
Stimuli responsive aggregation-induced emission of bis(4-((9H-fluoren-9-ylidene)methyl)phenyl)thiophene single crystals†
Maxim S.
Kazantsev
 *ab,
Alina A.
Sonina
ab,
Igor P.
Koskin
ab,
Peter S.
Sherin
bc,
Tatyana V.
Rybalova
ab,
Enrico
Benassi
*bd and
Evgeny A.
Mostovich
ab
*ab,
Alina A.
Sonina
ab,
Igor P.
Koskin
ab,
Peter S.
Sherin
bc,
Tatyana V.
Rybalova
ab,
Enrico
Benassi
*bd and
Evgeny A.
Mostovich
ab
aN.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry SB RAS, Lavrentieva 9, Novosibirsk, 630090, Russia. E-mail: maximkazantsev1988@gmail.com
bNovosibirsk State University, Pirogova 2, Novosibirsk, 630090, Russia
cInternational Tomography Center, Institutskaya 3A, Novosibirsk, 630090, Russia
dLanzhou Institute of Chemical Physics, Tianshui Middle Road, Lanzhou, 730000, China. E-mail: enrico.benassi@sns.it
First published on 22nd May 2019
Abstract
Aggregation induced emission (AIE) materials are in the spotlight of current materials development due to their unique properties and potential applications in light-emitting devices, bio-imaging and sensors. In this work we synthesised and thoroughly studied a novel luminogen based on bis(4-((9H-fluoren-9-ylidene)methyl)phenyl)thiophene (BFMPT) showing polymorph-dependent AIE. BFMPT crystallises in two polymorphs of different crystal shapes and colours, however both of them are highly-luminescent with the same photoluminescence (PL) quantum yield of 40%. Polymorph I can undergo irreversible phase transition to polymorph II. Deep quantum chemical study of BFMPT revealed that inter- and intramolecular interactions in both polymorphs are very similar and the main contributor to the optical difference between the polymorphs is a variation of BFMPT conformation. The thermal stress was demonstrated to negligibly affect the PL efficiency of BFMPT crystals. Due to high intrinsic torsional freedom and high solid-state PL efficiency, BFMPT could serve as a basis for the molecular design of high performance AIE materials.
Introduction
Highly emissive solid-state materials have attracted great interest due to various practical applications, such as organic light-emitting diodes1–3 and transistors,4–6 organic lasers,7,8 bio-imaging,9,10 and sensors.11–14 Despite high diversity of organic fluorophores they are usually highly emissive in solution, however they become low or non-emissive in the solid state due to aggregation-caused quenching.11,15 Nevertheless, a novel class of materials demonstrating the opposite effect, i.e. low emission in solution and high emission in the solid state, was developed.16,17 This phenomenon was named Aggregation Induced Emission (AIE) and has been extensively studied during the last few years.18–20 The main mechanism of this behaviour is deemed to be restriction of intramolecular rotations (RIR) in the solid phase to block the nonradiative decay channels, which are active in the monomeric state (in solution).18–20 Typically, AIE luminogens have a non-planar, propeller-like structure with rotor and stator building blocks having a sterical hindrance to allow intramolecular rotations. Some of the well-known representatives of these materials are: silole-,16 tetraphenylethylene-,19 and distyrylanthracene-20 derivatives and others.18 Mono-substituted dibenzofulvene (DBF) derivatives were also previously reported to be AIE-active luminogens21–23 and recently have attracted a keen interest as AIE materials.24–27 However, AIE luminogens based on two 9H-fluoren-9-ylidene units connected by a π-conjugated rotor have not yet been studied. This approach might not only allow AIE active species to be achieved but also a high conformational lability to be attained due to forbidden planarity and possible twist-ability of 4-(9H-fluoren-9-ylidene)methyls as bulky terminal groups. This might lead to polymorphism with a tuneable luminescence of the obtained materials. Moreover, such approach due to weak intra- and intermolecular interactions might lead to high sensitivity of the molecule to external stimuli, what could be exploited for optical recording or sensing applications.11–14,28The optoelectronic performance of organic highly-luminescent materials strongly depends not only on the chemical structure of the system, but also on crystal packing17 and molecular conformation;29 both were recently considered to be extremely important factors for material photophysics.17,30,31 Therefore, the study of polymorphic stimuli-responsive AIE-active materials especially with well-defined molecular and crystal structures is indispensable for the understanding of fundamental structure–property relationships to design novel high-performance luminescent materials.32–35
To obtain a polymorph-dependent photoluminescence (PL) various approaches were previously suggested:31 playing with N–H⋯N interactions and hydrogen bonding;36 conformational changes combined with π–π, C–H⋯N, C–H⋯F and C–H⋯π interactions;32,34,37,38 charge-assisted hydrogen bonding;39 O–H⋯O hydrogen bonding;40 π–π, C–H⋯O and C–H⋯π interactions;41 twisting, π–π and C–H⋯π interactions42,43 and even a new concept of Molecular Uniting Set Identified Characteristic (MUSIC) was proposed.31,44 Nevertheless, there is still a lack of accurate structure-packing–performance relationships and their systematic studies.30
In this work we have synthesised a novel AIE luminogen based on bis(4-((9H-fluoren-9-ylidene)methyl)phenyl)thiophene (BFMPT) and studied its crystallization, structure, photophysics (both in solution and solid state) and intrinsic properties by performing quantum chemical calculations. BFMPT was crystallized into two highly-emissive polymorphs with different PL colours and similar PL parameters. Form I represented needle-like crystals with orange emission (∼575 nm) whereas form II demonstrated plate-like crystals with pure green (∼555 nm) emission. Remarkably, form I was demonstrated to undergo irreversible transformation into form II upon thermal stress, which in turn has been shown to slightly affect the PL efficiency. Computational study of BFMPT revealed that weak positive solvatochromism is caused mainly by the change of quadrupole moment upon solvation. The difference in optical properties of BFMPT crystals was demonstrated to be mainly related to the change of BFMPT conformation, whereas intermolecular interactions are very similar in both forms.
Experimental
General
All chemicals and solvents were purchased from commercial sources (Sigma-Aldrich Co., USA and inner supply) and used without further purification. Elemental analysis was performed using a CHN-analyzer (EURO EA). Mass spectra were obtained using a Thermo Electron Corporation DFS mass spectrometer (70 eV) using direct injection, and the temperature of the ionisation chamber was 220–270 °C. IR spectra were recorded in transmission mode using a Varian 640-IR FT-IR spectrometer in potassium bromide pellets. Thermogravimetry and differential scanning calorimetry measurements were performed using a NETZSCH STA 449 instrument at a heating rate of 10 K min−1 in an Ar atmosphere. Cyclic voltammetry measurements were performed in CH2Cl2 solution using a computer controlled P-8 nano potentiostat/galvanostat (Elins, Russia) in combination with a three-electrode cell (Gamry), and 0.1 M tetrabutylammonium hexafluorophosphate was used as a supporting electrolyte. The Pt (3 mm), Pt wire and Ag/AgCl were used as working, counter and reference electrodes, respectively. The measurements were standardized by measuring the redox potential of ferrocene. HOMO and LUMO energy levels were estimated using onset oxidation and reduction potentials according to equations:| EHOMO = −(Eonsetox + 4.8) (eV) |
| ELUMO = −(Eonsetred + 4.8) (eV) |
UV-Vis absorption spectra of solutions were measured using an Agilent 8453 spectrophotometer. Photoluminescence (PL) spectra in solutions were recorded using an FLSP920 spectrofluorometer (Edinburgh Instruments, UK). All fluorescence spectra were corrected for wavelength-dependent sensitivity of detection. Liquid samples with the absorbance below 0.15 at the absorption maxima were placed in a 10 × 10 mm quartz cell and the solid samples were held on the surface of the glass plate. The photoluminescence quantum yields (PL QYs) of the samples in solution were determined relative to flavin mononucleotide (PL QY = 26%).45 The PL QY of single crystals was measured using a 3.3 inch-diameter integrating sphere (Newport 819C-SL-3.3) coupled with a UV-Vis spectrometer (Maya LSL, Ocean Optics); the detailed experimental procedure is described in ref. 46. The PL dynamics was recorded using a Time-Correlated Single-Photon Counting (TCSPC) unit of a FLSP920 spectrofluorometer. A pulsed diode laser (EPL-375, λ = 375 nm, and FWHM ≈ 100 ps) was used for the irradiation of both liquid and solid samples. The emission was collected at the magic angle relative to the polarisation of the diode laser. The time profiles were recorded over the whole fluorescence band for both liquid and crystalline samples. The obtained emission time profiles were globally analyzed using the numerical convolution of the instrument response function (IRF) with a sum of exponential functions. To build the Decay-Associated Spectra (DAS) the amplitudes of different components obtained by the global analysis at different wavelengths were rescaled with factor F(λ):
| F(λ) = S(λ)/∑ai(λ)τi |
At least three different crystals were used to check the reproducibility of the PL spectra and PL kinetics derived from solid samples. These tests have shown good coincidence of PL data for both types of polymorphs before and after heating.
Quantum chemical calculations were performed to provide a rationale to the experimental observations. Computational details are reported in the ESI† (Section 6).
Synthesis and material characterisation
Crystal growth and analysis
The crystals were grown by solvent–antisolvent crystallization. BFMPT powder was dissolved in toluene at a concentration of 0.15 g l−1. The solution was filtered through a 0.22 μm PTFE filter and placed into the closed vessel containing isopropanol for slow vapour diffusion. The growth time was 10–14 days. Single-crystal X-ray diffraction experiments were carried out using a Bruker KAPPA APEX II diffractometer equipped with a molybdenum X-ray tube (λ = 0.71073 Å). The integration and scaling of the intensity data were accomplished using the Bruker SAINT software.47 Absorption corrections were applied using SADABS.48 Crystal structures were solved by employing direct methods using SHELXS-97.49 Refinement was carried out using the full-matrix least-squares technique by employing SHELXL.50 All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined isotropically using the riding model. The crystal structures were analyzed for intermolecular interactions using PLATON.51 Crystal data and parameters of data collection and refinement are summarized in Table S1 (ESI†).The structural data for polymorphs have been deposited as CIFs at the Cambridge Crystallographic Data Base (CCDC 1881026 (form I) and 1881027 (form II)) which are also available in the ESI.† The X-ray powder diffraction patterns were recorded on a STOE STADI-MP diffractometer (CuKα1 radiation, curved Ge monochromator, transmission/Debye–Scherrer mode, and the MYTHEN 1K detector). For a high temperature powder X-ray diffraction experiment in an inert (N2) atmosphere form I was ground and placed in a glass capillary. The 2Θ range was from 3° to 35° and the heating rate was 5 °C min−1 from room temperature to 300 °C.
Results and discussion
Synthesis and characterisation
BFMPT was synthesised as shown in Scheme 1. The reaction between 9-H-fluorene 1 and 4-bromobenzaldehyde was performed under modified Knövenagel conditions. Addition of phase transfer catalyst – Bu4NI facilitated the condensation reaction yielding ylidene 2 with a high yield. At the final step, the Pd(PPh3)4-catalysed Stille cross-coupling reaction between bromide 2 and 2,5-bis(tri(n-butyl)stannyl)thiophene in DMF precipitated BFMPT as a light-yellow solid with greenish fluorescence. The obtained compound was characterized by IR and HR-MS spectroscopies and elemental analysis (Fig. S1 and S2, ESI†). Cyclic voltammetry measurements on BFMPT CH2Cl2 solution (Fig. S3, ESI†) revealed irreversible oxidation and quasi-reversible reduction processes. The HOMO and LUMO energy levels estimated using onset oxidation and reduction potentials are −5.50 eV and −2.82 eV, respectively.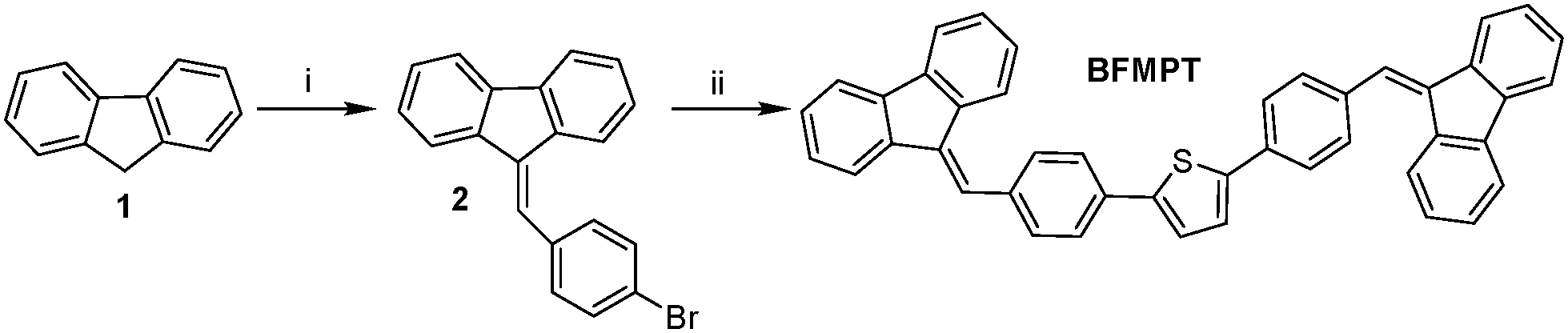 | ||
| Scheme 1 Synthesis of BFMPT. Reagents and conditions: (i) 4-bromobenzaldehyde, KOH, EtOH, n-Bu4NI, 2 h, reflux, 80%; (ii) 2,5-bis(tri(n-butyl)stannyl)thiophene, Pd(PPh3)4 (5 mol%), DMF, 80 °C, 6 h, 90%. | ||
Crystal growth and analysis
The crystals of BFMPT were grown by solvent–antisolvent crystallization resulting in two crystal forms – orange needles (form I, Fig. 1a) and yellow plates (form II, Fig. 1b). The optical images of the crystals in crossed polarisers are shown in the ESI,† Fig. S4a and c. In-plane rotation of both forms in crossed polarisers results in completely dark images demonstrating linear birefringence (Fig. S4b and d, ESI†). The crystals typically have a lateral size up to 1 mm. Under optical excitation (405 nm) both forms demonstrated bright photoluminescence: orange emission for the needles and green emission for the plates could clearly be observed (Fig. 1c and d). | ||
| Fig. 1 Optical images of BFMPT single crystals. In transmitted light: form I (a) and form II (b); under blue laser irradiation (405 nm): form I (c) and form II (d), (e) form I after heating at 280 °C for 5 seconds and (f) form II after heating at 280 °C for 5 seconds. | ||
To test the possible stimuli-response of the crystals we have performed an optical microscopy study after heating on a hot plate and subsequent cooling down to room temperature. Remarkably, form I was demonstrated to irreversibly change the PL colour from orange to green after heating at 280 °C, maintaining the crystal habit (Fig. 1e). Form II under the same conditions demonstrated no change of PL colour (Fig. 1f). To test whether the observed transformation represents a single-crystal-to-single-crystal transition, the needle-shaped crystals after the heating were analysed via crossed-polarised optical microscopy (Fig. S4e and f, ESI†). Some fading of the crystals in transmitted light was observed, which indicated partial maintenance of long range order. However, the resulting crystal demonstrated a lot of cracks which are clearly seen in the inset of Fig. 1e. As similar cracks were also observed for form II (Fig. 1f) we conclude that they are most likely originated from the thermal stress of BFMPT crystals rather than from the crystal structure transformation. The grinding of form I in a mortar was also found to turn the PL colour to green (Fig. S5, ESI†).
Thermal analysis on BFMPT powder containing the mixture of forms (Fig. S6, ESI†) demonstrated exothermal irreversible phase transition at temperatures 240–280 °C that should be attributed to the transition from form I to form II. The resulting form II melts at 326 °C; upon cooling the melt crystallizes at 230 °C into form II. The as synthesised powder was also demonstrated to be of form II. Therefore, form I is concluded to be a meta-stable form, irreversibly transforming to thermodynamically stable form II under heating at temperatures >240 °C or mechanical stress.
X-ray study
The structure of both forms was solved by X-ray analyses. The molecular structures of the polymorphs are principally different in torsional angles ϕ1(C9–C8–C17a–C18a) and ϕ2(C15–C14–C17b–C18b) being equal to −48.7° and 39.2° for form I and −71.9° and 56.4° for form II, respectively (Fig. 2a). Therefore, the conformation of the BFMPT backbone is closer to planar in form I. The torsional angles for form I are similar to those previously reported for mono-substituted dibenzofulvene derivatives,26 whereas the molecules in form II are considered to be highly twisted. Fig. 2 demonstrates the crystal packing and intermolecular interactions in BFMPT polymorphs. In form I C13–H⋯π, C23A–H⋯π and C26A–H⋯π interactions between fluorene cycles and phenyl rings form dimers that are bonded into chains along axis b by C16–H⋯π interactions (Fig. 2a and Table S2, ESI†). These chains are also bonded by interactions C20a–H⋯π(Th) and π(Cg2)⋯π(Cg3) to form layers parallel to the (a, b) plane. The layers, in turn, are connected by C28b–H⋯π interactions and π(Cg4b)⋯π(Cg4b) (Fig. 2c). Form II also demonstrated similar C13–H⋯π and C16–H⋯π interactions with the fluorene group (Fig. 2d) with partial reorientation of the central conjugated fragment (Fig. 2d). Similar interactions between thiophene and fluorenylidene (C20a–H⋯π and C20b–H⋯π), forming a chain along axis c (Fig. 2e), are also observed. Therefore, both forms demonstrate very similar intermolecular interactions, however the structure of form II is more ordered as compared with that of form I. We speculate that the conformational lability of the central thiophene–phenylene fragment allows rotation via external stimuli to attain more ordered and thermodynamically stable form II. Powder X-ray diffraction (XRD) analysis shows that, both after heating at 280 °C and grinding, XRD patterns of form I perfectly match those of form II (Fig. 3). This unambiguously confirms that form I upon heating and grinding transits to form II. Variable-temperature XRD for the powder of form I after grinding were also recorded indicating that form II formed as a result of grinding is stable up to 300 °C (Fig. S7, ESI†).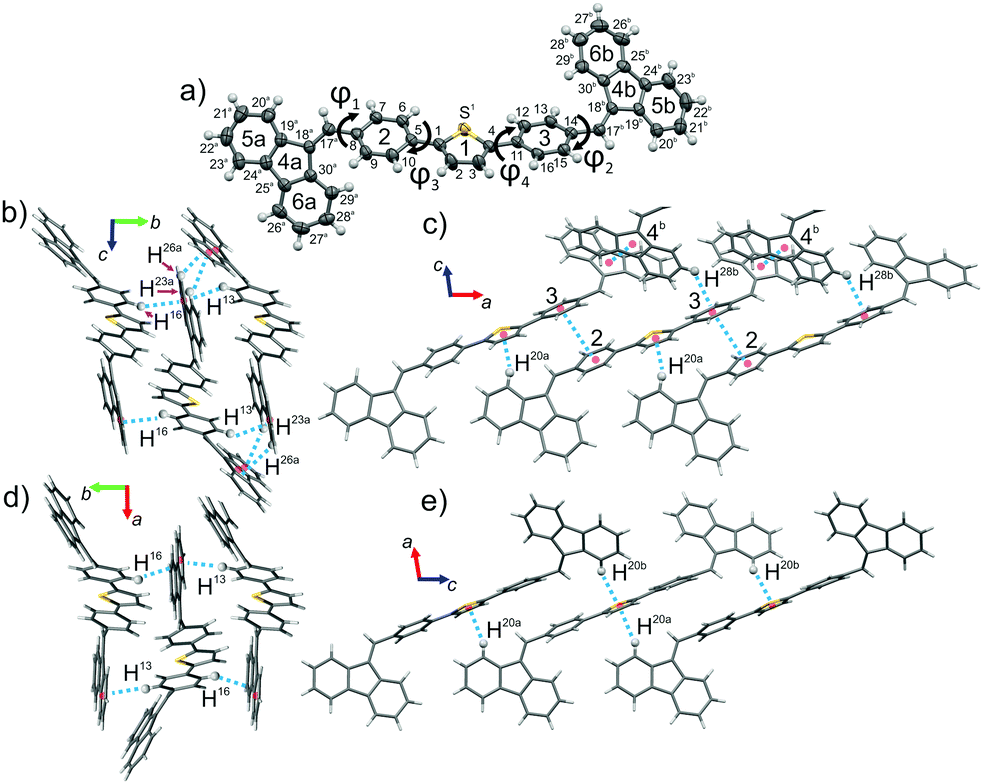 | ||
| Fig. 2 X-ray data: (a) molecular structure, atom and cycle numbering of BFMPT, (b) and (c) crystal structure and intermolecular interactions in form I, (d) and (e) crystal structure and intermolecular interactions in form II. Dashed blue lines represent noncovalent interactions. | ||
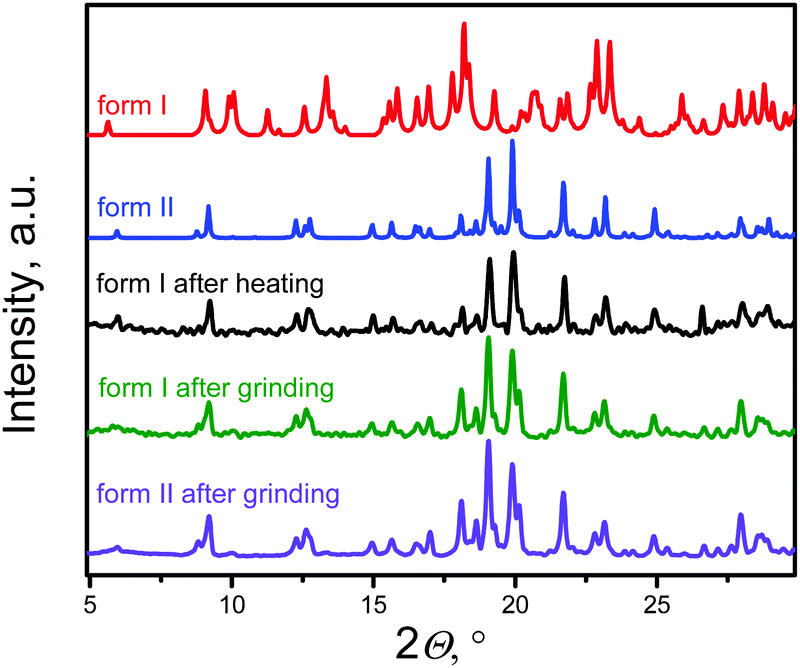 | ||
| Fig. 3 Room-temperature powder X-ray diffraction data. Patterns of: form I and form II (simulated from single crystal X-ray data, red and blue, respectively), form I after heating at 280 °C for 5 seconds (black) and grinding (olive), and form II after grinding (violet). | ||
Optical properties
The electronic absorption and emission spectra of BFMPT in four different solvents are shown in Fig. 4a. A change of solvent polarity leads to weak positive solvatochromism of both absorption and emission bands (Table 1): red shifts of absorption bands from 394 to 409 nm and fluorescence bands from 505 to 540 nm passing from cyclohexane to DMSO. This is assigned to the change of quadrupole moment upon solvation (vide infra). Noteworthily, BFMPT exhibits relatively large Stokes shifts (Table 1) in all solvents that indicates a substantial reorganization of molecular geometry in the excited state as compared with that of the ground state. This reorganisation includes large twists of fluorene moieties with respect to the central thiophene/phenylene fragment which was shown computationally (vide infra).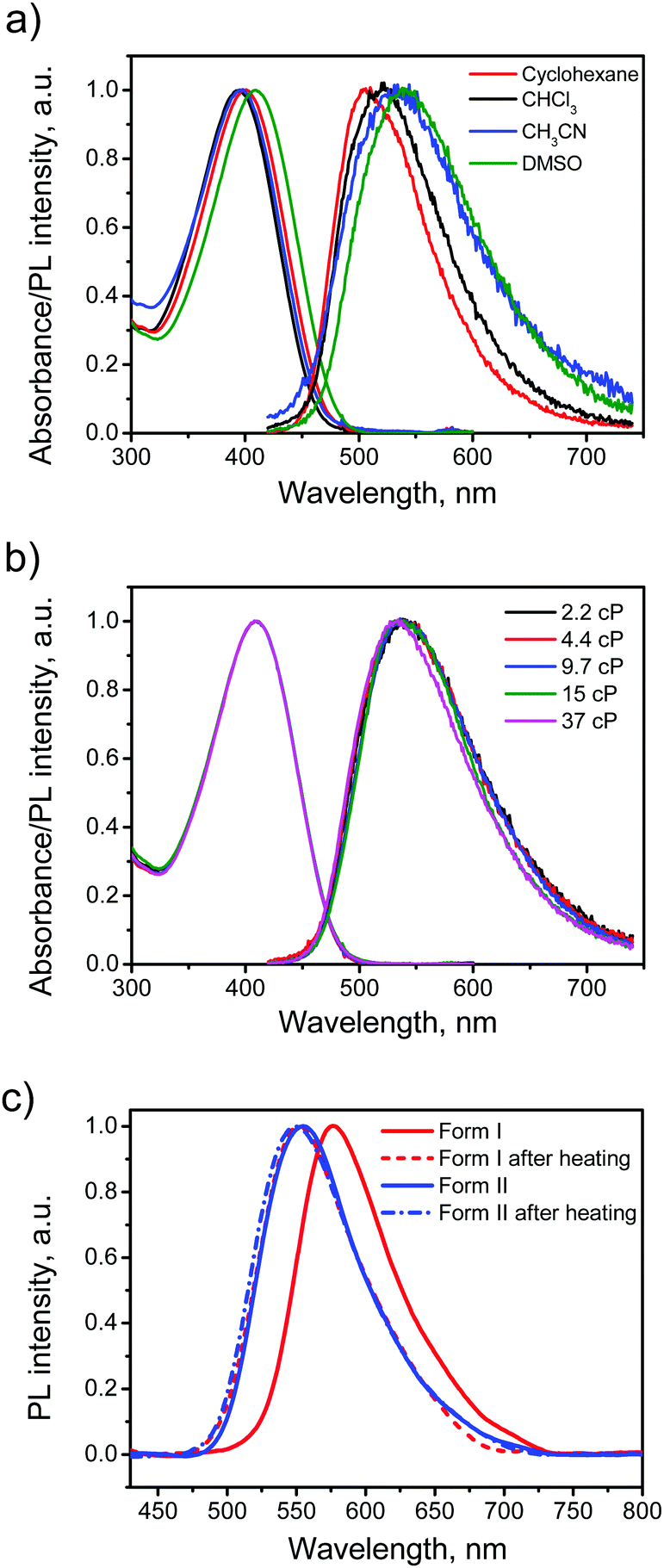 | ||
| Fig. 4 Steady-state optical data: absorption and PL spectra of BFMPT in different organic solvents (a) and DMSO:glycerol mixtures of different viscosity (2.2–37 cP) (b); PL spectra of BFMPT crystals measured by integrating sphere (c): form I before (red) and after heating at 280 °C (dashed red); form I before (blue) and after heating at 280 °C (dashed blue). | ||
| Solvent | λ abs (nm) | λ em (nm) | Stokes shift (eV) | PL QY (%) |
|---|---|---|---|---|
| a Absorption maximum wavelengths. b Emission maximum wavelengths; for the DMSO:Gly mixtures the viscosity is represented in parenthesis. | ||||
| Cyclohexane | 394 | 505 | 0.69 | 0.90 |
| CHCl3 | 396 | 520 | 0.75 | 0.51 |
| CH3CN | 400 | 532 | 0.76 | 0.19 |
| DMSO (2.2 cP) | 409 | 540 | 0.73 | 0.81 |
| DMSO:Gly (4.4 cP) | 409 | 540 | 0.73 | 1.0 |
| DMSO:Gly (9.7 cP) | 409 | 540 | 0.73 | 1.9 |
| DMSO:Gly (15 cP) | 409 | 536 | 0.72 | 2.3 |
| DMSO:Gly (33 cP) | 409 | 532 | 0.70 | 2.3 |
The PL kinetics of BFMPT in all used solvents demonstrated very fast decay (Fig. S8a, ESI†), which is below the time resolution of our setup (<50 ps). We expected that BFMPT would possess AIE and viscosity-dependent optical properties and measured a BFMPT PL quantum yield (QY) in different organic solvents and in binary glycerol/DMSO mixtures of different viscosity (Table 1). In organic solvents, BFMPT exhibits very low PL QY values, <1%, reaching the minimal value of 0.19% in CH3CN. Weak emission in solution can be attributed to a fast decay of the excited states due to intramolecular rotations of fluorene-9-ylidene and thiophene/phenylene groups (vide infra). In this respect, the increase of the PL QY value in DMSO should probably be attributed to significantly higher viscosity of this solvent (2.2 cP) as compared to that of CH3CN (0.3 cP). In order to check the influence of solvent viscosity on BFMPT optical properties, absorption and emission spectra were recorded at room temperature for BFMPT in a set of binary DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycerol (Gly) mixtures of different viscosities. Variation of mixture composition (DMSO:glycerol from 6:1 to 1:1 by mass) demonstrated no influence on the absorption band and results in a weak blue shift of the emission band (Fig. 4b). As expected, the PL QY linearly increased in the viscosity range of 2–10 cP due to slowing of intramolecular motions by the solvent media. The PL QY reaches a plateau of 2.3% at higher viscosities that could probably be explained by the S1 state decay via intermolecular hydrogen-bonding interactions,52,53 which became the major decay channel of S1 → S0 transition on the background of the restricted motions due to high viscosity.
:glycerol (Gly) mixtures of different viscosities. Variation of mixture composition (DMSO:glycerol from 6:1 to 1:1 by mass) demonstrated no influence on the absorption band and results in a weak blue shift of the emission band (Fig. 4b). As expected, the PL QY linearly increased in the viscosity range of 2–10 cP due to slowing of intramolecular motions by the solvent media. The PL QY reaches a plateau of 2.3% at higher viscosities that could probably be explained by the S1 state decay via intermolecular hydrogen-bonding interactions,52,53 which became the major decay channel of S1 → S0 transition on the background of the restricted motions due to high viscosity.
In order to reveal AIE properties of BFMPT we measured the spectra and PL QYs for single crystals by integrating sphere (Fig. 4c). Remarkably, both polymorphs demonstrated high PL QYs of ∼40%: form I has a PL maximum at 576 nm, whereas form II has a PL maximum at 554 nm. The PL spectra of the crystals after the thermal stress almost coincide with the PL spectrum of form II that also confirms the transition of form I to form II. Therefore, the solid state PL of BFMPT could irreversibly be changed from orange to pure green colour. To evaluate the optical changes upon phase transition more precisely, we also recorded the PL spectra of form I after stepwise heating at different temperatures (220–280 °C, Fig. S8b, ESI†). These data indicate a smooth shift of the PL maximum from form I (577 nm) to form II (552 nm) with an increase of annealing temperature. An important issue is whether the thermal stress and the phase transition affect the PL properties of the studied materials. Indeed, cracks and structural deformations might induce the exciton traps which should lower the PL efficiency.54 To address this issue we measured the PL QY of the crystals after the phase transformation for form I and as-grown crystals of form II after the heating under the same conditions (Fig. 4c). Both samples demonstrated the PL spectrum perfectly matching that of neat form II and almost the same PL QY of ∼40%. Therefore, we concluded that the phase transition and the thermal stress do not significantly affect the PL efficiency of BFMPT materials.
To further explore the PL properties of BFMPT polymorphs we recorded the PL kinetics over the whole emission bands for both polymorphs as well as for them after the heating. The representatives are shown in Fig. 5a and b and Fig. S9a and b†. The obtained fluorescence time profiles were well reproduced with four exponential functions numerically convoluted with the IRF. The time constants obtained from this global analysis are listed in Table 2.
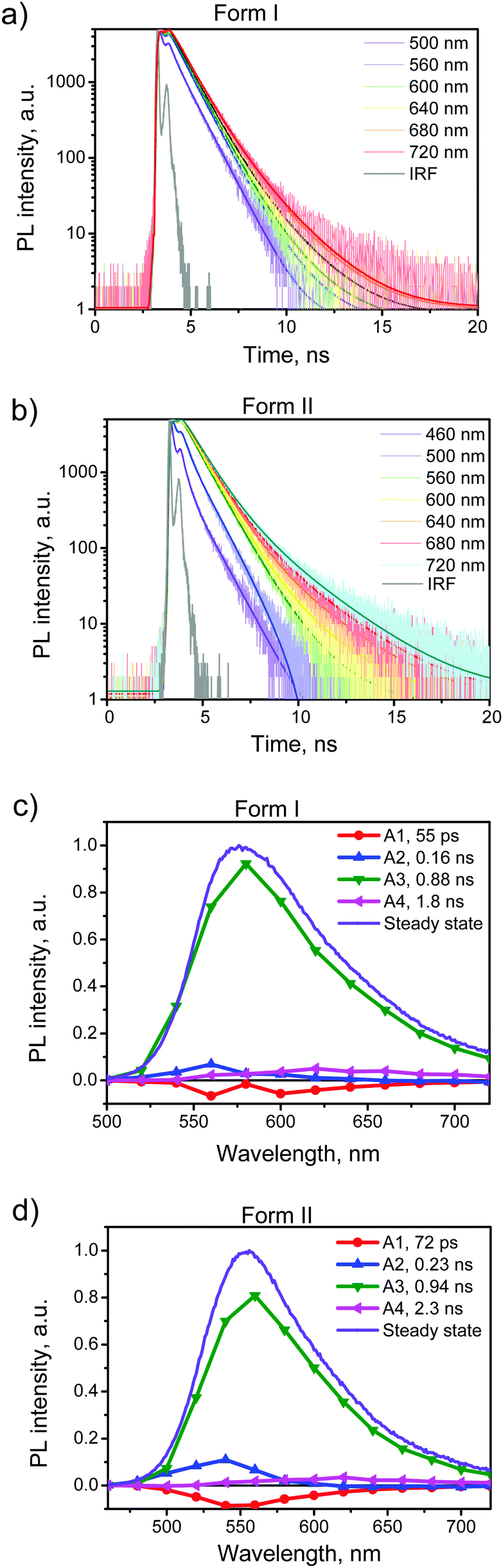 | ||
| Fig. 5 PL kinetics of BFMPT polymorphs: PL time profiles recorded using form I (a) and form II (b) over the whole emission band after the excitation at 375 nm; the solid lines represent 4-exponential fits convoluted with the instrument response function (IRF); decay associated spectra obtained from 4 exponential fits of fluorescence time profiles for form I (c) and form II (d) in comparison with steady-state spectra recorded using 375 nm laser excitation. | ||
To visualize the contributions of various components in the total PL we constructed Decay Associated Spectra (DAS) for both polymorphs (Fig. 5c and d). Positive amplitudes in DAS correspond to the decay of signals and negative to their rise. Spectra of the first two components have positive and negative bands indicating relaxation or exciton migration dynamics within a crystal. This dynamics is more pronounced for form II as compared with form I (Fig. 5a and b). The major component of decay (τ3) should be assigned to the PL decay of BFMPT in the solid state and it is nearly the same for both forms (∼0.9 ns). The latter could be explained by the similarity of the crystal structures and intermolecular interactions in both polymorphs (Fig. 2 and 7). The last component, due to low contribution in the observed dynamics, most likely should be attributed to impurities present in trace amounts in the crystals.
The PL kinetics of both types of crystals after the heating (Fig. S9, ESI†) demonstrated similar 4-exponential decay with the major τ3 time of ∼0.8 ns. This lowering of the PL lifetime of the heated crystals should indeed be attributed to the increase of the amount of defects in the crystals upon heating. Although the phase transformation processes and structural deformations of BFMPT single crystals slightly decrease the PL lifetime, these negative factors do not dramatically affect the PL QYs of the studied materials, which nevertheless demonstrated high emission efficiency even after the thermal stress (Table 2).
Quantum chemical calculations
In order to gain further insights into intrinsic properties of BFMPT we performed a quantum chemical study of its molecular structure, inter- and intramolecular interactions, excited states, solvatochromism and electronic energy transfer (EET). The molecular geometry in the gas phase was optimised starting from geometries adopted from form I and form II crystal structures. Optimisations resulted in identical structures (Table S3, ESI†) with characteristic dihedral angles: ϕ1, ϕ2 = ±42 deg, ϕ3, ϕ4 = ±22 deg (see Fig. 2 for angle designations). Molecular geometry optimised in three different solvents (Tables S4–S6, ESI†) showed a tendency to planarise the ϕ1 dihedral angle with the increase of solvent polarity: chloroform (42.5 deg), acetonitrile (38.3 deg), and dimethylsulfoxide (37.7 deg). According to Charges from ELectrostatic Potentials using a Grid-based (ChElPG)55 population analysis (Table S7, ESI†), dipole moment of the relaxed geometry increases with the increase of solvent polarity whereas quadrupole moment remains almost unchanged.The analysis of the potential energy surface (PES) profile as a function of ϕ1 dihedral angle (Fig. 6) showed the presence of three local maxima around −10 and ±85 degrees caused by strong sterical repulsion between H8(H13)/H29 atoms and conjugation breaking between fluorene and phenylene fragments, respectively. The energy minima are found at ∼±45 degrees; in correspondence of this, fluorene groups have a rather broad torsional freedom at the room temperature (∼±25 deg). This torsional freedom allows an adjustment of molecular geometry upon crystallization. Moreover, the values of dihedral angles in form I are almost coincident to the computed value corresponding to the minima, whereas those of form II are farther: in particular, the computed energy difference corresponding to the experimental value of ϕ2 is about 5 kJ mol−1. This difference in conformers’ torsional energy should be considered as one of the factors determining form I kinetic stability: in order to undergo transition from form I to form II BFMPT molecules must overcome a kinetic torsional barrier which is then thermodynamically compensated by the crystal field effect. The PES as a function of ϕ3 dihedral angle (Fig. S10, ESI†) showed the presence of three local maxima around 0, 180 and 90 degrees with 1, 14 kJ mol−1 barriers and ∼100 degrees torsional freedom which is in agreement with previously reported weak sterical repulsion between hydrogen and sulphur atoms in the thiophene/phenylene fragment.56
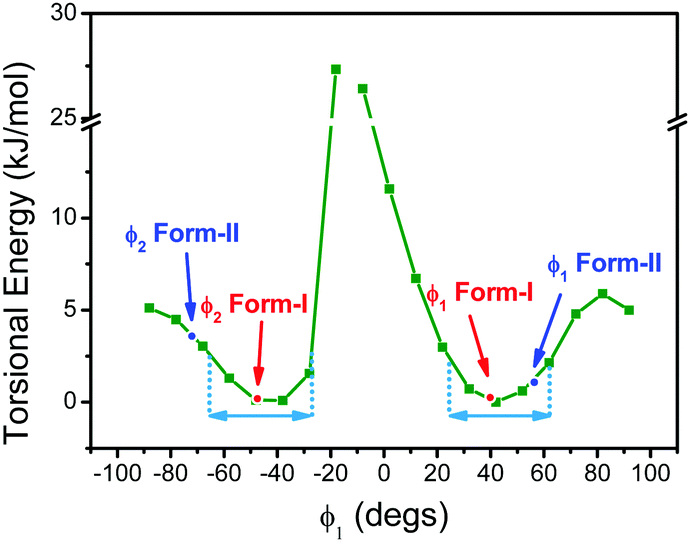 | ||
| Fig. 6 Potential energy surface of BFMPT as a function of ϕ1 dihedral angle. Red and blue dots and arrows show the values of ϕ1 and ϕ2 dihedral angles of form I and form II, respectively. Cyan arrows represent the value of torsional freedom at room temperature. | ||
In order to investigate the electronic excited states, the TD-DFT CAM-B3LYP/6-311++G** level of theory was shown to give the best match with the position of the brightest absorption band experimentally detected in the lowest polarity solvent (i.e. cyclohexane). Further simulation (Table S9, ESI†) of solvent effects of chloroform, dimethylsulphoxide, and acetonitrile solutions by means of IEFPCM (molecular geometry optimised at the B3LYP/6-311++G** level of theory with SMD solvent simulation) resulted in almost perfect correspondence with experimental data (∼0.01 eV margin of error).
Solvent effects on the relaxed PES of ϕ1 and ϕ3 dihedral angles (Fig. S11 and S12, ESI†) demonstrated a negligible effect on the positions of local maxima and minima, although torsional barriers slightly increase with the increase of solvent polarity. Upon rotation around the ϕ1 dihedral angle (Fig. S13, ESI†) S1v and T6v excited states became closer in energy (ΔE ∼ 0.03 eV) at about ±85 degrees local PES maxima. We are aware of the fact that TD-DFT does not represent the “golden standard” for this type of investigation, whereas other levels of theory (e.g. MR-CASSCF) would be more recommended to explore a region of the ES PES where the presence of singularity may be hypothesised. Nonetheless, due to the large dimension of the system under investigation, the most reliable methods would not be affordable. We should therefore be careful in the interpretation of the reported data as a mere indication of the proximity of these two states when the geometry becomes twisted. From the computed data, we also observe, that upon rotation around the ϕ3 dihedral angle (Fig. S14, ESI†) the S1v and T6v excited states tend to get close in energy when the dihedral angle tends to ±20 degrees. Therefore, these pathways might be one of the possible channels of luminescence quenching in solution via intersystem crossing, which is restrained in the solid state.
We attempted to rationalise the complex solvatochromic behaviour of BFMPT (Table S10, ESI†). The inductive and orientational contributions bound with the change of electric dipole and quadrupole moments were computed using the quantum mechanical values. The main contribution to the observed solvatochromism is the orientational one due to the change of the electric quadrupole moment (in the order of cents of eV), followed by the inductive one due to the change of the quadrupole (in the order of milli/cents of eV). The orientational contribution due to the change of electric dipole moment is comparable with the latter, whereas the inductive contribution due to the change of electric dipole moment is negligible.
The optimised geometry of the S1r state (Table S11, ESI†) was found to be more planar as compared to the ground state with characteristic dihedral angles: ϕ1, ϕ2 = ±26 deg, ϕ3, ϕ4 = 0 deg, demonstrating significant geometry reorganisation upon excitation, which is in accordance with experimental findings. Torsional barriers on the relaxed PES scan around the ϕ1 dihedral angle (Fig. S15, ESI†) were found to be ∼1.5 times higher as compared with the ground state. However, despite being more rigid in S1r BFMPT still exhibits distorted relaxed geometry which possibly leads to the absence of fine-structured PL spectra (Fig. 4a).
Absorption spectra were additionally evaluated in geometry adopted from X-ray data with and without the presence of classical charges, a simple model for the effect of the crystal surroundings. It was revealed that the shift of the brightest S0 → S1 absorption band due to geometry reorganization between form I and form II conformers (3.2637 eV vs. 3.5924 eV) is higher than the shift due to the effect of the crystal surroundings (3.7823 eV vs. 3.6241 eV). Thus, the difference of conformation between form I and form II is the main contributor to the difference of the crystals’ optical properties.
Reduced Density Gradient (RDG) analysis (Fig. S16, ESI†) of the ground state optimised geometry shows the presence of several non-covalent interaction regions: H6⋯S1 and H10⋯H2 being an equal combination of attraction and repulsion previously observed in the computational study of thiophene–phenylene co-oligomers;56 H29a⋯π(ring 2) and H29b⋯π(ring 3). Furthermore, Natural-Bond Orbital (NBO) analysis shows no presence of intramolecular charge transfer between interacting atoms suggesting that attraction should be attributed to electrostatic interaction. RDG analysis of form I and form II dimers (Fig. 7) revealed that previously shown short contacts between molecules are indeed representative regions of attractive dispersive interactions which are very similar for form I and form II. In addition, Hirshfeld surface analysis (Table S11, ESI†) of form I and form II crystal structures revealed that the statistical contribution of various intermolecular interactions is also almost identical in both forms.
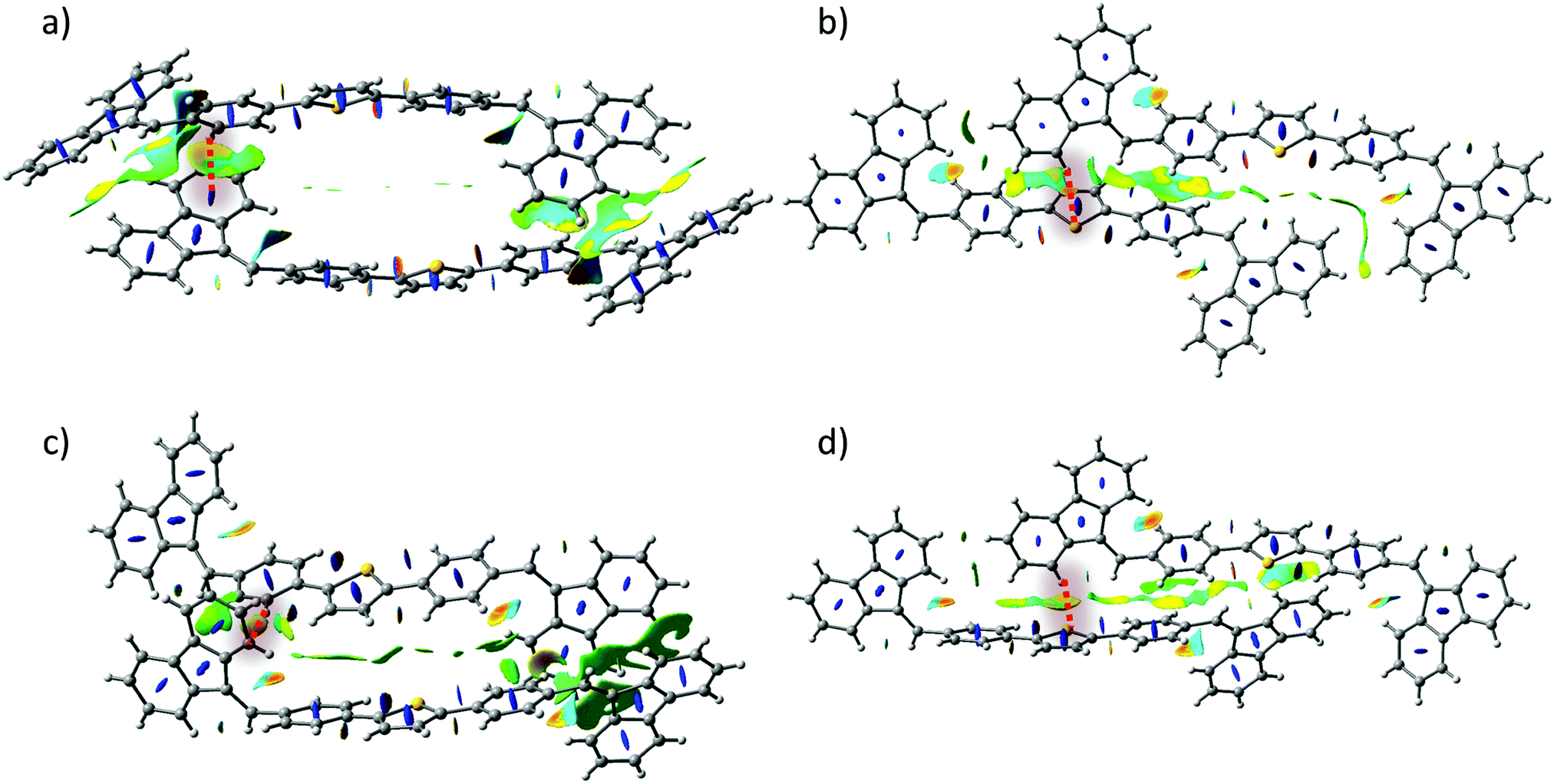 | ||
| Fig. 7 Plots of the RDG isosurfaces (s = 0.5 a.u. and a red-to-blue colour scale from −0.010 a.u. < sign(λ2) ρ(r) < +0.010 a.u.) calculated for (a), (b) – form I dimers, (c), (d) – form II dimers. Blue colour stands for repulsion, and yellow/orange for (weak/middle intensity) attraction. | ||
In order to gain a further insight into the intrinsic exciton migration dynamics of both forms, values of singlet exciton couplings were evaluated (Fig. 8) for the set of dimers adopted from form I and form II X-ray data. We observe that the computed values of EET coupling are rather small. The singlet exciton coupling for form I is nearly twice as high as that for form II. These findings support the experimental observations about the high PL in crystals. The large values of average diffusion lengths alongside the crystal axis are mainly due to the large phonon contribution bounded with the large size of the single molecule. Also in this case values found for form I (〈la〉 = 16.84 nm; 〈lb〉 = 3.65 nm; 〈lc〉 = 17.56 nm) are about two times higher than those found for form II (〈la〉 = 6.68 nm; 〈lb〉 = 0.84 nm; 〈lc〉 = 8.35 nm).
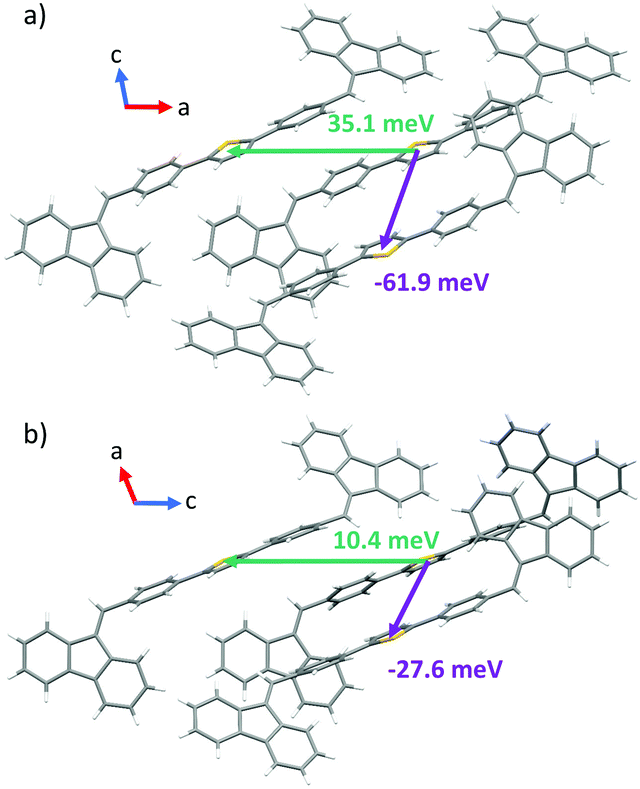 | ||
| Fig. 8 Values of exciton couplings calculated for nearest neighbours in the crystal structures of form I (a) and form II (b). Orientations of crystallographic axes are also represented. | ||
Conclusions
A novel AIE luminogen based on bis(4-((9H-fluoren-9-ylidene)methyl)phenyl)thiophene (BFMPT) was synthesised and characterised, both experimentally and quantum-chemically. BFMPT demonstrated polymorph-dependent two-colour aggregation induced emission. Two conformational polymorphs were crystallized and thoroughly studied; the orange emission of form I could irreversibly be changed to green by external stimuli, i.e. heating or grinding. The optical properties of BFMPT both in solutions and in single crystals have been evaluated: very low luminescence efficiency in solution (<1%) and high PL QYs (∼40%) for single crystals have been demonstrated. The PL kinetics for both forms revealed very similar decay time constants which is attributed to the similarity of intermolecular interactions of polymorphs, which was confirmed by performing quantum chemical calculations. Furthermore, the PL efficiency of BFMPT single crystals does not significantly get affected by the thermal stress. Despite the lowering of the photoluminescence lifetime caused by structural deformations, the heated crystals retained high luminescence efficiency. Computational study of BFMPT revealed that positive solvatochromism is mainly caused by the change of quadrupole moment upon solvation. The optical difference between BFMPT polymorphs was demonstrated to be mainly related to the change of BFMPT conformation, whereas intermolecular interactions are very similar in both forms. Altogether, due to high intrinsic torsional freedom BFMPT-related compounds could serve as a basis for further molecular design of novel high performance aggregation induced emissive materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by RSF project No 18-73-00081. We acknowledge the Ministry of Science and Higher Education of Russian Federation for access to the integrating sphere (4.7154.2017/8.9) and time-resolved fluorescence equipment (AAAA-A16-116121510087-5). The authors also acknowledge Novosibirsk State University program “5-100” and the Siberian Supercomputing Centre (http://www2.sscc.ru/). We thank A. F. Achkasov and E. A. Losev for their assistance with the recording of powder X-ray diffraction data. We thank V. A. Postnikov and V. V. Grebenev for DSC measurements. The Multi-Access Chemical Research Center SB RAS is acknowledged for spectral and analytical measurements.Notes and references
- R. Ding, J. Feng, W. Zhou, X.-L. Zhang, H.-H. Fang, T. Yang, H.-Y. Wang, S. Hotta and H.-B. Sun, Sci. Rep., 2015, 5, 12445 CrossRef PubMed.
- T. Miwa, S. Kubo, K. Shizu, T. Komino, C. Adachi and H. Kaji, Sci. Rep., 2017, 7, 284 CrossRef PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- S. Hotta, T. Yamao, S. Z. Bisri, T. Takenobu and Y. Iwasa, J. Mater. Chem. C, 2014, 2, 965–980 RSC.
- T. Komori, H. Nakanotani, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2014, 2, 4918–4921 RSC.
- C. Zhang, P. Chen and W. Hu, Small, 2016, 12, 1252–1294 CrossRef CAS PubMed.
- H.-H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H.-B. Sun, Laser Photon. Rev., 2014, 8, 687–715 CrossRef.
- J. Gierschner, S. Varghese and S. Y. Park, Adv. Optical. Mater, 2016, 2, 348–364 CrossRef.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- T. Han, X. Feng, B. Tong, J. Shi, L. Chen, J. Zhic and Y. Dong, Chem. Commun., 2012, 48, 416–418 RSC.
- Y. Zhang, W. Xu, L. Kong, B. Han, Z. Cai, J. Shi, B. Tong, Y. Dong and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1779 RSC.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhuc and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- J. Zhao, Z. Chi, Z. Yang, Z. Mao, Y. Zhang, E. Ubba and Z. Chi, Mater. Chem. Front., 2018, 2, 1595–1608 RSC.
- H. Tong, Y. Dong, M. Haubler, J. W. Y. Lam, H. H.-Y. Sung, I. D. Williams, J. Sun and B. Z. Tang, Chem. Commun., 2006, 1133–1135 RSC.
- Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, L. Ji, Z. Zhu, C. K. W. Jim, G. Yu, Q. Li, Z. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70–72 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H.-Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC.
- P. Gopikrishna and P. K. Iyer, J. Phys. Chem. C, 2016, 120, 26556–26568 CrossRef CAS.
- P. Gopikrishna, D. Das, L. R. Adil and P. K. Lyer, J. Phys. Chem. C, 2017, 121, 18137–18143 CrossRef CAS.
- P. Gopikrishna, L. R. Adil and P. K. Iyer, Mater. Chem. Front., 2017, 1, 2590–2598 RSC.
- G. A. Corrente, E. Fabiano, F. Manni, G. Chidichimo, G. Gigli, A. Beneduci and A.-L. Capodilupo, Chem. Mater., 2018, 30, 5610–5620 CrossRef CAS.
- C. Wang, Y. Yu, Z. Chai, F. He, C. Wu, Y. Gong, M. Han, Q. Lia and Z. Li, Mater. Chem. Front., 2019, 3, 32–38 RSC.
- H. Liu, Z. Lu, B. Tang, Z. Zhang, Y. Wang and H. Zhang, Dyes Pigm., 2018, 149, 284–289 CrossRef CAS.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef PubMed.
- Y. Ji, Z. Peng, B. Tong, J. Shi, J. Zhi and Y. Dong, Dyes Pigm., 2017, 139, 664–671 CrossRef CAS.
- R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Lia and T. Yi, Chem. Sci., 2014, 5, 3922–3928 RSC.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168–6173 CrossRef CAS PubMed.
- J. Jia and Y. Wu, Dyes Pigm., 2017, 136, 657–662 CrossRef CAS.
- H. Y. Zhang, Z. L. Zhang, K. Q. Ye., J. Y. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369–2372 CrossRef CAS.
- Y. Zhao, H. Gao, Y. Fan, T. Zhou, Z. Su, Y. Liu and Y. Wang, Adv. Mater., 2009, 21, 3165–3169 CrossRef CAS.
- S. Chen, H. Liu, Y. Li, D. Li and Y. Li, Adv. Electron. Mater., 2017, 3, 1700132 CrossRef.
- T. Hinoue, Y. Shigenoi, M. Sugino, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai, Chem. – Eur. J., 2012, 18, 4634–4643 CrossRef CAS PubMed.
- S. Hisamatsu, H. Masu, M. Takahashi, K. Kishikawa and S. Kohmoto, Cryst. Growth Des., 2015, 15, 2291–2302 CrossRef CAS.
- M. S. Yuan, D. E. Wang, P. Xue, W. Wang, J. C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2015, 26, 2467–2477 CrossRef.
- Y. Xu, K. Wang, Y. Zhang, Z. Xie, B. Zou and Y. Ma, J. Mater. Chem. C, 2016, 4, 1257–1262 RSC.
- J. Shi, S. J. Yoon, L. Viani, S. Y. Park, B. Milián-Medina and J. Gierschner, Adv. Optical Mater., 2017, 5, 1700340 CrossRef.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- W. Holzer, A. Penzkofer, M. Fuhrmann and P. Hegemann, Photochem. Photobiol., 2002, 75, 479–487 CrossRef CAS PubMed.
- M. S. Kazantsev, E. S. Frantseva, L. G. Kudriashova, V. G. Konstantinov, A. A. Mannanov, T. V. Rybalova, E. V. Karpova, I. K. Shundrina, G. N. Kamaev, M. S. Pshenichnikov, E. A. Mostovich and D. Y. Paraschuk, RSC Adv., 2016, 6, 92325–92329 RSC.
- SAINT, Data Reduction and Frame Integration Program for the CCD Area-Detector System, Bruker Analytical X-ray Systems, Madison, Wisconsin, USA, 1997–2006.
- G. M. Sheldrick, SADABS, Program for area detector adsorption correction, Institute for Inorganic Chemistry, University of Goettingen, Goettingen, Germany, 1996.
- G. M. Sheldrick, SHELX-97 – Program for Crystal Structure Analysis, Release 97-2 J, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, J. Appl. Cryst., 2003, 36, 7–13 CrossRef CAS.
- P. S. Sherin, J. Grilj, Y. P. Tsentalovich and E. Vauthey, J. Phys. Chem. B, 2009, 113, 4953–4962 CrossRef CAS PubMed.
- B. Dereka and E. Vauthey, Chem. Sci., 2017, 8, 5057–5066 RSC.
- J. Gierschner, L. Luer, B. Milian-Medina, D. Oelkrug and H.-J. Egelhaaf, J. Phys. Chem. Lett., 2013, 4, 2686–2697 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comp. Chem., 1990, 11, 361–373 CrossRef CAS.
- I. P. Koskin, E. A. Mostovich, E. Benassi and M. S. Kazantsev, J. Phys. Chem. C, 2017, 121, 23359–23369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1881026 and 1881027. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00198k |
| This journal is © the Partner Organisations 2019 |