
Aggregation induced emission enhancement by plasmon coupling of noble metal nanoparticles†
Jiangling
He
ab,
Shuang
Li
c,
Da
Lyu
ab,
Dingfeng
Zhang
c,
Xiao
Wu
c and
Qing-Hua
Xu
 *ab
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: chmxqh@nus.edu.sg
bNational University of Singapore (Suzhou) Research institute, Suzhou 215123, P. R. China
cInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P. R. China
First published on 9th September 2019
Abstract
Materials that display aggregation induced emission (AIE) can offer large contrast ratio for highly sensitive fluorescence based applications such as sensing and imaging. Conventional AIE chromophores are generally based on improved emission quantum yield due to reduced nonradiative decay rates by restriction of intramolecular motion. In this work we present another type of AIE phenomenon with a totally different working mechanism. It is based on aggregation induced plasmon coupling of metal nanoparticles (NPs) to simultaneously enhance the excitation efficiency and radiative decay rate of chromophores. The unique surface plasmon resonance (SPR) properties of noble metal NPs can give rise to a significantly enhanced local electric field to modulate the optical properties of nearby chromophores. Plasmon coupling interactions between adjacent metal NPs can cause giant enhancement in the local electric field and consequently even stronger enhancing capability. The working principle was demonstrated by using rhodamine B isothiocyanate (RiTC) as the model chromophore and cysteine as the coupling agent to induce aggregation of noble metal NPs. The fluorescence of RiTC was pre-quenched by attaching to the surface of Au and core–shell Au@Ag NPs. Upon addition of cysteine to induce the aggregation of RiTC conjugated Au@Ag NPs, the fluorescence of RiTC was significantly enhanced to a level much beyond that of fluorescence recovery. A series of core–shell Au@Ag NPs with different Ag shell thicknesses have been prepared to optimize the performance of aggregation induced plasmon coupling enhanced fluorescence. The optimum enhancement effect was achieved for Au@Ag NPs with a 5.6 nm Ag shell, which enhanced the fluorescence intensity of RiTC in the coupled nanostructures to be 44.8 times stronger than that of pre-quenched RiTC and 7.6 times that of free RiTC. Based on this method, the limit of detection of 3.4 pM was obtained for the detection of cysteine, which is highly selective and more sensitive than most previously reported methods. This quenched-enhanced (off–on) method is highly sensitive, simple, straightforward and universal, and can be easily extended for the detection of other analytes.
Introduction
Fluorescence methods have been widely used in chemical and biological sensing applications because of their high sensitivity, simplicity, and diversity.1,2 Integration of fluorescence based detection with microscopy technology has revolutionized the bio-imaging technology, allowing non-invasive visualization of biological events at the cellular and even molecular level.3,4 Detection sensitivity of fluorescence methods is determined by the contrast ratio of signal strength over the noise level. The sensitivity could be improved by increasing the signal strength and/or decreasing the noise level.5,6 A lot of effort has been devoted to developing chromophores with strong fluorescence or assays with dramatic change in fluorescence intensity in response to the analytes.Aggregation of chromophores or nanoparticles generally results in fluorescence quenching, which is known as aggregation-caused quenching and poses a big problem for biological applications. Aggregation-induced emission (AIE) breaks this common belief and has attracted vigorous attention since its discovery in 2001 by Tang et al.7 It provides a new concept for lots of potential applications.8–12 The mechanism of AIE generally arises from improved quantum yield (QY) due to reduction of nonradiative decay rates by restriction of intramolecular motion.13,14 Lots of chromophores have been developed to display the AIE phenomenon.13–17 AIE chromophores are attractive probes for fluorescence based applications as they offer large contrast ratio upon aggregate formation.
Here we present another type of AIE phenomenon but with a totally different working mechanism. It is based on aggregation induced plasmon coupling of metal nanoparticles (NPs) to enhance the fluorescence of chromophores by improving both the excitation efficiency and radiative decay rate. Noble metal NPs, such as gold (Au) and silver (Ag) NPs, have been known to display unique properties of localized surface plasmon resonance (SPR), arising from collective oscillation of conduction band electrons induced by light radiation.18–20 Plasmon resonance will result in a significantly enhanced local electric field at the surface of metal NPs, which will modulate optical properties of chromophores in close proximity. Metal–chromophore interactions will result in increased excitation efficiency, radiative decay and non-radiative rates of the chromophores, all of which depend on the separation distance between the metal and chromphores.6,21–24 Generally the fluorescence of chromophores in direct contact with metal NPs will be quenched. As the separation distance between metals and chromophores increases, a transition from fluorescence quenching to enhancement could be observed, with the optimum fluorescence enhancement at an intermediate separation distance of 5 to 30 nm, depending on the nature of the chromophore and metal NPs.6,22 Plasmon coupling interactions between closely spaced metal NPs will result in giant enhancement in the electric field in the gap region,6,25,26 which has been known to be responsible for significantly enhanced optical responses such as surface enhanced Raman scattering27,28 and two-photon photoluminescence.29–31 It has also been reported that coupled metal nanostructures displayed larger enhancement in fluorescence of chromophores compared to the mono-dispersed metal NPs.6,32 Ingenious design of assembled plasmon coupled nanostructures is of great importance to modify optical responses of nearby small molecules.
Different enhancing capability of aggregated and non-aggregated metal NPs could be utilized to develop schemes to achieve aggregation enhanced emission to display significantly different fluorescence intensities before and after formation of aggregated metal NPs. Lots of chemically and biologically important species can cause aggregation of metal NPs, which could be further utilized to develop various chemical and biological sensors and bio-imaging applications.
As one of the most important amino acids, cysteine is important for many physiological processes.30 A deficiency of cysteine would lead to many diseases. It is important to detect cysteine with high sensitivity. Cysteine molecules have been known to cause aggregation of metal NPs in an acidic environment.30,33 Taking advantage of this phenomenon, we demonstrate a scheme of aggregation induced plasmon coupling enhanced fluorescence as shown in Scheme 1. Rhodamine B isothiocyanate (RiTC) was chosen as the model fluorescent probe. Its fluorescence was pre-quenched by attaching it to the surface of Au and Au@Ag NPs. Upon addition of cysteine to induce the aggregation of RiTC modified Au and Au@Ag NPs, the fluorescence of RiTC was significantly enhanced due to significantly enhanced excitation efficiency and radiative decay rates resulting from stronger interactions with plasmon resonance of aggregated metal NPs. A series of core–shell Au@Ag NPs with different Ag shell thicknesses have been prepared to optimize the aggregation induced plasmon coupling enhanced fluorescence. The optimum result was achieved for Au@Ag NPs with a 5.6 nm Ag shell, which enhances the fluorescence intensity of RiTC in the coupled nanostructures to be 44.8 times stronger than that of pre-quenched RiTC and 7.6 times that of free RiTC. Based on this method, the detection limit of 3.4 pM was obtained for the detection of cysteine, which is highly selective and more sensitive than most previously reported methods. This method is simple, straightforward and universal, and can be easily extended for the detection of other analytes.
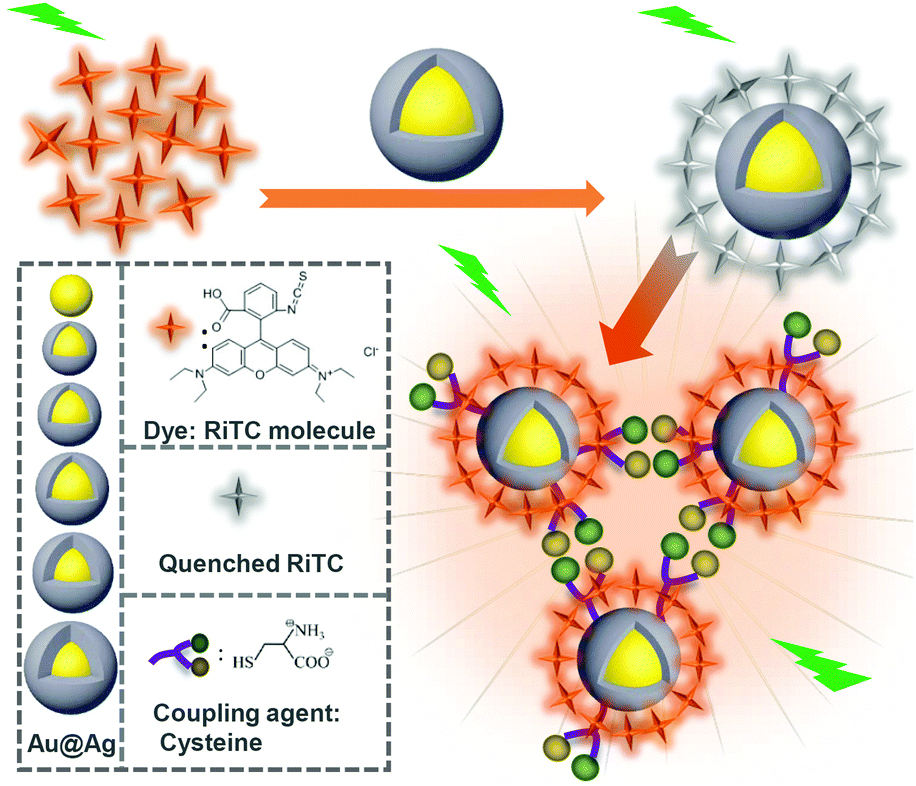 | ||
| Scheme 1 Illustration of aggregation induced plasmon coupling enhanced emission of pre-quenched fluorophores. | ||
Experimental section
Materials and characterization
Cetyltrimethylammonium chloride solution (CTAC, 25 wt% in H2O), silver nitrate (AgNO3), sodium borohydride (NaBH4), ascorbic acid, tri-sodium citrate dehydrate (99%), gold(III) chloride trihydrate (HAuCl4·3H2O), rhodamine B isothiocyanate (RiTC), DL-dithiothreitol (DTT), L-alanine (Ala), L-arginine (Arg), L-asparagine (Asn), L-aspartic acid (Asp), L-cysteine (Cys), L-glutamic acid (Glu), L-glutamine (Gln), L-glutathione (GSH), glycine (Gly), L-histidine (His), L-isoleucine (Ile), L-leucine (Leu), L-lysine (Lys), L-methionine (Met), L-phenylalanine (Phe), L-proline (Pro), L-serine (Ser), L- threonine (Thr), L-tryptophan (Trp), L-tyrosine (Try), L-valine (Val), and sodium dodecyl sulfate (SDS) were purchased from Sigma Aldrich. All chemicals were of analytical purity. All aqueous solutions were prepared in de-ionized (DI) water.Transmission electron microscopy (TEM) images were taken on a JEOL 2010 microscope. FT-IR spectra were recorded on a Bruker Vector 33 FT-IR spectrometer. Fluorescence and extinction spectra were measured on a Horiba Jobin-Yvon FluoroMax-4 Spectrophotometer and a Hitachi UH5300 UV-vis spectrophotometer, respectively. Fluorescence lifetime decay curves were measured by the time-correlated single photon counting (TCSPC) method using the Hydraharp 400 module under excitation at 520 nm. The second harmonic generation of the output of a Ti:sapphire oscillator (Spectra Physics, Mai Tai HP) was utilized as the excitation source.
Preparation of Au@Ag NPs with various shell thicknesses
19 nm Au NPs and Au@Ag NPs with different thicknesses of Ag shell were prepared by following a previously reported method with some slight modifications.34 10 mL of as-prepared 19 nm Au NP solution was purified by centrifugation, washed using aqueous CTAC solution, and then re-dispersed into 0.08 M CTAC aqueous solution with a total volume of 10 mL. Upon addition of different amounts (25, 50, 100, 150, and 200 μL) of 10 mM AgNO3 solution, 0.3 mL of 0.1 M ascorbic acid solution was introduced into the mixture. The resultant solutions were heated at 60 °C for 1 h. The solution changed the color from wine red to orange, indicating successful formation of core–shell Au@Ag NPs.Preparation of Au–RiTC and Au@Ag–RiTC NPs
SDS was first added to stabilize the obtained metal NPs. Upon addition of 100 μL of SDS solution (1.0 wt%), 1 mL of metal NP solution was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 rpm for 15 min to remove excess surfactant, re-dispersed in 1 mL of deionized water, and centrifuged again to remove supernatant. After adding 50 μL of freshly prepared RiTC solution (100 μM), the solution mixture was kept overnight under magnetic stirring. The obtained deep purple solution was centrifuged and washed with deionized water until the solution turned colorless, indicating no residue of RiTC in the solution. The obtained Au–RiTC and Au@Ag–RiTC NP suspensions were diluted to 1 mL using deionized water, and then stored in a refrigerator for further use.
200 rpm for 15 min to remove excess surfactant, re-dispersed in 1 mL of deionized water, and centrifuged again to remove supernatant. After adding 50 μL of freshly prepared RiTC solution (100 μM), the solution mixture was kept overnight under magnetic stirring. The obtained deep purple solution was centrifuged and washed with deionized water until the solution turned colorless, indicating no residue of RiTC in the solution. The obtained Au–RiTC and Au@Ag–RiTC NP suspensions were diluted to 1 mL using deionized water, and then stored in a refrigerator for further use.
Cysteine induced aggregation of Au–RiTC and Au@Ag–RiTC NPs in solution
Cysteine was utilized as the coupling agent to form aggregates of Au–RiTC and Au@Ag–RiTC NPs through its amine and thiol functional groups (Scheme 1). Different amounts (0.8, 1.2, 1.7, 2.2, 2.8, 4.0, 5.7, 8.3, 11.7, and 16.7 μM) of cysteine solution were added into the Au–RiTC and Au@Ag–RiTC NP solution. The pH of the solution was adjusted to ∼2.3 by using HCl to initiate the coupling process. The resultant solution was kept for more than 3 min to ensure complete reaction before spectral measurements.DTT treatment of metal–RiTC to release free RiTC
To release RiTC from the metal–RiTC NPs, 100 μL of DTT (0.5 M) was added into 1 mL of as-prepared metal–RiTC NP solution. The resultant mixture was incubated in an oven at 50 °C for 3 h. After centrifugation at 13200 rpm for 15 min, the resultant supernatant is a solution of free RiTC that is cleaved from the metal NPs.
Results and discussion
Au@Ag core–shell NPs with various shell thicknesses were prepared according to a previously reported method by using CTAC as the capping surfactant and ascorbic acid as the reducing agent. After coating with an Ag shell of increasing shell thickness, the obtained Au@Ag core–shell NPs solution changed color from wine-red to orange (Fig. 1a). The original SPR band of Au NPs at ∼520 nm gradually blue-shifted and a new SPR band appeared at ∼396 nm for Au@Ag NPs with thicker Ag shells, which is consistent with the previous report.35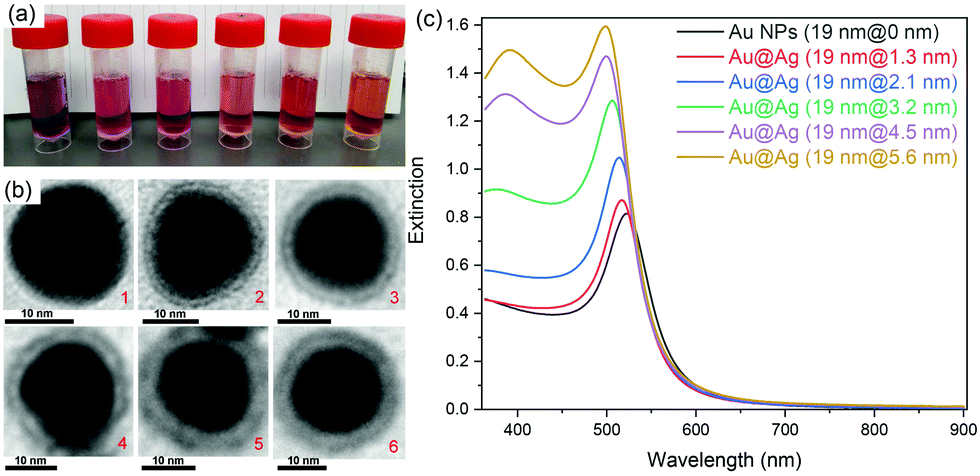 | ||
| Fig. 1 Photographs (a), typical TEM images (b), and the corresponding extinction spectra (c) of Au@Ag core–shell NPs with various silver shell thicknesses (0, 1.3, 2.1, 3.2, 4.5, and 5.6 nm). 0 nm refers to Au NPs. | ||
Rhodamine B isothiocyanate (RiTC) was chosen as the model fluorophore to demonstrate aggregation induced plasmon coupling enhanced fluorescence. The fluorescence emission spectrum of RiTC aqueous solution displayed a characteristic band peaking at ∼580 nm with quantum yield (QY) of 13% (Fig. S1, ESI†). Upon surface conjugation with RiTC, extinction spectra of Au NPs and Au@Ag NPs became slightly redshifted and broadened (Fig. S2, ESI†). On the other hand, the fluorescence of RiTC was quenched by a factor of 4.6 to 8.1-fold by Au NPs and Au@Ag NPs with different Ag shell thicknesses. The detailed quenching factors are summarized in Table S1 (ESI†), which were obtained by comparing the difference in fluorescence intensities of metal–RiTC NPs before and after DTT treatment to release free RiTC to the solution. The observed fluorescence quenching arises from non-radiative energy transfer from RiTC to metal NPs.6 Au NPs without the Ag shell displayed the largest quenching efficiency due to the best spectral overlap between its absorption and the fluorescence of RiTC.
Cysteine was chosen as the coupling agent to induce aggregation of metal NPs. Cysteine molecules can bind to the surface of Au or Ag NPs through their thiol groups and induce the aggregation of cysteine modified metal NPs through their amino and carboxyl groups in an acidic environment.36–38Fig. 2 shows the evolution of extinction spectra of metal–RiTC NP solutions during the aggregate formation process. Upon gradual addition of cysteine, the original SPR band from 380 to 520 nm gradually decreased while another broad band appeared in the longer wavelength region, suggesting formation of an anisotropic assembly of metal–RiTC NPs.39 Cysteine induced aggregation of metal NPs has also been confirmed by TEM images (Fig. S3, ESI†), which showed that monodispersed metal–RiTC NPs became clumped together after addition of cysteine. FT-IR spectra of the aggregated metal NPs (Fig. S4, ESI†) displayed a sharp peak at 1404 cm−1 arising from the symmetric COO stretching mode and a broad band around 3200 cm−1 arising from the stretching modes of OH and NH, which are absent from FTIR spectra of isolated metal NPs. These results further confirm the role of cysteine in the formation of aggregated metal NPs.
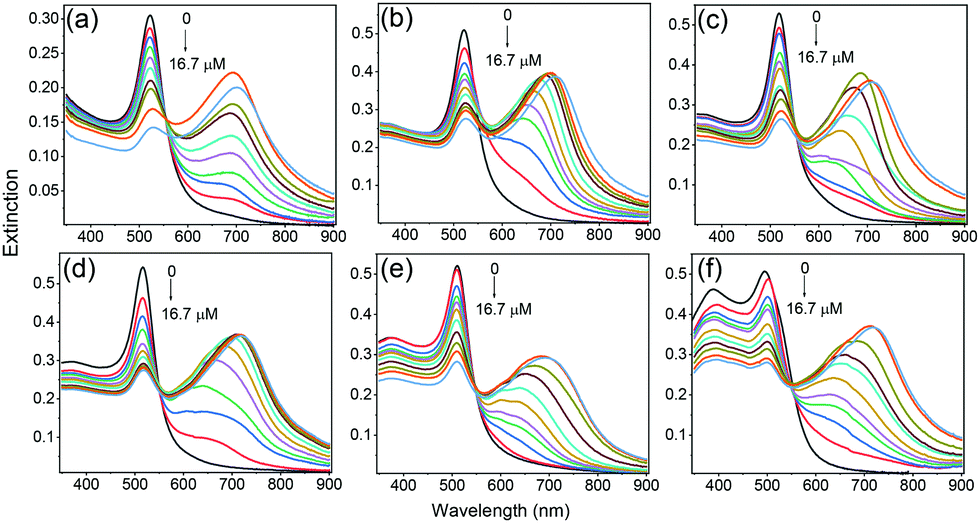 | ||
| Fig. 2 Extinction spectra of Au@Ag–RiTC NP solution with various Ag shell thicknesses upon gradual addition of cysteine: 0 nm (a), 1.3 nm (b), 2.1 nm (c), 3.2 nm (d), 4.5 nm (e), and 5.6 nm (f); 0 nm refers to Au NPs. | ||
The corresponding fluorescence spectra are shown in Fig. 3. Upon gradual addition of cysteine to induce aggregation of Au–RiTC and Au@Ag–RiTC NPs, the pre-quenched fluorescence of RiTC in Au–RiTC and Au@Ag–RiTC NPs steadily recovered and increased to a level even beyond that of free RiTC. The fluorescence intensity reached the optimum and then decreased slightly. The fluorescence spectra with optimum enhancement of coupled Au@RiTC and Au@Ag–RiTC NPs versus free RiTC molecules and isolated NPs without addition of cysteine are summarized in Fig. 4. The optimum fluorescence of coupled Au–RiTC NPs was 24.3 times that of uncoupled Au–RiTC NPs, and 3.0 times that of free RiTC molecules. For Au@Ag–RiTC NPs, these enhancement factors gradually increased with increasing Ag shell thickness until 5.6 nm (see enhancement factors for different Au@Ag NPs in Table S1, ESI†). Coupled Au@Ag–RiTC NPs with a 5.6 nm Ag shell display optimum fluorescence intensity and the largest fluorescence enhancement versus pre-quenched RiTC (44.8-fold) and free RiTC (7.6-fold).
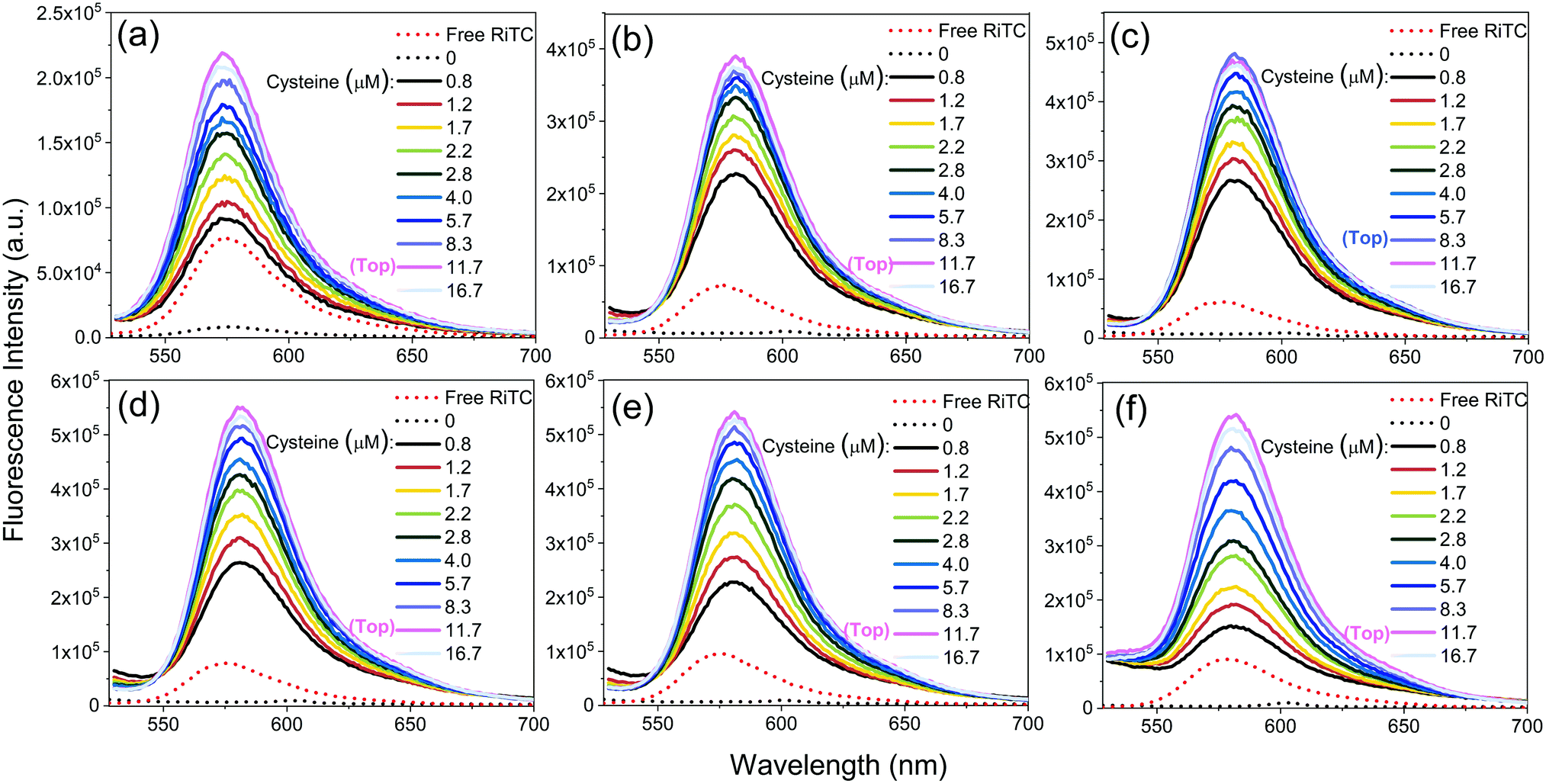 | ||
| Fig. 3 Fluorescence spectra of free RiTC and Au@Ag–RiTC NPs with different Ag shell thicknesses upon addition of different amounts of cysteine: 0 nm (a), 1.3 nm (b), 2.1 nm (c), 3.2 nm (d), 4.5 nm (e), and 5.6 nm (f); 0 nm refers to Au NPs. λEX = 520 nm. | ||
 | ||
| Fig. 4 (a)–(f) Fluorescence spectra of free RiTC (green), isolated metal–RiTC NPs (black) and optimum coupled metal–RiTC NPs (red) with different Ag shell thicknesses (0, 1.3, 2.1, 3.2, 4.5, and 5.6 nm). (g) The corresponding fluorescence intensity of optimum coupled Au@Ag–RiTC NPs (red bar), free RiTC (green bar) and isolated Au@Ag–RiTC NPs (black bar). (h) Enhancement factor versus free RiTC (Icoupled metal–RiTCNPs/IfreeRiTC, green bar) and enhancement factor versus pre-quenched RiTC (Icoupledmetal–RiTCNPs/IIsolatedmetal–RiTCNPs, black bar) for different Au@Ag NPs. | ||
The effects of pH and ionic strength on fluorescence enhancement have been checked by using Au@Ag NPs with the optimum performance (Ag shell thickness of 5.6 nm) (Fig. S5, ESI†). The results indicated that fluorescence enhancement arising from cysteine induced aggregation of metal NPs was only observed in the acidic environment with a low pH. The fluorescence enhancement remained nearly unaffected when the concentration of inert NaCl was varied from 0 to 4.0 M to change the ionic strength of the solution.
Significantly enhanced fluorescence upon aggregate formation can be ascribed to both enhanced excitation efficiency and QY. Fluorescence intensity of chromophores is determined by the product of excitation efficiency and emission QY, both of which will be modulated by the metal–chromophore interactions. When a chromophore is placed near metal NPs, the excitation efficiency will be enhanced as a result of local electric field amplification due to plasmon resonance. On the other hand, the emission QY will be modulated as both the radiative and non-radiative decay rates of the chromophores will be enhanced. Generally metal–chromophore interactions will result in reduced QY for high QY molecules, while improved QY for molecules with extremely low QY. The QY of RiTC in H2O (13%) is reasonably high. Its fluorescence became significantly quenched upon attaching to the surface of Au or Au@Ag NPs due to reduced QY arising from the dominant contribution from the increased non-radiative energy transfer process, despite the improved excitation efficiency. Upon formation of aggregates, the local electric field will be significantly further enhanced in the gap region compared to the un-aggregated metal NPs as confirmed by the simulation results shown in Fig. S6 (ESI†). The numerical simulation also suggests that local electric field increases with increasing size of the aggregates. As the QY of pre-quenched RiTC in Au–RiTC and Au@Ag–RiTC molecules is very low, metal–chromophore interaction is favourable for improving the QY. Metal aggregates with larger sizes have stronger weightage of scattering components over the absorption components compared to isolated metal NPs, which is favourable for improving the radiative decay rates.40 Consequently the QY is expected to be improved upon forming aggregates of metal NPs. On the other hand, further enhanced local electric field will result in further enhancement in the excitation efficiency. These two factors (enhanced excitation efficiency and QY) add up to result in significantly enhanced overall fluorescence intensities in these aggregated metal nanostructures. Au@Ag NPs with thicker Ag shell display larger enhancement because Ag NPs generally display stronger local electric effects compared to Au NPs. Numerical simulation confirmed that Au@Ag NPs display a stronger local electric field compared to Au NPs, which increases with increasing thickness of the Ag shell (Fig. S6a, ESI†).
The possibility of observed fluorescence enhancement in Fig. 3 and 4 due to detaching RiTC from the surface of metal NPs could be excluded by the fluorescence lifetime measurements (Fig. S7, ESI†). The fluorescence lifetime of free RiTC in solution was measured to be ∼2.2 ns. Upon attaching to the surface of metal NPs, the fluorescence lifetime of RiTC was reduced to within the instrument response function time of ∼150 ps and was not well resolved. The fluorescence lifetime decay of the aggregated metal–RiTC molecules remained within the instrument response, opposite to increased emission lifetime as expected for detached RiTC molecules. The observation of shortened emission lifetime and significantly enhanced fluorescence intensity manifests the plasmon resonance enhanced excitation efficiency and improved radiative decay rates. When RiTC was attached to the isolated metal Au or Au@Ag NPs, fluorescence quenching was observed. In contrast, fluorescence intensity was enhanced when RiTC was attached to the aggregated metal NPs. The difference between the two cases arises from significantly enhanced local electric fields in-between coupled metal NPs, which will significantly enhance the excitation efficiency and radiative decay rates of chromophores in these regions to overcome the negative contribution from the non-radiative energy transfer process.
Fig. 3 shows that the fluorescence intensity exhibited a sharp increase upon the addition of cysteine. This phenomenon could be utilized to develop a platform for sensitive detection of cysteine. Among different Au and Au@Ag NPs, Au@Ag–RiTC with a 5.6 nm Ag shell displayed optimum enhancement and is expected to give the best sensitivity for the detection of cysteine. As shown in Fig. 5a, the fluorescence intensity is linearly proportional to [cysteine] in the low concentration range. The limit of detection (LOD) was estimated to be 3.4 pM based on a signal-to-noise ratio of 3.0. The obtained LOD is more sensitive compared to the previously reported methods: 10 nM to 0.27 μM for the electrochemical method,41 5 μM for the 2PPL method,30 5 μM for the fluorescent-dye-based fluorometric method,42 and 10 μM for the colorimetric method.43 This method offers high sensitivity as it is based on turning on pre-quenched fluorescence with reduced background noise to a level significantly higher than the intensity of original fluorescence.
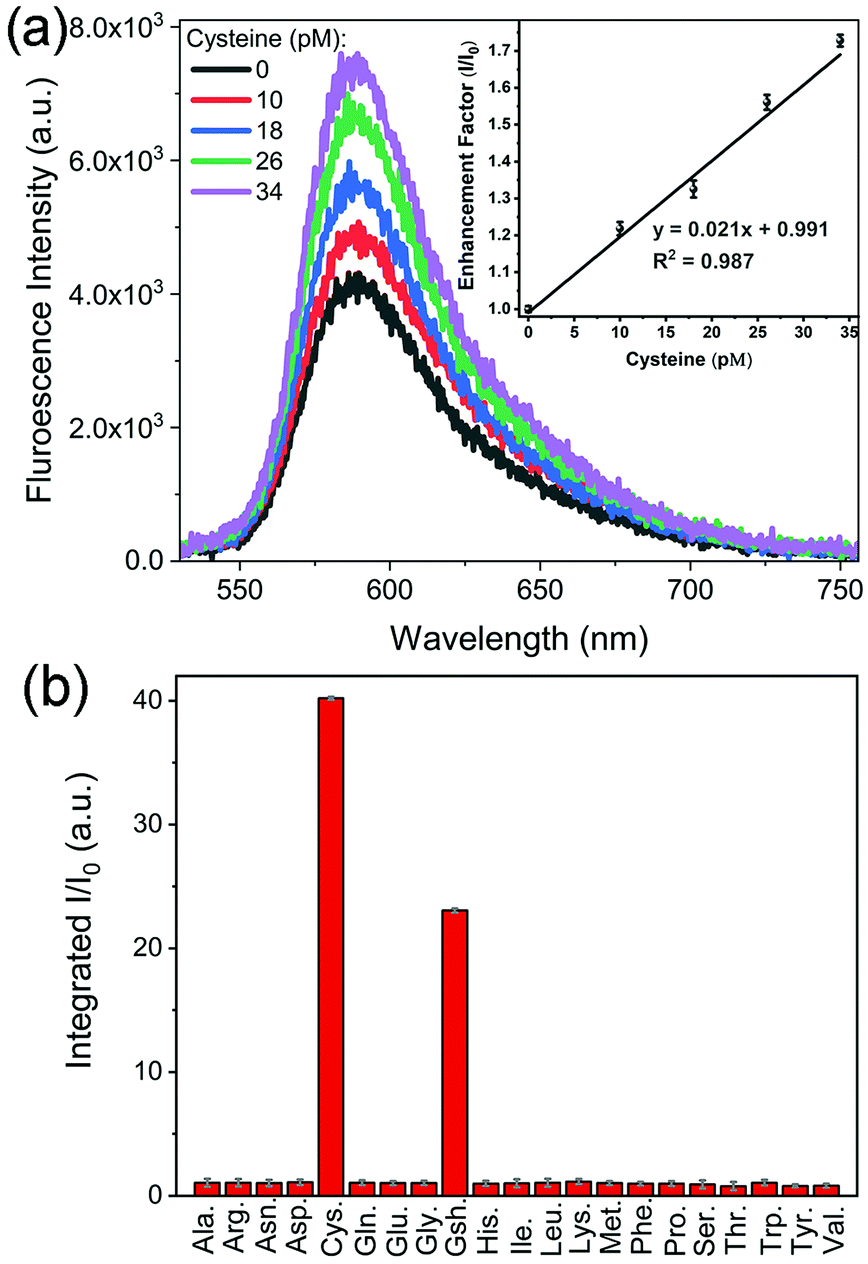 | ||
| Fig. 5 Fluorescence emission spectra of Au@Ag(5.6 nm)–RiTC NP solution upon addition of different concentrations of cysteine in the low concentration range. Inset is the corresponding enhancement factor I/I0versus [cysteine], where I0 and I represent the fluorescence intensity of Au@Ag(5.6 nm)–RiTC NPs without and with addition of cysteine. (b) Enhancement factor for coupled Au@Ag(5.6 nm)–RiTC NPs in the presence of various amino acids conducted in laboratory tap water. [amino acid] = 10 μM. | ||
Detection of cysteine has also been tested in laboratory tap water to mimic a real sample. Similar results were obtained with a LOD of 3.8 pM (Fig. S8a, ESI†), indicating its potential for detection in real samples. The selectivity of this method in discriminating 21 common amino acids has been further tested in laboratory tap water as shown in Fig. 5b and Fig. S8b (ESI†). Upon addition of different amino acids with a concentration of 10 μM, only cysteine and glutathione molecules displayed aggregation induced fluorescence enhancement, while no obvious fluorescence enhancement was observed for other amino acids. Cysteine displayed a slightly stronger fluorescence enhancement effect compared to glutathione. These results indicated that this aggregation induced Plamson coupling enhanced fluorescence method can effectively identify cysteine and glutathione from others. Other amino acids cannot induce aggregation enhanced fluorescence as they do not contain thiol functional groups.30 As this scheme is simply based on cysteine induced aggregation enhanced fluorescence, it is as simple and straightforward as the colorimetric method. This method could be easily extended to the detection of other analytes by changing the surface modification of the metal NPs to render analyte-induced aggregation of metal NPs.
Conclusions
In summary, a simple scheme that displays novel aggregation induced emission has been developed based on plasmon coupling enhanced pre-quenched fluorescence. In this scheme, fluorescence of a model chromophore, RiTC, was pre-quenched (turn off) by attaching it to the surface of metal NPs. Cysteine induced aggregation of metal NPs results in significantly enhanced local electric field, which allows enhanced excitation efficiency and stronger modulation of the radiative decay rates of RiTC. The combined effects result in significantly enhanced fluorescence intensity that is much beyond the level of fluorescence recovery. A series of core–shell Au@Ag NPs with Ag shell thickness varying from 0 to 5.6 nm were prepared to demonstrate the working principle and optimize the performance. The optimum enhancement was observed for Au@Ag NPs with Ag shell thickness of 5.6 nm, where the fluorescence intensity of aggregated Au@Ag–RiTC NPs is 44.8 times that of the pre-quenched RiTC (Au@Ag–RiTC) and 7.6 times that of free RiTC, respectively. This method could be utilized to develop a sensing scheme for the detection of cysteine, which gave excellent sensitivity with LOD of 3.4 pM. It is more sensitive than most previously reported methods. The method can effectively identify cysteine and glutathione from 19 other common amino acids. This aggregation induced plasmon coupling enhanced fluorescence method is simple and straightforward. It can be easily extended for the detection of other analytes by surface modification of metal NPs with proper analyte sensitive functional groups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work is supported by the Ministry of Education, Singapore (Tier 1 R-143-000-A41-114 and Tier 2 R-143-000-A68-114) and the National Natural Science Foundation of China (Grant No. 21673155).Notes and references
- K. L. Diehl and E. V. Anslyn, Chem. Soc. Rev., 2013, 42, 8596–8611 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- R. Brock, G. Vàmosi, G. Vereb and T. M. Jovin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10123–10128 CrossRef CAS.
- F.-M. Boldt, J. Heinze, M. Diez, J. Petersen and M. Börsch, Anal. Chem., 2004, 76, 3473–3481 CrossRef CAS.
- K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 2161–2195 CrossRef CAS.
- S. Li, T. Zhang, Z. Zhu, N. Gao and Q.-H. Xu, RSC Adv., 2016, 6, 58566–58572 RSC.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Dong, J. W. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- H. Tong, Y. Hong, Y. Dong, M. Häußler, J. W. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705–3707 RSC.
- H. Shi, J. Liu, J. Geng, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 9569–9572 CrossRef CAS PubMed.
- Y. Chen, J. W. Lam, R. T. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- C. Fang, Y. Xie, M. R. Johnston, Y. Ruan, B. Z. Tang, Q. Peng and Y. Tang, J. Phys. Chem. A, 2015, 119, 8049–8054 CrossRef CAS.
- F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 14492–14497 CrossRef CAS PubMed.
- Y. Ren, J. W. Lam, Y. Dong, B. Z. Tang and K. S. Wong, J. Phys. Chem. B, 2005, 109, 1135–1140 CrossRef CAS PubMed.
- W. Z. Yuan, P. Lu, S. Chen, J. W. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed.
- H. Wang, Y. Li, Y. Zhang, J. Mei and J. Su, Chem. Commun., 2019, 55, 1879–1882 RSC.
- W. Huang, M. Bender, K. Seehafer, I. Wacker, R. R. Schröder and U. H. Bunz, Macromol. Rapid Commun., 2019, 40, 1800774 CrossRef PubMed.
- T. R. Jensen, M. D. Malinsky, C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2000, 104, 10549–10556 CrossRef CAS.
- S. Zhang, K. Bao, N. J. Halas, H. Xu and P. Nordlander, Nano Lett., 2011, 11, 1657–1663 CrossRef CAS PubMed.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed.
- F. Tam, G. P. Goodrich, B. R. Johnson and N. J. Halas, Nano Lett., 2007, 7, 496–501 CrossRef CAS.
- D. Cheng and Q.-H. Xu, Chem. Commun., 2007, 248–250 RSC.
- L. Zhao, T. Ming, H. Chen, Y. Liang and J. Wang, Nanoscale, 2011, 3, 3849–3859 RSC.
- T. Ming, H. Chen, R. Jiang, Q. Li and J. Wang, J. Phys. Chem. Lett., 2012, 3, 191–202 CrossRef CAS.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS PubMed.
- S. Campione, S. M. Adams, R. Ragan and F. Capolino, Opt. Express, 2013, 21, 7957–7973 CrossRef PubMed.
- X. Qian, X. Zhou and S. Nie, J. Am. Chem. Soc., 2008, 130, 14934–14935 CrossRef PubMed.
- H. Gehan, L. Fillaud, M. M. Chehimi, J. Aubard, A. Hohenau, N. Felidj and C. Mangeney, ACS Nano, 2010, 4, 6491–6500 CrossRef CAS PubMed.
- Z. Guan, L. Polavarapu and Q. H. Xu, Langmuir, 2010, 26, 18020–18023 CrossRef CAS PubMed.
- Z. Guan, S. Li, P. B. Cheng, N. Zhou, N. Gao and Q. H. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 5711–5716 CrossRef CAS PubMed.
- Z. Guan, N. Gao, X. F. Jiang, P. Yuan, F. Han and Q. H. Xu, J. Am. Chem. Soc., 2013, 135, 7272–7277 CrossRef CAS PubMed.
- T. Zhang, N. Gao, S. Li, M. J. Lang and Q. H. Xu, J. Phys. Chem. Lett., 2015, 6, 2043–2049 CrossRef CAS.
- P. Sudeep, S. S. Joseph and K. G. Thomas, J. Am. Chem. Soc., 2005, 127, 6516–6517 CrossRef CAS PubMed.
- P. Yuan, R. Ma, N. Gao, M. Garai and Q.-H. Xu, Nanoscale, 2015, 7, 10233–10239 RSC.
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2004, 108, 5882–5888 CrossRef CAS.
- Z. Guan, N. Gao, X.-F. Jiang, P. Yuan, F. Han and Q.-H. Xu, J. Am. Chem. Soc., 2013, 135, 7272–7277 CrossRef CAS.
- S. Zhang, X. Kou, Z. Yang, Q. Shi, G. D. Stucky, L. Sun, J. Wang and C. Yan, Chem. Commun., 2007, 1816–1818 RSC.
- Z. Sun, W. Ni, Z. Yang, X. Kou, L. Li and J. Wang, Small, 2008, 4, 1287–1292 CrossRef CAS.
- N. J. Halas, S. Lal, W.-S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Plasmonics, 2007, 2, 107–118 CrossRef CAS.
- M. N. Abbas, A. A. Saeed, B. Singh, A. A. Radowan and E. Dempsey, Anal. Methods, 2015, 7, 2529–2536 RSC.
- M. Zhang, M. Yu, F. Li, M. Zhu, M. Li, Y. Gao, L. Li, Z. Liu, J. Zhang, D. Zhang, T. Yi and C. Huang, J. Am. Chem. Soc., 2007, 129, 10322–10323 CrossRef CAS PubMed.
- Z. Zhong, S. Patskovskyy, P. Bouvrette, J. H. Luong and A. Gedanken, J. Phys. Chem. B, 2004, 108, 4046–4052 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00455f |
| This journal is © the Partner Organisations 2019 |