
Phosphine-catalyzed fixation of CO2 with γ-hydroxyl alkynone under ambient temperature and pressure: kinetic resolution and further conversion†
Yao-Liang
Sun
ab,
Yin
Wei
 *a and
Min
Shi
*abc
*a and
Min
Shi
*abc
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: weiyin@sioc.ac.cn; mshi@mail.sioc.ac.cn
bKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
cState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 21st May 2019
Abstract
Multiple skeleton derived γ-hydroxyl alkynones could be activated by phosphine, which then underwent cycloaddition with CO2 to afford functionalized carbonate products under ambient temperature and pressure. These functionalized carbonate products could easily release CO2 under heating conditions, giving a diversity of furanones in excellent yields. The optically active functionalized cyclic carbonates could be afforded through the kinetic resolution of propargyl alcohols via carbon dioxide fixation catalyzed by a new series of sterically hindered and highly nucleophilic bifunctional amino acid-derived phosphine catalysts with moderate to excellent selectivities. Plausible mechanisms were proposed and supported by isotope-labeling experiments and DFT calculations.
Introduction
Due to global warming associated with greenhouse gas emissions, effectively reducing carbon dioxide (CO2) in the atmosphere has attracted a lot of attention from the scientific community; direct conversion of CO2 to useful chemical products not only reflects the exploration of environmental protection issues, but also has many advantages such as low-cost, easy availability, abundance, nontoxicity, and inherent renewability.1 Among the carbon dioxide fixation products, cyclic carbonates2 are quite important in industrial materials, which are frequently used as predominant monomers for polycarbonates or electrolytes in lithium-ion-battery half-cells.3 Furthermore, cyclic carbonates have great potential in synthetic transformations; their synthetic value and relevant mechanistic studies have been continuously improved in these years.4 Meanwhile, along with the increasing requirement of CO2 as a useful renewable carbon resource participating in the stereo-controlled preparation of value-added commodities,5 a cobalt complex was firstly and widely used in catalytic enantioselective reactions for the synthesis of chiral carbonates and polycarbonates through kinetic resolution of racemic epoxides via CO2 fixation.6 Transition metals (Ni,7 Pd,8 Rh,9 Ir,10 Cu,11 and Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12) were also frequently used in catalytic asymmetric CO2 fixation, affording different useful compounds like chiral alcohols, acids, esters, carbonates and carbamates.
12) were also frequently used in catalytic asymmetric CO2 fixation, affording different useful compounds like chiral alcohols, acids, esters, carbonates and carbamates.
α-Alkylidene cyclic carbonates are a class of carbonates with special properties, which could be produced by the reaction of CO2 with propargylic alcohols under metal13 or metal-free14,15 catalysis. Recently, many efforts have been devoted to increase the efficiency of this green route to fix CO2, which means to look for mild reaction conditions and to achieve a wide range of substrate scopes. Yamada and co-workers first developed silver catalytic systems to combine CO2 with different propargyl alcohols to afford α-alkylidene cyclic carbonates and carbonyl compounds under 1.0 MPa of CO2.16 Ikariya17 and Lu18 independently reported N-heterocyclic carbenes (NHCs) and NHC–CO2 adducts served as potent organocatalysts to promote carbon dioxide fixation reaction under pressurized and heating conditions. Further transformations of α-alkylidene cyclic carbonates have also been studied. Using metal catalysts such as Pd,19 Pt,20 Rh,21 and Ni22 could easily transform α-alkylidene cyclic carbonates to oxygen heterocycles or carboxylic acid compounds.
In recent years, the nucleophilic phosphine-catalyzed reactions23 have been recognized as reliable methods which could combine olefins,24 allenes,25 alkynes,26 or Morita–Baylis–Hillman adducts (MBHADs)27 with other electrophilic or nucleophilic reagents to afford highly functionalized products. As early as 1989, Dixneuf et al. synthesized α-alkylidene cyclic carbonates by using tributylphosphine as a catalyst under harsh reaction conditions with 5.0 MPa pressure of CO2 at 100 °C for 8 h (Scheme 1).15 They proposed that tertiary phosphine either acted as a Brønsted base catalyst to undergo proton abstraction from the alcohol or as a nucleophilic catalyst to generate a zwitterionic intermediate before incorporating CO2. Based on the commonly accepted mechanisms of nucleophilic phosphine-catalyzed reactions in recent years, we hypothesized that introducing an electron deficient group on the alkyne's terminal position of substrate might lead to the very stable zwitterionic intermediate A, which could facilitate the following process of CO2 fixation since the oxygen nucleophilic site of intermediate A would directly attack carbon dioxide and the subsequent cyclization step would be easier to proceed due to the presence of a highly electrophilic center (Scheme 1). Recently, Yamada disclosed a Lewis-acid catalyzed decarboxylative28 Nazarov cyclization of cyclic carbonate derivatives, which underwent a cationic intermediate directed 4π-ring-closure process to afford 2-cyclopentenones.29 We anticipate that the α,β-unsaturated 4π system with a carbonyl group of functionalized cyclic carbonates could also participate in a decarboxylative ring-closure process to provide a range of novel furanone derivatives (Scheme 1).
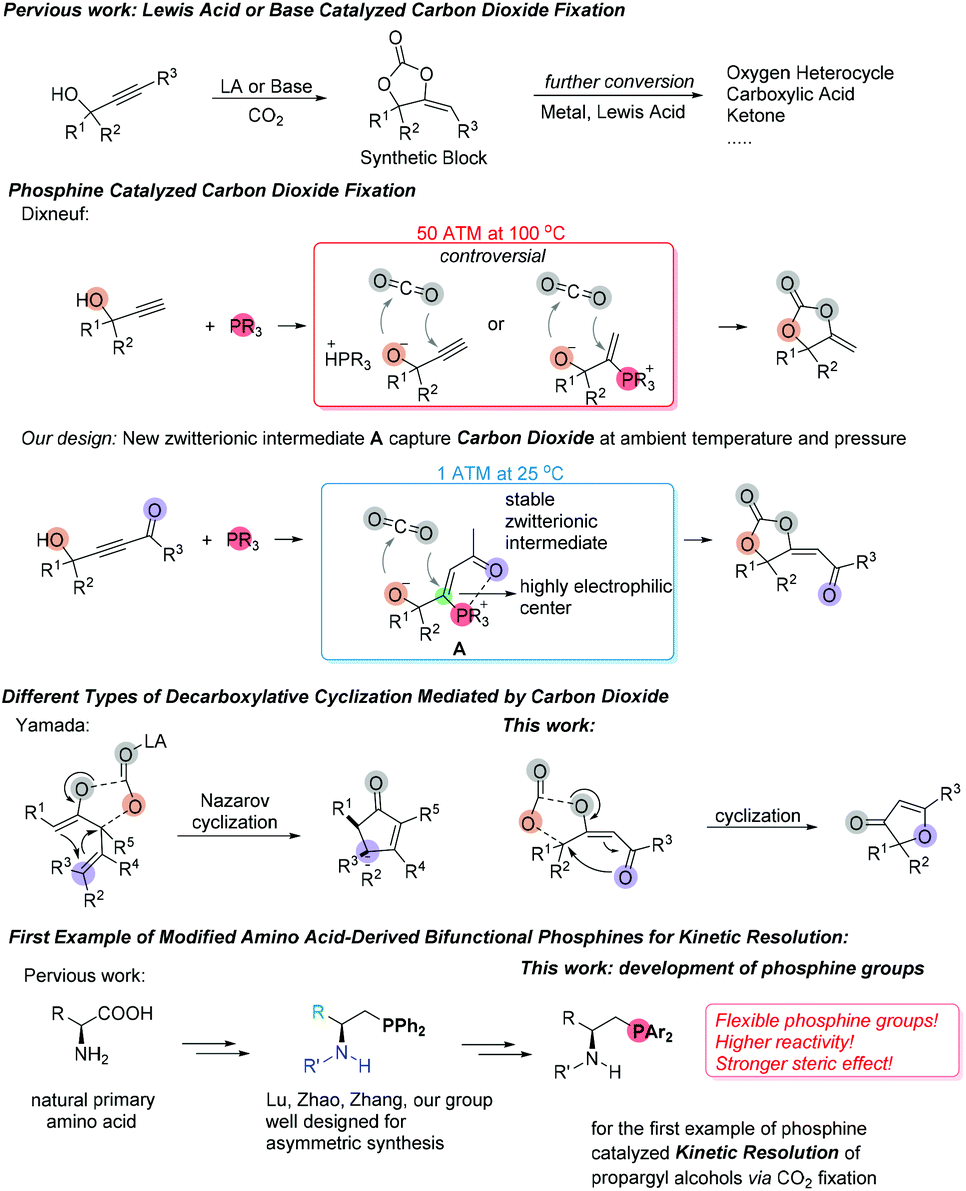 | ||
| Scheme 1 Newly designed phosphine-catalyzed reactions of alkynones with other substrates. | ||
In the past decade, Lu established a family of bifunctional amino acid-derived phosphine catalysts that were efficient catalysts in asymmetric reactions for the enantioselective construction of all-carbon quaternary stereogenic centers.30,31 Zhao,31 Zhang32 and our group33 also made the corresponding contributions in this area. The amino acid-derived phosphine catalysts have modular, tunable, dipeptide scaffolds and abundant derived amino groups as H-bond donors to adapt a variety of catalytic requirements. Although many highly efficient chiral phosphine catalysts have been frequently applied in asymmetric reactions, the use of chiral phosphine catalysts for kinetic resolution of racemic compounds was still a challenge. Vedejs and co-workers first developed kinetic resolution of secondary alcohols via chiral phosphine-catalyzed acylation.34 However, there is no report on the kinetic resolution of propargyl alcohols via CO2 fixation in the catalysis of chiral phosphines. In order to obtain optically active functionalized cyclic carbonates, we investigated a series of chiral phosphine catalysts to realize the kinetic resolution of propargyl alcohols via CO2 fixation and found that substituents on the phosphine center had a significant effect on the stereoselectivity of the reaction. Thus, we studied the performance of a new series of sterically hindered and highly nucleophilic bifunctional amino acid-derived phosphine catalysts in this reaction. Herein, we wish to report the first example of chiral phosphine catalyzed kinetic resolution of propargyl alcohols via carbon dioxide fixation in good yields with moderate to good selectivities (Scheme 1). We also conducted the mechanistic studies to reveal the detailed mechanisms through isotope-labeling experiments and DFT calculations.
Results and discussion
Experimental investigations
We initially tested the reaction outcomes employing 3-hydroxy-1-methyl-3-(3-oxobut-1-yn-1-yl)indolin-2-one 1a and carbon dioxide (balloon, 1 atm) as substrates in the presence of phosphine, and subsequently optimized the reaction conditions. The results are shown in Table 1. It was found that isatin-derived spirocarbonate 2a was obtained in 81% yield with 0.2 equiv. of PBu3 in THF at room temperature for 3 h (entry 1). The solvent effect was first investigated, and it was identified that THF was better than other solvents such as DCM, ether, MTBE and ethyl acetate (entries 2–6). It is interesting to note that when MeOH was used as a solvent, only isatin-derived furanone 3a was obtained in 49% yield (entry 3). Decreasing the concentration of the reaction mixture retarded the reaction, and 1a was recovered in 56% yield (entry 7). Employing other phosphine catalysts did not significantly improve the yield of 2a, giving 2a in yields ranging from trace to 66% (entries 8–12). Using other Lewis base catalysts such as DMAP and DABCO only gave 2a in yields of 20% and 31%, respectively (entries 13 and 14). The reaction could not take place in the presence of K2CO3, suggesting that the reaction could not be initiated by the deprotonation process with a Brønsted base catalyst (entry 15). The structures of 2a and 3a have been assigned by X-ray diffraction. Their ORTEP drawings are shown in Tables 2 and 3 and the CIF data are also summarized in the ESI.†|
|
|||||
|---|---|---|---|---|---|
| Entrya | Cat. | Solvent | Temp (°C) | Time (h) | Yield % of 2ab |
| a The reaction was carried out using 1a (0.1 mmol) and cat. (0.02 mmol) in the indicated solvent (0.5 mL) in a Schlenk tube at the indicated temperature under a CO2 atmosphere. b Determined by 1H NMR analysis of the crude reaction mixture by using 1,3,5-trimethoxybenzene as an internal standard. c Only product 3a was obtained in 49% yield. d 1 mL THF was used and 56% 1a was recovered. | |||||
| 1 | PBu 3 | THF | rt | 3 | 81 |
| 2 | PBu3 | DCM | rt | 3 | 64 |
| 3 | PBu3 | MeOH | rt | 3 | —c |
| 4 | PBu3 | Et2O | rt | 3 | Trace |
| 5 | PBu3 | MTBE | rt | 3 | 40 |
| 6 | PBu3 | EA | rt | 3 | 56 |
| 7 | PBu3 | THFd | rt | 3 | 32 |
| 8 | PMe3 | THF | rt | 3 | Complex |
| 9 | PMe3Ph | THF | rt | 3 | 66 |
| 10 | PMePh2 | THF | rt | 3 | 53 |
| 11 | PEtPh2 | THF | rt | 3 | 60 |
| 12 | PPh3 | THF | rt | 3 | Trace |
| 13 | DMAP | THF | rt | 3 | 20 |
| 14 | DABCO | THF | rt | 3 | 31 |
| 15 | K2CO3 | THF | rt | 3 | nr |

| a The reaction was carried out using 1 (0.3 mmol) and PBu3 (0.06 mmol), in THF (1.5 mL) in a Schlenk tube at the indicated temperature. |
|---|
|
a
| a The reaction was carried out using 2 (0.2 mmol) in DCM (2.0 mL) in a Schlenk tube at the indicated temperature. |
|---|

|
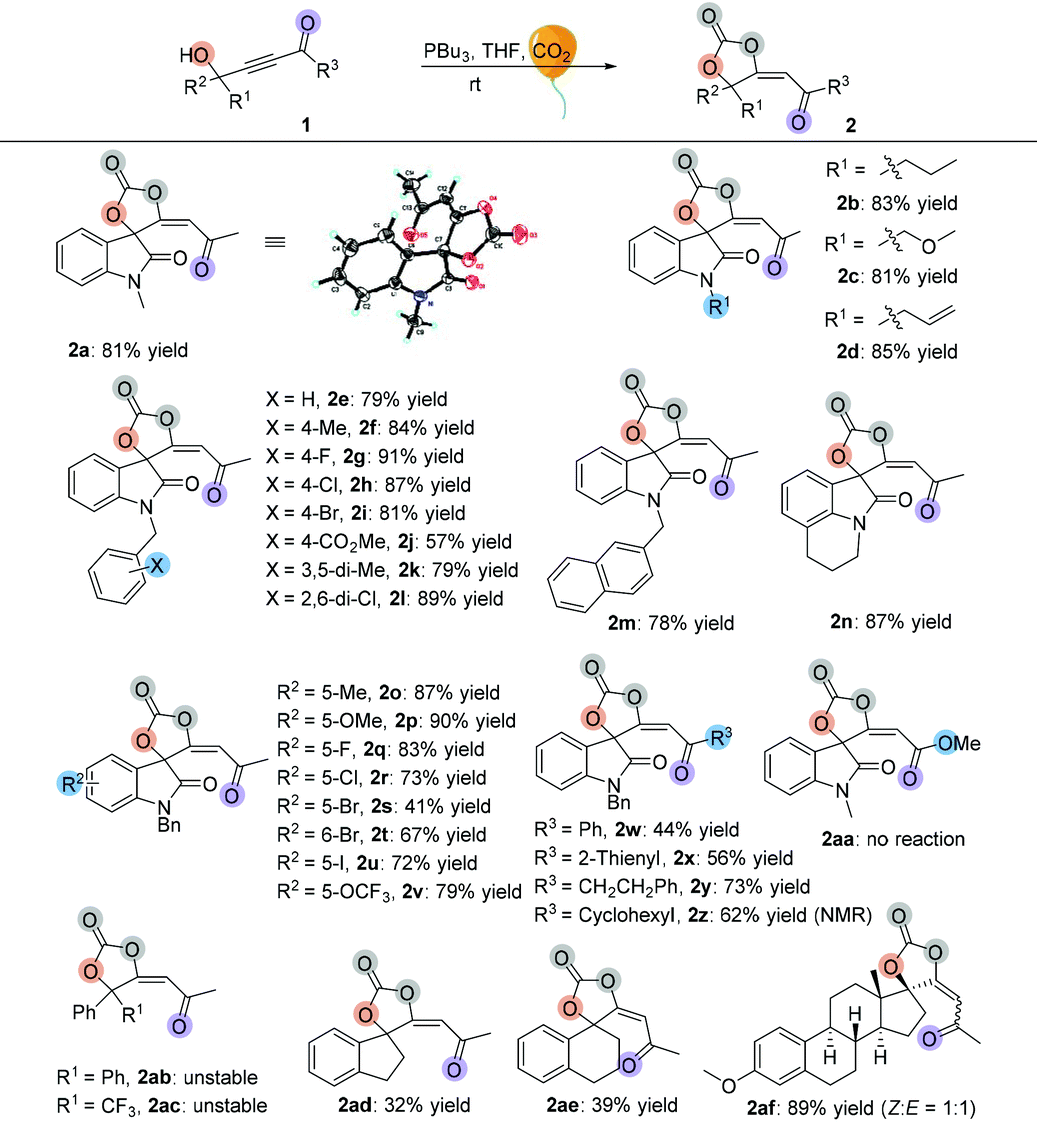
Having established the optimal reaction conditions, we next surveyed the substrate scope of the reaction by using a range of γ-hydroxyl alkynones 1 (Table 2). First, we examined isatin-derived substrates 1 with different N-alkyl groups. It was found that methyl, n-propyl, methyloxymethyl and allyl group substituted 3-hydroxy-3-(3-oxobut-1-yn-1-yl)indolin-2-ones 1a–1d afforded the desired spirocarbonates 2a–2d in the range of 81–85% yields. Using the benzyl group substituted 3-hydroxy-3-(3-oxobut-1-yn-1-yl)indolin-2-one 1e as the substrate also gave the corresponding product 2e in 79% yield. Changing the substituents on the benzyl moiety afforded the corresponding products 2f–2l in yields ranging from 57% to 91%; only the 4-CO2Me substituted product 2j was isolated in 57% yield. Replacing the benzyl group by a naphthalen-2-ylmethyl group (1m) did not significantly influence the reaction, giving 2m in 78% yield. 4H-Pyrrolo[3,2,1-ij]quinoline-derived substrate 1n was also tolerated, affording the corresponding product 2n in 87% yield.
Next, substrates 1o–1v having different substituents on the isatin moiety were examined. Substrates 1o and 1p containing electron-donating groups (Me, OMe) afforded the corresponding products in higher yields (87% yield and 90% yield) than substrates 1q–1v containing electron-withdrawing groups such as F, Cl, Br, I and OCF3 groups (41% yield to 83% yield). When R3 groups were phenyl and 2-thienyl groups, the desired products 2w and 2x were only obtained in 44% and 56% yields, respectively; using the CH2CH2Ph group instead of the methyl group could also give the desired product 2y in 73% yield. However, it is hard to isolate cyclohexyl group substituted product 2z from the reaction mixture, perhaps due to its instability. The NMR spectrum of the crude reaction solution indicated that 62% of 2z was produced. Propiolate-derived substrate 1aa failed to give the desired product 2aa. Other types of γ-hydroxyl alkynones were also investigated. Firstly, acyclic substrates 1ab and 1ac were synthesized, however, the corresponding products 2ab and 2ac were unstable and easily converted to furanones 3ab and 3ac (see Table 3). Under the standard conditions, both carbocyclic products 2ad and 2ae were successfully obtained in 32% and 39% yields, respectively, with exclusive E-configurations. Interestingly, using ethinylestradiol-derived substrate 1af smoothly gave a pair of separable products containing a cis isomer and a trans isomer in 89% total yield, presumably due to the steric effect.
After investigation of the substrate scope, we noted that the cyclic carbonate products 2 could easily release CO2 upon heating and obtained a diversity of furanones 3 in excellent yields. Subsequently, we examined the decarboxylative cyclization of cyclic carbonate products 2 at 60 °C. As shown in Table 3, changing the groups on the oxindole moiety with methyl, n-propyl, methyloxymethyl and allyl groups did not affect the product yields, giving the desired products 3a–3d in the range of 85%–92% yields. Most of the benzyl group substituted isatin-derived spirocarbonates could release CO2 and give the corresponding isatin-derived furanones in excellent yields. However, cyclic carbonate products 2 having sterically hindered substituents could not undergo the reaction; spirocarbonates 2l and 2af were stable upon heating, and none of the desired products 3l and 3af were detected. Naphthalen-2-ylmethyl substituted substrate 2m could smoothly give 3m in 91% yield; however, the transformation of 4H-pyrrolo[3,2,1-ij]quinoline-derived spirocarbonate 2n failed under identical conditions. These results suggested that the more rigid multicyclic systems or the larger sterically hindered groups would lead to less degree of rotational freedom of these non-reactive substrates and subsequently block out their rearrangements. Fortunately, either substrates containing electron-donating groups (Me, OMe) or substrates containing electron-withdrawing groups (F, Cl, Br, I and OCF3) could give the corresponding products in excellent yields. On changing the R3 groups, furanones 3w–3y containing a phenyl group, 2-thienyl group and CH2CH2Ph substituted group were obtained in 73%–91% yields; even cyclohexyl group substituted furanone 3z could be isolated in 59% yield after two-step transformations. Similarly, acyclic furanones 3ab and 3ac were isolated in 69% yield and 46% yield, respectively, suggesting that the furanones are thermodynamically more stable than spirocarbonates. Five- or six-membered carbocyclic furanones 3ad and 3ae were also obtained in 79% yield and 75% yield under the standard conditions, respectively.
Kinetic resolution investigations
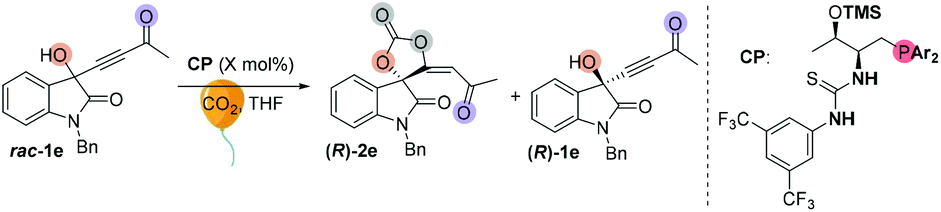
We initially screened a variety of chiral phosphines CP1–CP14 by using racemic 1e with CO2 (balloon, 1 atm) in THF. The results are summarized in Tables S1 and S2.† Preliminary investigations showed that Lu's catalyst CP14 gave the highest yield; however, as compared with other catalysts (Table SI†), the enantioselectivity remained low (s < 5) (Table 4, entry 1). We then focused our attention on improving the stereoselectivity by developing new chiral phosphine catalysts. Based on Lu and Pfaltz's work,23k we introduced a series of different substituents into the aryl group substituents on the phosphine atom and synthesized a series of new bifunctional chiral phosphines. When 30 mol% N-CP1 was used as a catalyst, the extra two methyl groups could significantly improve the enantioselectivity of spirocarbonate (up to 80% ee) and the s-factor was up to 7.8; reducing the amount of catalyst would result in lower conversion rates and lower s-factors (entries 2–4). Due to the excess steric hindrance, tert-butyl group substituted catalyst N-CP2 could not promote the reaction either at −40 °C or at −20 °C (entries 5 and 6). Next, we introduced methoxy groups into the phenyl ring to improve the nucleophilicity of catalysts. However, the DTBM type catalyst N-CP3 and 4-methoxyl group substituted N-CP4 showed almost the same selectivities as that of using CP14 as the catalyst (entries 7 and 8). The highly nucleophilic 3,5-dimethoxy groups substituted phosphine catalyst N-CP5 could catalyze the reaction at −60 °C, affording (R)-2e in 32% yield with 84% ee; however, (R)-1e was isolated with only 84% ee, which showed a low selectivity (s = 2.9) (entries 9–11). Finally, using 4-methoxy-3,5-dimethylphenyl group substituted catalyst N-CP6 could increase the selectivity slightly; (R)-2e was isolated in 42% yield with 86% ee, (R)-1e was simultaneously isolated in 51% yield with 64% ee and the s-factor was up to 9.5 (entries 12 and 13). Interestingly, employing 2-MeTHF as a solvent could further improve the enantioselectivity of (R)-1e up to 90% ee, which encouraged us to continue to explore the way to improve the selectivity of the reaction. Adding 2.0 mL 2-MeTHF was essential to avoid 1e from precipitating at low temperature (entry 14). After further screening of catalyst loading (entries 15 and 16), we found that the use of 15 mol% of N-CP-6 [1-((2S,3R)-1-(bis(4-methoxy-3,5-dimethylphenyl)phosphanyl)-3-((trimethylsilyl)oxy)butan-2-yl)-3-(3,5-bis(trifluoromethyl)phenyl)thiourea] could improve this kinetic resolution process to a higher selectivity (s = 24.6) as well as 54% conversion; unreacted (R)-1e could be recovered in 46% yield with 91% ee, and (R)-2e was isolated in 49% yield with 81% ee. The investigations on solvent effects are summarized in Table S3 in the ESI.†|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | CP | Temp (°C) | Time (h) | Conv. (%) | Yieldb (%) | eec (%) | s-Factord | |||
| CP (X mol%) | Ar | (R)-2e | (R)-1e | (R)-2e | (R)-1e | |||||
| a The reaction was carried out using rac-1e (0.1 mmol) and cat. (0.015–0.03 mmol), in THF (0.5 mL) in a Schlenk tube at the indicated temperature. b Isolated yields. c The ee value was determined by HPLC analysis using a chiral stationary phase. d Selectivity (s-factor) calculated as s = ln[(1 − C)(1-eeSM)]/ln[(1 − C)(1 + eeSM)]. e 2.0 mL 2-MeTHF was used. | ||||||||||
| 1 | CP14 (20) |

|
−40 | 12 | 31 | 29 | 69 | 68 | 20 | 3.1 |
| 2 | N-CP1 (10) |

|
−40 | 12 | 37 | 29 | 63 | 85 | 22 | 2.7 |
| 3 | N-CP1 (20) | −40 | 12 | 49 | 40 | 51 | 84 | 55 | 5.5 | |
| 4 | N-CP1 (30) | −40 | 12 | 57 | 48 | 43 | 80 | 75 | 7.8 | |
| 5 | N-CP1 (20) |
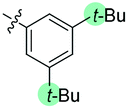
|
−40 | 12 | Trace | — | — | — | — | — |
| 6 | N-CP1 (20) | −20 | 24 | Trace | — | — | — | — | — | |
| 7 | N-CP1 (20) |
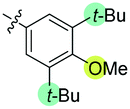
|
−40 | 12 | 49 | 41 | 51 | 61 | 43 | 3.9 |
| 8 | N-CP1 (20) |
|
−40 | 12 | 48 | 40 | 52 | 70 | 40 | 3.7 |
| 9 | N-CP1 (10) |

|
−60 | 12 | Trace | — | — | — | — | — |
| 10 | N-CP1 (20) | −60 | 12 | 39 | 32 | 61 | 84 | 25 | 2.9 | |
| 11 | N-CP1 (20) | −40 | 12 | 49 | 42 | 51 | 77 | 45 | 4.2 | |
| 12 | N-CP1 (20) |

|
−40 | 12 | 33 | 33 | 67 | 86 | 42 | 2.2 |
| 13 | N-CP1 (30) | −60 | 12 | 49 | 42 | 51 | 86 | 64 | 9.5 | |
| 14e | N-CP1 (20) | −40 | 12 | 63 | 52 | 37 | 80 | 90 | 5.4 | |
| 15 | N-CP1 (15) | −40 | 12 | 54 | 49 | 46 | 81 | 91 | 24.6 | |
| 16e | N-CP1 (10) | −40 | 12 | 53 | 46 | 47 | 80 | 88 | 23.2 | |
The generality of this chiral phosphine-catalyzed kinetic resolution process was then investigated. A wide range of substrates having different substituents on the oxindole backbones were also well tolerated in our catalytic system (Table 5). We first tested the effects of protecting groups on the nitrogen atom (Table 5, entries 1–3); substrates 1b–1d containing n-propyl, methyloxymethyl and allyl groups worked as well as the Bn protected substrate 1e, giving the desired products (R)-1b–1d and (R)-2b–2d with good selectivities (s = 17.0–20.6). On changing the substituents on the benzyl group, the reactions proceed to about 50% conversion, which afforded the corresponding products 2e–2i in ee values ranging from 81% to 90%, and the best s-factor was up to 24.5 (entries 4–8). Next, substrates 1o, 1p and 1v with various substituents on the oxindole moiety were examined. Substrates 1o and 1p containing electron-donating groups (Me, OMe) afforded the corresponding products in higher selectivities (s = 22.9 and 20.2) than substrate 1v containing an electron-withdrawing group such as OCF3 (entries 9–11). The substrates containing electron-withdrawing groups showed lower conversion and reactivity, probably due to the difficulty in regenerating the catalyst. 4H-Pyrrolo[3,2,1-ij]quinoline-derived substrate 1n was recovered with an excellent enantioselectivity in up to 96% ee, and (R)-2n was isolated with 92% ee, which proceeded to 44% conversion with s = 38.6 (entry 12). Other substrates could not produce the desired products with excellent selectivities and the results are summarized in Table S4 of the ESI.†
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | Starting materials | Time (h) | Conv. (%) | Yieldb (%) | eec (%) | s-Factord | ||
| (R)-2 | (R)-1 | (R)-2 | (R)-1 | |||||
| a The reaction was carried out using rac-1 (0.1 mmol) and cat. (0.015 mmol) in the indicated solvent (2.0 mL) in a Schlenk tube at the indicated temperature. b Isolated yields. c The ee value was determined by HPLC analysis using a chiral stationary phase. d Selectivity (s-factor) calculated as s = ln[(1 − C)(1-eeSM)]/ln[(1 − C)(1 + eeSM)]. | ||||||||
| 1 |
|
1b: 12 | 59 | 49 | 41 | 82 | 97 | 20.6 |
| 2 | 1c: 12 | 51 | 41 | 49 | 87 | 81 | 17.0 | |
| 3 | 1d: 12 | 60 | 52 | 40 | 79 | 97 | 18.6 | |
| 4 |

|
1e: 12 | 54 | 49 | 46 | 81 | 91 | 24.6 |
| 5 | 1f: 12 | 51 | 42 | 49 | 85 | 80 | 18.5 | |
| 6 | 1g:12 | 55 | 46 | 45 | 90 | 93 | 24.5 | |
| 7 | 1h: 12 | 49 | 40 | 51 | 84 | 76 | 19.4 | |
| 8 | 1i: 12 | 53 | 41 | 47 | 85 | 83 | 19.6 | |
| 9 |
|
1o: 12 | 54 | 40 | 46 | 85 | 90 | 22.9 |
| 10 | 1p: 12 | 53 | 42 | 47 | 90 | 86 | 20.2 | |
| 11 | 1v: 12 | 50 | 41 | 50 | 86 | 65 | 9.3 | |
| 12 |

|
1n: 12 | 44 | 47 | 46 | 92 | 96 | 38.6 |

Several optically active spirocarbonates were used to investigate whether the configuration retention of the chiral quaternary carbon center could be realized during decarboxylative cyclization, because it could afford optically active furanones. The results are summarized in Table 6. Allyl group protected furanone 3d was obtained in 89% yield with 82% ee, which completely maintained the chirality. Moreover, 4-methylbenzyl and 4-fluorobenzyl groups protected furanones 3f and 3g were also obtained in high yields with high ee values. However, partial racemization was observed during the transformation of 4-bromobenzyl spirocarbonates, suggesting that the decarboxylative cyclization might go through a carbocationic intermediate. Finally, 2o gave the desired product 3o in 81% ee with 95% es. 2v also underwent partial racemization and afforded the corresponding product 3v in 76% ee with 88% es.
| a The reaction was carried out using 2 (0.05 mmol) in DCM (1.0 mL) in a Schlenk tube at the indicated temperature. bIsolated yields. cThe ee value was determined by HPLC analysis using a chiral stationary phase. dThe enantiospecificity (es) = (ee of product/ee of starting material) × 100%. |
|---|

|
Mechanistic studies
To gain some insights into the reaction mechanism, several deuterium-labeling experiments were performed at different temperatures. The reaction of 1a with CO2 was first carried out in the presence of D2O (10 equiv.) under the standard reaction conditions. The reaction became sluggish to give the corresponding partially deuterated addition product [D]-2a in 65% yield along with 84% deuterium incorporation at the olefinic position (Scheme 2a). After heating, [D]-3a was obtained in 86% yield along with 80% deuterium incorporation at the olefinic position. Under the standard reaction conditions, using 53% O–H deuterated [D]-1a as a substrate produced the corresponding deuterated product [D]-2a′ in 81% yield with 52% D content incorporated at the olefinic position, respectively (Scheme 2a). The deuterium incorporation at the olefinic position proved the H-shift process shown in Scheme 2b. Then, we performed an 18O-labeling experiment with 18O-1a under the standard reaction conditions and obtained 18O-2a in 80% yield with 2% 18O incorporation, which afforded 3a with no 18O incorporation after heating. These results indicated that the oxygen atom of the carbonyl group in furanones 3 originated from CO2.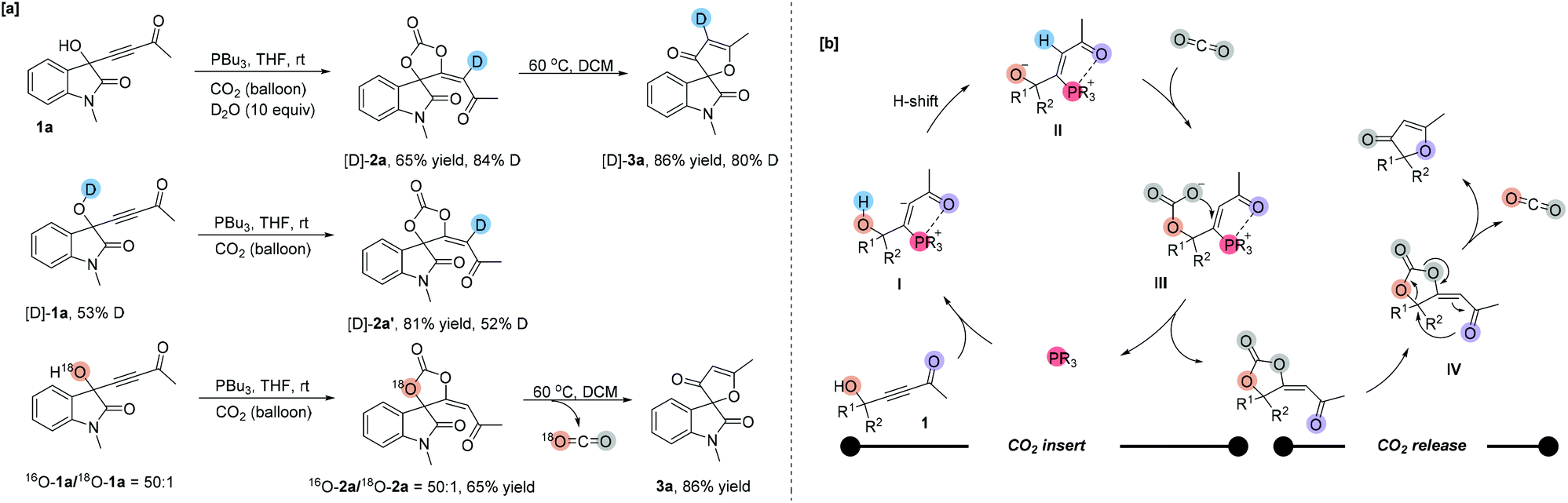 | ||
| Scheme 2 Isotope-labeling experiments and proposed mechanism. | ||
We proposed the following mechanism depicted in Scheme 2b to account for the reaction outcomes. The reaction starts with the addition of tertiary phosphine to the γ-hydroxyl alkynone 1 to afford a zwitterionic intermediate I. Then the carbanion abstracts the proton from the hydroxyl group to form another zwitterionic intermediate II. The oxygen anion moiety of intermediate II directly attacks carbon dioxide, generating an intermediate III, which undergoes a cyclization to furnish product 2a and the phosphine catalyst. Upon heating, the C–O bond of carbonate is cleaved and the oxygen atom of the carbonyl group attacks the quaternary carbon center simultaneously. With the release of CO2, the furanone 3 is generated.
In order to understand the detailed reaction mechanism, we performed DFT calculations on the suggested reaction pathways. We first investigated the proposed reaction pathway for the PBu3-catalyzed reaction of 1a and CO2 to produce 2a, and the solvation Gibbs free energy profiles in THF are shown in Scheme 3a. As shown in Scheme 3a, the addition of catalyst PBu3 to 1a leads to the formation of zwitterionic intermediate INT1, which is exergonic by 0.9 kcal mol−1. For comparison, we calculated the relative stability of INT1′ without any EWG group, which is highly endergonic and indicates that the intermediate INT1′ is unstable at room temperature. These calculation results indicated that the substrate having an EWG group plays a key role in stabilizing the important zwitterionic intermediates and facilitates the following CO2 fixation process. The proton from the hydroxyl group is transferred to the carbanion moiety of intermediate INT1 to generate an intermediate INT2viaTS1, which is located 6.2 kcal mol−1 above the intermediate INT1; converting INT1 to INT2 is exergonic by 1.7 kcal mol−1, indicating that this step is thermodynamically favorable. This result agrees with the finding in deuterium-labeling experiments in which the deuterium incorporation can be found at the olefinic position of product 2a (see Scheme 2a). Subsequently, INT2 is associated with CO2 to produce a complex INT3. On passing through transition state TS2 with an activation free energy of 9.9 kcal mol−1, INT3 undergoes addition to furnish an intermediate INT4, which is endergonic by 8.4 kcal mol−1. INT4 undergoes cyclization viaTS3 with an energy barrier of 10.1 kcal mol−1 to afford a product complex INT5; the energy barrier of the cyclization step is not very high, probably due to the fact that the carbanion moiety in INT4 is the highly electrophilic center. The cleavage of complex INT5 gives product 2a and PBu3. The calculation results revealed that the phosphine catalyst acts as a nucleophilic organocatalyst and the CO2 fixation is a stepwise cyclization process. All energy barriers along the proposed reaction pathway are not larger than 16.9 kcal mol−1, which are in line with the experiment performed at room temperature. The proposed reaction pathway for the reaction of 2a to afford 3a was also investigated, and the solvation Gibbs free energy profiles in DCM are shown in Scheme 3b. The carbonyl group of 2a attacks the quaternary carbon center and the C–O bond of the carbonate moiety undergoes concerted cleavage via transition state TS4 to afford the product complex INT6. Transition state TS4 is located 22.0 kcal mol−1 above 2a. The structure of transition state TS4 clearly demonstrates that the carbon–oxygen bond is extended to 2.07 Å, and the distance is about 1.99 Å between the quaternary carbon center and oxygen atom, indicating that the carbonyl group attacking the quaternary carbon center and the cleavage of the carbonate moiety is a concerted process. Finally, cleavage of the complex INT6 to yield the separate components 3a and CO2 is exothermic by 15.3 kcal mol−1. This transition state TS4 may account for a partial racemization process in the reaction of chiral cyclic carbonates 2; it is hard to control the stereoselectivity in the process of carbonyl group attacking due to the opening of the oxygen heterocycle.
 | ||
| Scheme 3 Mechanistic studies on the proposed reaction pathways. | ||
In order to understand the origin of enantioselectivity, we investigated the relative energies of key transition states (R)-TS3 and (S)-TS3 using (R)-1a and (S)-1a as substrates, respectively (Scheme 3c). The energy of transition states (S)-TS3 involving (S)-1a is lower than that of (R)-TS3 and of the kinetic resolution of cycloaddition of CO2 involving (R)-1a by 1.3 kcal mol−1, indicating that (S)-1a reacts faster than (R)-1a. The calculation results agree with the stereochemical findings in experiments. Sterically hindered benzene ring substituents on the phosphine atom probably provide more powerful stereocontrol. In transition state (R)-TS3, the steric hindrance between the benzene moiety of (R)-1a and the tertiary phosphine moiety in chiral phosphine probably leads to its higher energy. On the other hand, in transition state (S)-TS3, the benzene moiety of (S)-1a was far away from the tertiary phosphine moiety in chiral phosphine and this transition state was more favored to give the corresponding product (R)-2.
Conclusions
In conclusion, we have first disclosed the phosphine-catalyzed CO2 fixation of γ-hydroxyl alkynones, affording a series of spirocarbonates 2 in moderate to good yields. The CO2 fixation process could perform at room temperature under ambient pressure and the very broad substrate generality brought simple and quick synthesis of a variety of skeleton spirocarbonates. Moreover, the spirocarbonates 2 could easily release CO2 under heating conditions and obtain diverse furanones 3 in excellent yields. In addition, we have developed a new series of sterically hindered and highly nucleophilic bifunctional amino acid-derived phosphine catalysts and realized the first example of chiral phosphine catalyzed kinetic resolution of propargyl alcohols via carbon dioxide fixation, which has the s-factor up to 38.6. The detailed mechanistic studies were conducted by isotope-labeling experiments and DFT calculations. Further applications of the phosphine-catalyzed CO2 fixation reactions are underway in our lab.Computational methods
All DFT calculations were performed with the Gaussian 09 program. The geometries of all minima and transition states have been optimized at the M06/6-31+G(d) level of theory.35 The subsequent frequency calculations on the stationary points were carried out at the same level of theory to ascertain the nature of the stationary points as minima or first-order saddle points on the respective potential energy surfaces. All transition states were characterized by one and only one imaginary frequency pertaining to the desired reaction coordinate. The intrinsic reaction coordinate (IRC) calculations were carried out at the same level of theory to further authenticate the transition states. The conformational space of flexible systems has first been searched manually. Thermochemical corrections at 298.15 K have been calculated for all minima from unscaled vibrational frequencies obtained at this same level. The solvent effect was estimated by the IEFPCM method with radii and nonelectrostatic terms for the SMD solvation model36 in THF (ε = 7.4257) or DCM (ε = 8.93). Solution-phase single point energy calculations were performed at the M06/6-311+G(d,p) level based on the gas phase optimized structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Basic Research Program of China [(973)-2015CB856603], the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000) and sioczz201808, and the National Natural Science Foundation of China (No. 20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008, 21772037, 21772226 and 21861132014), and the Fundamental Research Funds for the Central Universities 222201717003. We are grateful for the facility support from the Shanghai Supercomputer Center.References
- (a) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS; (b) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC; (c) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC; (d) F. Frédéric-Georges, C. Marc-André and L. Marc-André, Chem. – Eur. J., 2014, 20, 2990–2996 CrossRef; (e) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC; (f) F. Tan and G. Yin, Chin. J. Chem., 2018, 36, 545–554 CrossRef CAS; (g) C. Yu, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644–659 CrossRef; (h) P. H. Dixneuf, C. Bruneau and J. Fournier, in Enzymatic and Model Carboxylation and Reduction Reactions for Carbon Dioxide Utilization, ed. M. Aresta and J. V. Schloss, Springer Netherlands, Dordrecht, 1990, pp. 65–77 Search PubMed; (i) I. Dimitriou, P. Garcia-Gutierrez, R. H. Elder, R. M. Cuellar-Franca, A. Azapagic and R. W. K. Allen, Energy Environ. Sci., 2015, 8, 1775–1789 RSC; (j) P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC; (k) Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 RSC; (l) N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC; (m) J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS; (n) A. Dibenedetto, A. Angelini and P. Stufano, J. Chem. Technol. Biotechnol., 2014, 89, 334–353 CrossRef CAS.
- (a) N. Kindermann, T. Jose and A. W. Kleij, Top. Curr. Chem., 2017, 375, 15 CrossRef; (b) H. Zhang, H.-B. Liu and J.-M. Yue, Chem. Rev., 2014, 114, 883–898 CrossRef CAS PubMed.
- (a) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS; (b) B. Carry, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2016, 55, 6257–6260 CrossRef CAS PubMed.
- (a) A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y.-J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6439–6442 CrossRef CAS PubMed; (b) X. Wei, D. Liu, Q. An and W. Zhang, Org. Lett., 2015, 17, 5768–5771 CrossRef CAS PubMed; (c) S. Sharma, S. H. Han, Y. Oh, N. K. Mishra, S. Han, J. H. Kwak, S.-Y. Lee, Y. H. Jung and I. S. Kim, J. Org. Chem., 2016, 81, 2243–2251 CrossRef CAS PubMed; (d) W. Guo, J. E. Gomez, A. Cristofol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS PubMed.
- (a) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed; (b) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC; (c) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- (a) X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS; (b) R. L. Paddock and S. T. Nguyen, Chem. Commun., 2004, 1622–1623 RSC; (c) A. Berkessel and M. Brandenburg, Org. Lett., 2006, 8, 4401–4404 CrossRef CAS PubMed; (d) T. Chang, L. Jin and H. Jing, ChemCatChem, 2009, 1, 379–383 CrossRef CAS; (e) M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS; (f) S. Liu, N. Suematsu, K. Maruoka and S. Shirakawa, Green Chem., 2016, 18, 4611–4615 RSC.
- (a) M. Takimoto and M. Mori, J. Am. Chem. Soc., 2002, 124, 10008–10009 CrossRef CAS; (b) M. Takimoto, Y. Nakamura, K. Kimura and M. Mori, J. Am. Chem. Soc., 2004, 126, 5956–5957 CrossRef CAS PubMed.
- M. Yoshida, M. Fujita, T. Ishii and M. Ihara, J. Am. Chem. Soc., 2003, 125, 4874–4881 CrossRef CAS PubMed.
- (a) M. Ishii, F. Mori and K. Tanaka, Chem. – Eur. J., 2014, 20, 2169–2174 CrossRef CAS PubMed; (b) S. Kawashima, K. Aikawa and K. Mikami, Eur. J. Org. Chem., 2016, 3166–3170 CrossRef CAS.
- (a) M. Zhang, X. Zhao and S. Zheng, Chem. Commun., 2014, 50, 4455–4458 RSC; (b) S.-C. Zheng, M. Zhang and X.-M. Zhao, Chem. – Eur. J., 2014, 20, 7216–7221 CrossRef CAS PubMed.
- Y.-Y. Gui, N. Hu, X.-W. Chen, L. L. Liao, T. Ju, J.-H. Ye, Z. Zhang, J. Li and D.-G. Yu, J. Am. Chem. Soc., 2017, 139, 17011–17014 CrossRef CAS PubMed.
- S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2010, 132, 4072–4073 CrossRef CAS PubMed.
- (a) C. Bruneau and P. H. Dixneuf, J. Mol. Catal., 1992, 74, 97–107 CrossRef CAS; (b) I. Yoshio, I. Jiro, T. Masaaki and H. Harukichi, Bull. Chem. Soc. Jpn., 1987, 60, 1204–1206 CrossRef; (c) H.-S. Kim, J.-W. Kim, S.-C. Kwon, S.-C. Shim and T.-J. Kim, J. Organomet. Chem., 1997, 545–546, 337–344 CrossRef CAS; (d) Y. Gu, F. Shi and Y. Deng, J. Org. Chem., 2004, 69, 391–394 CrossRef CAS PubMed; (e) H.-F. Jiang, A.-Z. Wang, H.-L. Liu and C.-R. Qi, Eur. J. Org. Chem., 2008, 2309–2312 CrossRef CAS; (f) O. Lu, X. Tang, H. He, C. Qi, W. Xiong, Y. Ren and H.-F. Jiang, Adv. Synth. Catal., 2015, 357, 2556–2565 CrossRef; (g) T.-J. Kim, K.-H. Kwon, S.-C. Kwon, J.-O. Baeg, S.-C. Shim and D.-H. Lee, J. Organomet. Chem., 1990, 389, 205–217 CrossRef CAS; (h) J. Hu, J. Ma, Q. Zhu, Q. Qian, H. Han, Q. Mei and B. Han, Green Chem., 2016, 18, 382–385 RSC.
- (a) H. Zhou, G.-X. Wang, W.-Z. Zhang and X.-B. Lu, ACS Catal., 2015, 5, 6773–6779 CrossRef CAS; (b) C. N. Della, G. Bartolo, R. Giuseppe, V. Lucia, Z. Tito and C. Mirco, Adv. Synth. Catal., 2011, 353, 133–146 CrossRef; (c) K. Uemura, T. Kawaguchi, H. Takayama, A. Nakamura and Y. Inoue, J. Mol. Catal. A: Chem., 1999, 139, 1–9 CrossRef CAS; (d) K. Toshihiro, K. Keigo and M. Noritaka, Angew. Chem., Int. Ed., 2012, 51, 6700–6703 CrossRef PubMed.
- (a) J. Fournier, C. Bruneau and P. H. Dixneuf, Tetrahedron Lett., 1989, 30, 3981–3982 CrossRef CAS; (b) J. M. Joumier, J. Fournier, C. Bruneau and P. H. Dixneuf, J. Chem. Soc., Perkin Trans. 1, 1991, 3271–3274 RSC; (c) Y. Kayaki, M. Yamamoto and T. Ikariya, J. Org. Chem., 2007, 72, 647–649 CrossRef CAS PubMed.
- (a) W. Yamada, Y. Sugawara, H. M. Cheng, T. Ikeno and T. Yamada, Eur. J. Org. Chem., 2007, 2604–2607 CrossRef CAS; (b) S. Kikuchi, S. Yoshida, Y. Sugawara, W. Yamada, H.-M. Cheng, K. Fukui, K. Sekine, I. Iwakura, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2011, 84, 698–717 CrossRef CAS; (c) S. Kikuchi, K. Sekine, T. Ishida and T. Yamada, Angew. Chem., Int. Ed., 2012, 51, 6989–6992 CrossRef CAS PubMed; (d) X. Tang, C. Qi, H. He, H.-F. Jiang, Y. Ren and G. Yuan, Adv. Synth. Catal., 2013, 355, 2019–2028 CrossRef CAS; (e) Q.-W. Song, B. Yu, X.-D. Li, R. Ma, Z.-F. Diao, R.-G. Li, W. Li and L.-N. He, Green Chem., 2014, 16, 1633–1638 RSC; (f) Z.-Z. Yang, Y. Zhao, H. Zhang, B. Yu, Z. Ma, G. Ji and Z. Liu, Chem. Commun., 2014, 50, 13910–13913 RSC.
- Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194–4197 CrossRef CAS PubMed.
- (a) Y.-B. Wang, Y.-M. Wang, W.-Z. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2013, 135, 11996–12003 CrossRef CAS PubMed; (b) Y.-B. Wang, D.-S. Sun, H. Zhou, W.-Z. Zhang and X.-B. Lu, Green Chem., 2014, 16, 2266–2272 RSC.
- (a) K. Ohe, H. Matsuda, T. Ishihara, S. Ogoshi, N. Chatani and S. Murai, J. Org. Chem., 1993, 58, 1173–1177 CrossRef CAS; (b) P. Toullec, A. Carbayo Martin, M. Gio-Batta, C. Bruneau and P. H. Dixneuf, Tetrahedron Lett., 2000, 41, 5527–5531 CrossRef CAS.
- K. Ohe, H. Matsuda, T. Morimoto, S. Ogoshi, N. Chatani and S. Murai, J. Am. Chem. Soc., 1994, 116, 4125–4126 CrossRef CAS.
- (a) P. Le Gendre, T. Braun, C. Bruneau and P. H. Dixneuf, J. Org. Chem., 1996, 61, 8453–8455 CrossRef CAS; (b) P. L. Gendre, P. Thominot, C. Bruneau and P. H. Dixneuf, J. Org. Chem., 1998, 63, 1806–1809 CrossRef; (c) Y. Hara, S. Onodera, T. Kochi and F. Kakiuchi, Org. Lett., 2015, 17, 4850–4853 CrossRef CAS PubMed.
- N. Ryo, Y. Tatsuya, O. Gen and K. Masanari, Angew. Chem., Int. Ed., 2017, 56, 208–211 CrossRef PubMed.
- (a) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140–1152 RSC; (b) A. Marinetti and A. Voituriez, Synlett, 2010, 174–194 CrossRef CAS; (c) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005–1018 CrossRef CAS PubMed; (d) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588–11619 RSC; (e) C. Gomez, J.-F. Betzer, A. Voituriez and A. Marinetti, ChemCatChem, 2013, 5, 1055–1065 CrossRef CAS; (f) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed; (g) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927–2940 RSC; (h) Y. Wei and M. Shi, Chem. – Asian J., 2014, 9, 2720–2734 CrossRef CAS PubMed; (i) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657–1677 RSC; (j) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369–1378 CrossRef CAS PubMed; (k) P. G. Isenegger, F. Bächle and A. Pfaltz, Chem. Eur. J., 2016, 22, 17595–17599 CrossRef CAS PubMed.
- (a) J. Chen, J. Li, J. Wang, H. Li, W. Wang and Y. Guo, Org. Lett., 2015, 17, 2214–2217 CrossRef CAS PubMed; (b) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621–1624 CrossRef CAS PubMed; (c) Y. Li, X. Su, W. Zhou, W. Li and J. Zhang, Chem. – Eur. J., 2015, 21, 4224–4228 CrossRef CAS PubMed; (d) X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874–6877 CrossRef CAS PubMed; (e) M. Yang, T. Wang, S. Cao and Z. He, Chem. Commun., 2014, 50, 13506–13509 RSC; (f) X.-N. Zhang, G.-Q. Chen, X.-Y. Tang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 10768–10773 CrossRef CAS PubMed.
- (a) V. R. Gandi and Y. Lu, Chem. Commun., 2015, 51, 16188–16190 RSC; (b) Y. Gu, P. Hu, C. Ni and X. Tong, J. Am. Chem. Soc., 2015, 137, 6400–6406 CrossRef CAS PubMed; (c) S. Y. Lee, Y. Fujiwara, A. Nishiguchi, M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 4587–4591 CrossRef CAS PubMed; (d) X. Han, W.-L. Chan, W. Yao, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2016, 55, 6492–6496 CrossRef CAS PubMed; (e) E. Li, H. Jin, P. Jia, X. Dong and Y. Huang, Angew. Chem., Int. Ed., 2016, 55, 11591–11594 CrossRef CAS PubMed; (f) H. Liu, Y. Liu, C. Yuan, G.-P. Wang, S.-F. Zhu, Y. Wu, B. Wang, Z. Sun, Y. Xiao, Q.-L. Zhou and H. Guo, Org. Lett., 2016, 18, 1302–1305 CrossRef CAS PubMed.
- (a) C. T. Mbofana and S. J. Miller, ACS Catal., 2014, 4, 3671–3674 CrossRef CAS; (b) D. T. Ziegler and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 12069–12072 CrossRef CAS PubMed.
- L. Zhang, H. Liu, G. Qiao, Z. Hou, Y. Liu, Y. Xiao and H. Guo, J. Am. Chem. Soc., 2015, 137, 4316–4319 CrossRef CAS PubMed.
- (a) Y. Chen, M. Chen and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6493–6497 CrossRef CAS PubMed; (b) R. Shen, J. Yang, S. Zhu, C. Chen and L. Wu, Adv. Synth. Catal., 2015, 357, 1259–1269 CrossRef CAS.
- K. Komatsuki, Y. Sadamitsu, K. Sekine, K. Saito and T. Yamada, Angew. Chem., Int. Ed., 2017, 56, 11594–11598 CrossRef CAS PubMed.
- (a) X. Han, F. Zhong, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 767–770 CrossRef CAS PubMed; (b) H. Ni, X. Tang, W. Zheng, W. Yao, N. Ullah and Y. Lu, Angew. Chem., Int. Ed., 2017, 56, 14222–14226 CrossRef CAS PubMed; (c) F. Zhong, X. Han, Y. Wang and Y. Lu, Chem. Sci., 2012, 3, 1231–1234 RSC.
- H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467–4470 CrossRef CAS PubMed.
- (a) X. Su, W. Zhou, Y. Li and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6874–6877 CrossRef CAS PubMed; (b) W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853–14857 CrossRef CAS PubMed; (c) W. Zhou, P. Chen, M. Tao, X. Su, Q. Zhao and J. Zhang, Chem. Commun., 2016, 52, 7612–7615 RSC.
- (a) H.-P. Deng and M. Shi, Eur. J. Org. Chem., 2012, 183–187 CrossRef CAS; (b) H.-P. Deng, Y. Wei and M. Shi, Eur. J. Org. Chem., 2011, 1956–1960 CrossRef CAS; (c) X.-N. Zhang, G.-Q. Chen, X. Dong, Y. Wei and M. Shi, Adv. Synth. Catal., 2013, 355, 3351–3357 CrossRef CAS.
- (a) E. Vedejs, O. Daugulis and S. T. Diver, J. Org. Chem., 1996, 61, 430–431 CrossRef CAS; (b) E. Vedejs and O. Daugulis, J. Am. Chem. Soc., 1999, 121, 5813–5814 CrossRef CAS; (c) E. Vedejs and O. Daugulis, J. Am. Chem. Soc., 2003, 125, 4166–4173 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) W. J. Hehre, R. Ditchfield and J. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1524876, 1519502 and 1856499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9QO00642G |
| This journal is © the Partner Organisations 2019 |