8.17 (d, J = 7.8 Hz, 2H), 5.62 (d, J = 9.6 Hz, 1H), 5.46 (d, J = 6 Hz, 1H), 5.33 (d, J = 4.8 Hz, 1H), 5.18 (d, J = 6 Hz, 1H), 4.62 (appt, J = 6 Hz, 1H), 3.82–3.78 (m, 1H), 3.73 (dd, J = 5.4, 11.4 Hz, 1H), 3.53–3.43 (m, 3H), and 3.28–3.25 (m, 1H); 13C NMR (DMSO-d6, 150 MHz): δ 147.2, 144.9, 137.4, 126.4, 124.9, 123.1, 88.3, 80.4, 77.2, 72.8, 72.0, and 61.2; HR-ESI-MS m/z [M + Na]+ calc'd for C14H16N4NaO7+ 375.0916, found 375.0919.
Compound 14
A solution of 1 (25 mg, 0.0121 mmol, 1 eq.) and 4-cyano-phenylacetylene 1m (23 mg, 0.182 mmol, 1.5 eq.) in water/tert-butanol (1:5, v/v, 3 mL) was heated in a water bath until the reaction mixture became homogeneous. Then, CuSO4·5H2O (3 mg, 0.0121 mmol, 0.1 eq.) and sodium ascorbate (5 mg, 0.0242 mmol, 0.2 eq.) were added and the resulting colorless reaction mixture was stirred at room temperature overnight. TLC indicated the complete conversion of the starting material into a polar compound. The reaction mixture was filtered through celite pad, washed with methanol (20 mL), and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (94:6, v/v) as solvent afforded compound 14 (30 mg, 75% yield) as a white solid. Silica gel TLC (ran twice), demonstrated an Rf = 0.20 (with EtOAc/MeOH, 9:1, v/v, as solvent); 1H NMR (DMSO-d6, 600 MHz): δ 9.05 (s, 1H), 8.09 (d, J = 7.8 Hz, 2H), 7.94 (d, J = 7.8 Hz, 2H), 5.61 (d, J = 9 Hz, 1H), 5.45 (d, J = 5.4 Hz, 1H), 5.33 (d, J = 2.4 Hz, 1H), 5.17 (d, J = 4.8 Hz, 1H), 4.63 (d, J = 5.4 Hz, 1H), 3.81–3.77 (m, 1H), 3.72 (dd, J = 5.4, 10.8 Hz, 1H), 3.52–3.42 (m, 3H), and 3.29–3.25 (m, 1H); 13C NMR (DMSO-d6, 150 MHz): δ 145.3, 135.5, 133.5, 126.2, 122.7, 119.2, 110.7, 88.2, 80.4, 77.2, 72.7, 70.0, and 61.2; HR-ESI-MS m/z [M + Na]+ calc'd for C15H16N4NaO5 355.10184, found 355.10254.
Compound 15
To a solution of 1 (20 mg, 0.097 mmol, 1 eq.) and mono-propargylated fluorescein 1n (20 mg, 0.048 mmol, 0.5 eq.) in water/tert-butanol (1:1, v/v, 2 mL) were added CuSO4·5H2O (3 mg, 0.0097 mmol, 0.2 eq.) and sodium ascorbate (4 mg, 0.019 mmol, 0.4 eq.), and the resulting yellow colored reaction mixture was stirred at room temperature overnight. TLC indicated the complete conversion of the starting material into a fluorescent polar compound. The reaction mixture was filtered through celite pad, washed with methanol (20 mL) and adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (94:6, v/v) as solvent afforded compound 15 (23 mg, 82% yield) as a yellow solid. Silica gel TLC demonstrated an Rf = 0.22 (with EtOAc/MeOH, 8.5:1.5, v/v, as solvent); 1H NMR (DMSO-d6, 600 MHz): δ 10.15 (s, 1H), 8.47 (s, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.80 (appt, J = 7.2 Hz, 1H), 7.73 (appt, J = 7.2 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.12 (s, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.72 (s, 1H), 6.66 (d, J = 8.4 Hz, 1H), 6.58 (s, 2H), 5.56 (d, J = 9.6 Hz, 1H), 5.39 (d, J = 6 Hz, 1H), 5.26 (d, J = 4.2 Hz, 1H), 5.23 (s, 2H), 4.62–4.61 (m, 1H), 3.80–3.76 (m, 1H), 3.69 (dd, J = 5.4, 9 Hz, 1H), 3.46–3.37 (m, 4H), and 3.26–3.23 (m, 1H); 13C NMR (DMSO-d6, 150 MHz): δ 169.1, 160.2, 160.0, 152.9, 152.3, 152.2, 142.6, 136.1, 130.6, 129.5, 129.4, 126.5, 125.1, 124.5, 124.4, 113.3, 112.8, 111.8, 109.9, 102.7, 102.0, 88.0, 83.1, 80.4, 77.4, 72.5, 70.0, 61.8, and 61.2; HR-ESI-MS m/z [M + H]+ calc'd for C29H26N3O10 576.16182, found 576.16227.
Compound 16
To a solution of 1 (44 mg, 0.214 mmol, 2 eq.) and dipropargylated-hydroquinone 1o (20 mg, 0.107 mmol, 1 eq.) in water/tert-butanol (1:2, v/v, 2 mL), were added CuSO4·5H2O (5.3 mg, 0.0214 mmol, 0.2 eq.) and sodium ascorbate (8.5 mg, 0.0428 mmol, 0.4 eq.), and the resulting reaction mixture was stirred at room temperature for 48 h. TLC indicated the formation of a polar compound. The reaction mixture was filtered through a celite pad, which was washed with methanol (40 mL), and the solution adsorbed on silica gel. Purification by silica gel chromatography using ethyl acetate/methanol (80:20, v/v) as solvent afforded compound 16 (51 mg, 79% yield) as a white solid. Silica gel TLC demonstrated an Rf = 0.28 (with EtOAc/MeOH, 6:4, v/v, as solvent); 1H NMR (D2O, 600 MHz): δ 8.25 (s, 2H), 6.99 (s, 4H), 5.71 (d, J = 9 Hz, 2H), 5.20 (s, 4H), 3.96 (appt, J = 9 Hz, 2H), 3.87 (d, J = 12 Hz, 2H), 3.76–3.66 (m, 6H), and 3.59 (appt, J = 9 Hz, 2H); 13C NMR (D2O, 150 MHz): 152.3, 143.7, 124.5, 116.9, 87.4, 79.9, 75.9, 72.2, 68.9, 62.0, 60.4, 54.4, and 42.5; HR-ESI-MS m/z [M + Na]+ calc'd for C24H32N6NaO12 619.21975, found 619.19861.
Conflicts of interest
The authors have filed a patent application for the process described in this paper (TH1801005294), but otherwise have no conflicts of interest to declare.Acknowledgements
The authors are thankful to Dr Ravi Shanker L. and Dr Yanling Hua for assistance with HRMS and NMR, respectively. Financial support from Suranaree University of Technology (SUT) and the Thailand Research Fund (TRF), Thailand, grant BRG5980015, and the National Research University project grant from the Commission on Higher Education of Thailand to SUT is gratefully acknowledged.References
- F. W. Lichtenthalar, Angew. Chem., Int. Ed. Engl., 1994, 33, 2364–2374 CrossRef.
- R. Wombacher, Angew. Chem., Int. Ed. Engl., 2006, 45, 2469–2472 CrossRef CAS PubMed.
- J. R. Knowles, Nature, 1991, 350, 121–124 CrossRef CAS PubMed.
- Q. Wang and S. G. Withers, J. Am. Chem. Soc., 1995, 117, 10137–10138 CrossRef CAS.
- Q. Wang, D. Trimbur, R. Graham, R. A. J. Warren and S. G. Withers, Biochemistry, 1995, 34, 14554–14562 CrossRef CAS.
- R. Charoenwattanasatien, S. Pengthaisong, I. Breen, R. Mutoh, S. Sansenya, Y. Hua, A. Tankrathok, L. Wu, C. Songsiriritthigul, H. Tanaka, S. J. Williams, G. J. Davies, G. Kurisu and J. R. K. Cairns, ACS Chem. Biol., 2016, 11, 1891–1900 CrossRef CAS PubMed.
- Y. Hua, S. Sansenya, C. Saetang, S. Wakuta and J. R. Ketudat Cairns, Arch. Biochem. Biophys., 2013, 537, 39–48 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-I. Shoda, Chem. Commun., 2009, 3378–3379 RSC.
- D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907–11911 CrossRef CAS PubMed.
- S. Shoda, A. Kobayashi, M. Noguchi, T. Tanaka, H. Nagai and T. Matsumoto, JP2009292790A, 2009.
- M.-L. Larabi, C. Frechou and G. Demailly, Tetrahedron Lett., 1994, 35, 2175–2178 CrossRef CAS.
- A. E. Meslouti, D. Beaupere, G. Demailly and R. Uzan, Tetrahedron Lett., 1994, 35, 3913–3916 CrossRef.
- R. Beniazza, N. Bayo, F. Molton, C. Duboc, S. Massip, N. McClenaghan, D. Lastecoueres and J.-M. Vincent, Beilstein J. Org. Chem., 2015, 11, 1950–1959 CrossRef CAS PubMed.
- R. Beniazza, R. Lambert, L. Harmand, F. Molton, C. Duboc, S. Denisov, G. Jonusauskas, N. D. McClenaghan, D. Lastecoueres and J. M. Vincent, Chem.–Eur. J., 2014, 20, 13181–13187 CrossRef CAS PubMed.
- A. Pathigoolla, R. P. Pola and K. M. Sureshan, Appl. Catal., A, 2013, 453, 151–158 CrossRef CAS.
- (a) B. L. Wilkinson, L. F. Bornaghi, S.-A. Poulsen and T. A. Houston, Tetrahedron, 2006, 62, 8115–8125 CrossRef CAS; (b) G. C. Feast, O. E. Hutt, X. Mulet, C. E. Conn, C. J. Drummond and G. P. Savage, Chem.–Eur. J., 2014, 20, 2783–2792 CrossRef CAS PubMed; (c) S. Muthana, H. Yu, H. Cao, J. Cheng and X. Chen, J. Org. Chem., 2009, 74, 2928–2936 CrossRef CAS PubMed.
- S. Dedola, D. L. Hughes, S. A. Nepogodiev, M. Rejzek and R. A. Field, Carbohydr. Res., 2010, 345, 1123–1134 CrossRef CAS PubMed.
- N. Anand, N. Jaiswal, S. K. Pandey, A. K. Srivastava and R. P. Tripathi, Carbohydr. Res., 2011, 346, 16–25 CrossRef CAS PubMed.
- L. L. Rossi and A. Basu, Bioorg. Med. Chem. Lett., 2005, 15, 3596–3599 CrossRef CAS PubMed.
- E. D. Chrysina, E. Bokor, K.-M. Alexacou, M.-D. Charavgi, G. N. Oikonomakos, S. E. Zographos, D. D. Leonidas, N. G. Oikonomakos and L. Somsak, Tetrahedron: Asymmetry, 2009, 20, 733–740 CrossRef CAS.
- E. Bokor, T. Docsa, P. Gergely and L. Somsak, Bioorg. Med. Chem., 2010, 18, 1171–1180 CrossRef CAS PubMed.
- E. Bokor, E. Kyriakis, T. G. A. Solovou, C. Koppany, A. L. Kantsadi, K. E. Szabo, A. Szakacs, G. A. Stravodimos, T. Docsa, V. T. Skamnaki, S. E. Zographos, P. Gergely, D. D. Leonidas and L. Somsak, J. Med. Chem., 2017, 60, 9251–9262 CrossRef CAS PubMed.
- R. P. Tanpure, B. Ren, T. S. Peat, L. F. Bornaghi, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2015, 58, 1494–1501 CrossRef CAS PubMed.
- B. L. Wilkinson, A. Innocenti, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2008, 51, 1945–1953 CrossRef CAS PubMed.
- S. G. Gouin, E. Vanquelef, J. M. G. Fernandez, C. O. Mellet, F. Y. Dupradeau and J. Kovensky, J. Org. Chem., 2007, 72, 9032–9045 CrossRef CAS PubMed.
- A. Massarotti, S. Aprile, V. Mercalli, E. D. Grosso, G. Grosa, G. Sorba and G. C. Tron, ChemMedChem, 2014, 9, 2497–2508 CrossRef CAS PubMed.
- P. Stefan, P. Oliver, T. Norbert, W. Ulrich and E. Frank, WO/2017/207754, 2017.
- S. Cecioni, V. Oerthel, J. Iehl, M. Holler, D. Goyard, J.-P. Praly, A. Imberty, J. F. Nierengarten and S. Vidal, Chem.–Eur. J., 2011, 17, 3252–3261 CrossRef CAS PubMed.
- D. H. Kim, Y. S. Choe, K.-H. Jung, K.-H. Lee, J. Y. Choi, Y. Choi and B.-T. Kim, Arch. Pharmacal Res., 2008, 31, 587–593 CrossRef CAS PubMed.
- S. Maschauer, R. Haubner, T. Kuwert and O. Prante, Mol. Pharmaceutics, 2014, 11, 505–515 CrossRef CAS PubMed.
- Z. Lei, J. Wang, G. Mao, Y. Wen, Y. Tian, H. Wu, Y. Li and H. Xu, J. Agric. Food Chem., 2014, 62, 6065–6071 CrossRef CAS PubMed.
- C. J. Carroux, G. M. Rankin, J. Moeker, L. F. Bornaghi, K. Katneni, J. Morizzi, S. A. Charman, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2013, 56, 9623–9634 CrossRef CAS PubMed.
- M. Lopez, L. F. Bornaghi, A. Innocenti, D. Vullo, S. A. Charman, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2010, 53, 5913–5926 CrossRef PubMed.
- S. Ballet, C. Betti, A. Novoa, C. Tomboly, C. U. Nielsen, H. S. Helms, A. Lesniak, P. Kleczkowska, N. N. Chung, A. W. Lipkowski, B. Brodin, D. Tourwe and P. W. Schiller, ACS Med. Chem. Lett., 2014, 5, 352–357 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed.
- G. J. Roth, B. Liepold, S. G. Muller and H. J. Bestmann, Synthesis, 2004, 1, 59–62 Search PubMed.
- D. Goyard, T. Docsa, P. Gergely, J.-P. Praly and S. Vidal, Carbohydr. Res., 2015, 402, 245–251 CrossRef CAS PubMed.
- I. Delso, J. Valero-Gonzalez, F. Gomollon-Bel, J. Castro-Lopez, W. Fang, I. Navratilova, D. M. F. van Aalten, T. Tejero, P. Merino and R. Hurtado-Guerrero, ChemMedChem, 2018, 13, 128–132 CrossRef CAS PubMed.
- D. Goyard, A. S. Chajistamatiou, A. I. Sotiropoulou, E. D. Chrysina, J.-P. Praly and S. Vidal, Chem. - Eur. J., 2014, 20, 1–11 CrossRef PubMed.
- G. Yu, Y. Ma, C. Han, Y. Yao, G. Tang, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2013, 135, 10310–10313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary material contains Fig. S1–S6, including purification, 1H NMR spectra of 1–16, 13C NMR spectra of 1–16, and HRMS spectrum of compound 16. See DOI: 10.1039/c9ra00736a |
| This journal is © The Royal Society of Chemistry 2019 |