DOI:
10.1039/C9QM00105K
(Research Article)
Mater. Chem. Front., 2019,
3, 1199-1208
Syntheses, crystal structures, chirality and aggregation-induced phosphorescence of stacked binuclear platinum(II) complexes with bridging Salen ligands†
Received
18th February 2019
, Accepted 17th April 2019
First published on 18th April 2019
Abstract
A novel class of cyclometalated binuclear Pt(II) complexes, [(C^N)Pt(μ-Salen)Pt(C^N)] (C^N = 2-phenylpyridine or benzo[h]quinoline, Salen = N,N′-bis(salicylidene)ethylenediamine), have been synthesized and characterized. Owing to the blockage of intramolecular rotation in the bridging Salen ligands, all the double-layer stacked complexes exhibit strong aggregation-induced phosphorescence with quantum yields up to 0.35 through well-tuned intramolecular Pt–Pt interactions (3.37–4.48 Å) and strong multiple intermolecular interactions, such as H–H, C–H, O–H, Cl–H, and Pt–H interactions. Interestingly, if chiral Salen ligands are adopted, it is facile to prepare (R,R)/(S,S) enantiopure Pt(II) complexes, which were characterized by X-ray diffraction, circular dichroism spectra and time-dependent density functional theory calculations. This is the first example of stacked binuclear Pt(II) complexes with well-known tetradentate Salen ligands as bis-bidentate-bridging ligands and would render a new structural motif for bridging ligands and chiral binuclear complexes.
Introduction
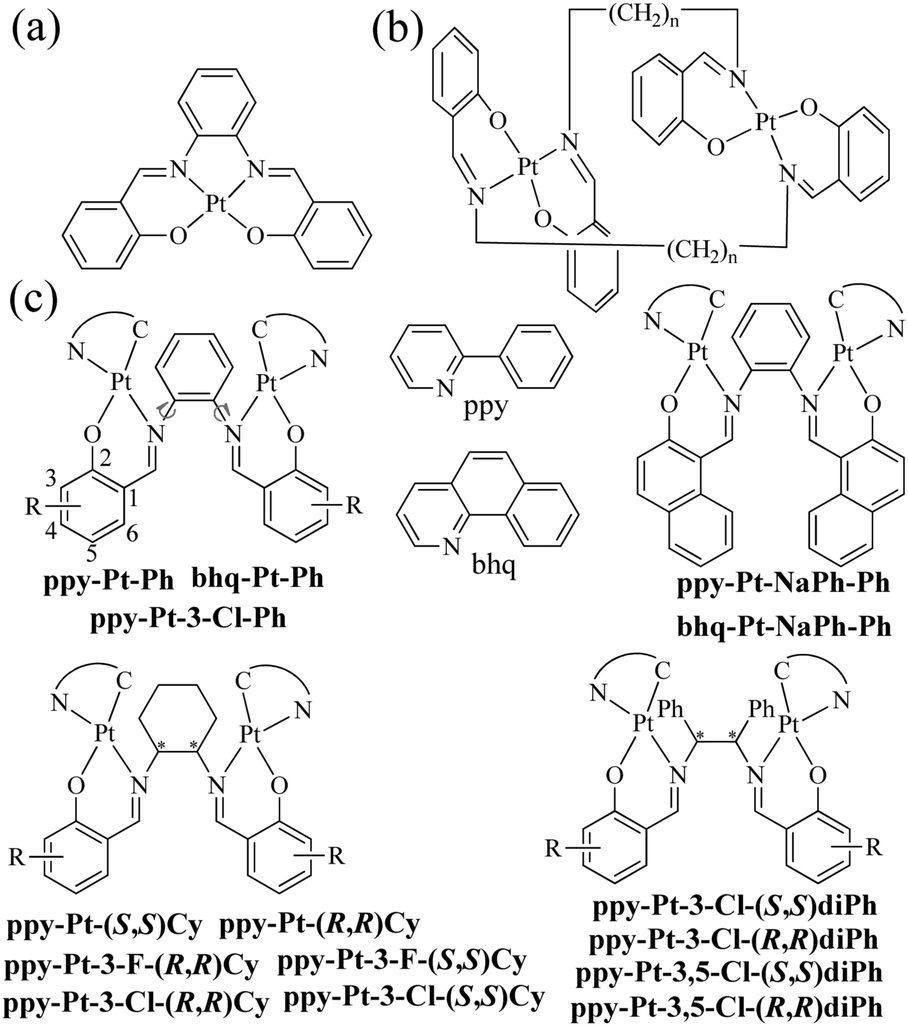
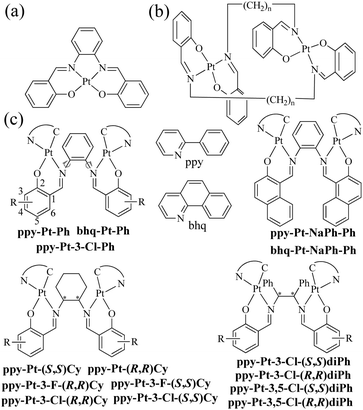
The class of cyclometalated Pt(II) complexes has been extensively investigated in the past two decades owing to their interesting and rich phosphorescence properties.1–8 Unlike other well-known octahedral phosphorescent d6 Ru(II)9,10 and Ir(III)11,12 complexes, d8 Pt(II) complexes adopt a square-planar geometry with a vacant coordination site at the Pt(II) center, which allows axial substrate-binding interactions and might lead to strong intramolecular and intermolecular interactions consequently. Therefore, Pt(II) complexes have rich transition states including not only (π–π*) intraligand charge transfer (3ILCT) and (d–π*) triplet metal-to-ligand charge transfer (3MLCT) common to normal transition metal complexes, but also metal–metal-to-ligand charge transfer (3MMLCT) through strong intramolecular and intermolecular Pt–Pt interactions. Moreover, strong intermolecular Pt–Pt interactions combined with intermolecular π–π stacking interactions have been widely used to promote molecular self-assembly and supramolecular gelation.13,14 On the other hand, Pt(II) complexes are far more likely to encounter the problem of emission “aggregation-caused quenching” (ACQ),15 because they easily form excimers through these strong intermolecular interactions in high concentrations and the solid state. In order to solve the ACQ problem, a lot of effort has been made to develop aggregation-induced emission (AIE)15–18 or phosphorescence (AIP)19-active materials.
In the literature, there are a lot of phosphorescent stacked binuclear Pt(II) complexes. The cyclometalated or auxiliary ligands of these stacked binuclear Pt(II) complexes usually are C^N,20–24 C^N^N,25,26 N^N^N,27–29 and C^C* (N-heterocyclic carbenes),30,31 whose mononuclear Pt(II) complexes are highly emissive in dilute solution. The simple bidentate pyrazolate (pz),21–25,28,31 pyridine-2-thiolate (pyt),20 bis(diphenylphosphino)methane (dppm),25,26 disulfur,29 and amidinate30 ligands with strong coordination ability are used as bridging ligands. It has been proved that the 3MMLCT transition of stacked binuclear Pt(II) complexes is strongly dependent on the intramolecular Pt–Pt distances (2.8–4.0 Å) through the interacting 5dz2 Pt(II) orbitals.20–31 For example, the transition energy or emission band (λem) of binuclear [(C^N)Pt(μ-pyt)Pt(C^N)] complexes can be well tuned in the whole region of visible light by the presence of steric bulk at the 3,5-positions of the bridging pz ligands, which in turn alters the Pt–Pt distance within the complex.21,22 Our research group has a longstanding interest in the synthesis and optical properties of N,N′-bis(salicylidene)ethylenediamine (Salen),32–38 a particular class of tetradentate N^O^O^N chelating bis-Schiff base ligands, and their complexes (Scheme 1a),39,40 due to their facile preparation, good stabilities, biological activities, rich photophysical properties, high emission quantum yields (Φ), and wide applications in catalysts,41,42 DNA cleavage,43,44 optical and magnetic materials,45,46 supramolecular materials,47,48 cell imaging,49,50 and organic light-emitting diodes.51–55 When Salen ligands react with Pt2+ ions directly, they can act as tetradentate chelating reagents to form phosphorescent mononuclear Pt(Salen) complexes (Φ up to 0.27 in dilute MeCN) (Scheme 1a).52–55 Moreover, Komiya et al.56 reported some separate binuclear Pt2(Salen)2 complexes (Scheme 1b) which are linked by non-conjugated long heptyl and consequently exhibit AIP through intermolecular Pt–Pt interactions rather than intramolecular Pt–Pt interactions. Huang's57 and our group58 recently demonstrated that mononuclear [(C^N)Pt(N^O)] complexes (Φ up to 0.38 in the crystal state) and propeller-type binuclear [(C^N)Pt(μ-SA)Pt(C^N)] (SA = salicylaldehyde azine) complexes (Φ up to 0.14 in the solid state) have neither intramolecular nor intermolecular Pt–Pt interactions but show strong AIP as well. Herein we present a series of novel neutral and chiral binuclear Salen-bridged [(C^N)Pt(μ-Salen)Pt(C^N)] complexes (C^N: ppy = 2-phenylpyridine and bhq = benzo[h]quinolone; Salen: Ph = N,N′-bis(salicylidene)-1,2-phenylenediamine, Cy = N,N′-bis(salicylidene)-1,2-cyclohexanediamine, and diPh = N,N′-bis(salicylidene)-1,2-diphenylethane-1,2-diamine) (Scheme 1c). Through the blockage of the intramolecular rotation of the bridging Salen ligands and well-tuned intramolecular Pt–Pt interactions (3.37–4.48 Å), these double-layer stacked complexes exhibit strong AIP (Φ up to 0.35 in the solid state). To the best of our knowledge, they are the first examples of stacked binuclear Pt(II) complexes with well-known tetradentate Salen ligands as bis-bidentate-bridging ligands.
 |
| | Scheme 1 Reported mononuclear Pt(Salen) complexes (a), reported binuclear Pt2(Salen)2 complexes (b), and stacked binuclear [(C^N)Pt(μ-Salen)Pt(C^N)] complexes (c) in this work. | |
Results and discussion
Synthesis and characterization
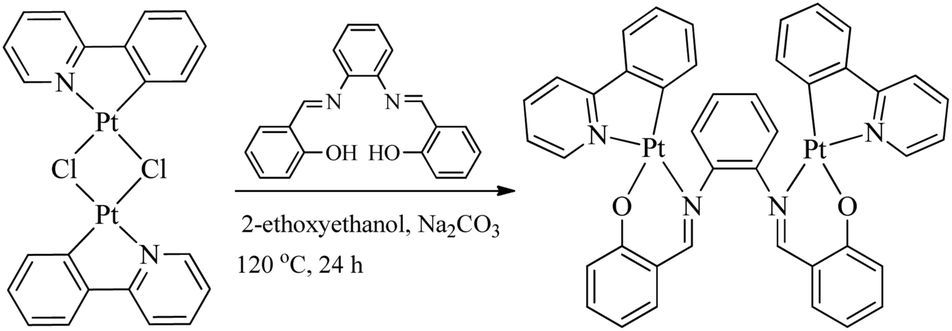
All bridging Salen ligands were reasonably easy to synthesize by the condensation of a primary diamine with 2 equivalents of salicylaldehyde precursor in ethanol under reflux conditions according to our previous reports.34,38 The reason why we chose auxiliary C^N ligands is that their Pt(II) complexes are highly phosphorescent and [(C^N)Pt(μ-Cl)2Pt(C^N)] (Scheme 2) is an ideal precursor to prepare bis-cyclometalated Pt(II) complexes.23,59 The bridging Salen ligands were adopted due to their simple, chiral, and rotatable structures (Scheme 1c) that would help to achieve chiral and AIE properties for some special applications in chiral recognition60,61 and circularly polarized luminescence (CPL).62,63 The binuclear Pt(II) complexes were straightforwardly synthesized according to Scheme 2. The complexes with conjugated μ-Ph are weakly soluble in CH2Cl2, benzene, and THF but not soluble in water, ether, ethanol, and hexane, and thus we didn’t obtain their nuclear magnetic resonance (NMR) spectra. The complexes with non-conjugated μ-Cy or μ-diPh have much better solubility in CH2Cl2, benzene, and THF.
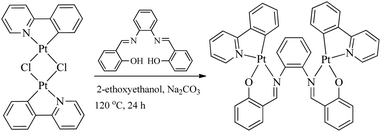 |
| | Scheme 2 Synthesis of binuclear [(C^N)Pt(μ-Salen)Pt(C^N)] complexes. | |
Absorption and AIP properties
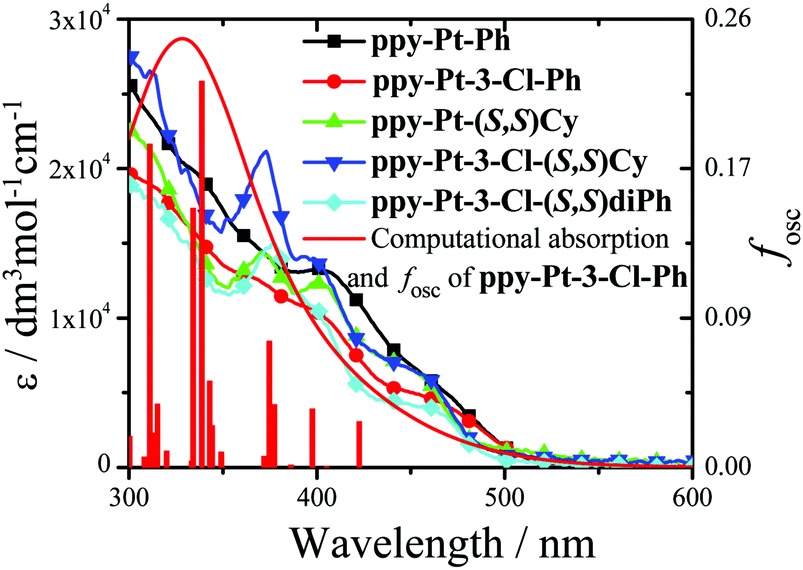
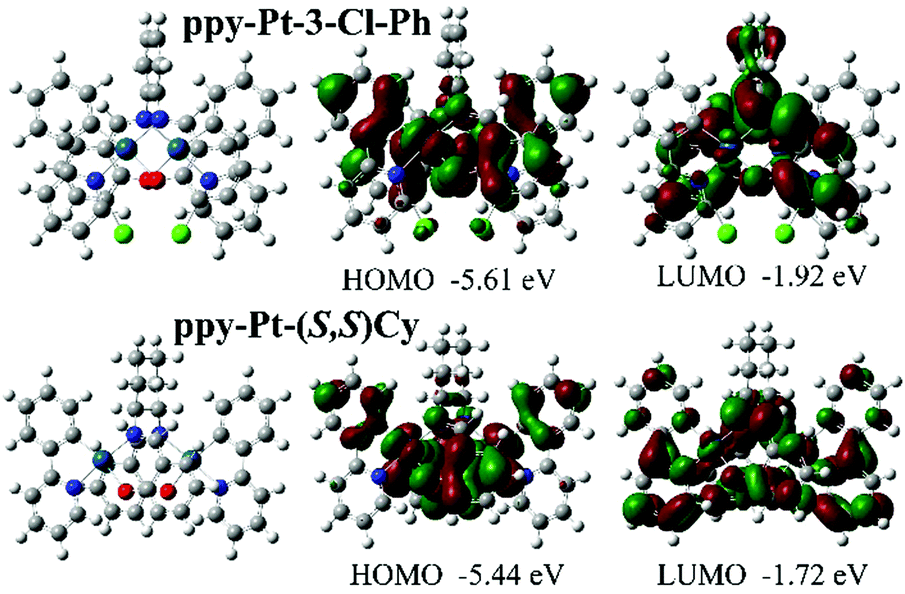
The room-temperature UV/visible absorption and AIP properties of the Pt(II) complexes are listed in Table 1, Fig. 1–5 and Fig. S1–S12 (ESI†). These Pt(II) complexes have similar absorption spectra, even though they bear different bridging ligands and C^N auxiliary ligands. To gain insight into the nature of the excited states and transitions, density functional theory (DFT) and time-dependent-DFT (TD-DFT) calculations were carried out for ppy-Pt-3-Cl-Ph and ppy-Pt-(S,S)Cy with the Gaussian 09 program package (PBE0/SDD/6-31G).64 The computational absorption spectra are almost identical to the experimental absorption spectra (Fig. 1 and Fig. S6, ESI†). The lower energy absorption band of ppy-Pt-3-Cl-Ph (λabs = 445 nm in THF) is reproduced well by the computation, which predicts one absorption band at 436 nm. The lower energy absorption is due to the highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) (oscillator strength fOSC = 0.0275, 91%) transition. The energy level and frontier molecular orbitals of ppy-Pt-3-Cl-Ph (Fig. 2) reveal that its HOMO is composed primarily of π-conjugated units of the bridging ligand (iminomethylphenol units), auxiliary ppy ligands (phenyl units), and Pt(II) ions, which might be due to their electron-donating nature. On the contrary, its LUMO is mainly made up of the whole π-functions of the bridging ligand. Therefore, the lower energy absorption can mainly be assigned to singlet intraligand charge transfer (1ILCT) in the bridging ligand, ligand-to-ligand charge transfer (1LLCT) from auxiliary ligands to the bridging ligand, and metal-to-ligand charge transfer (1MLCT). The Cl atoms in the bridging ligand and pyridine rings in auxiliary ligands have little contribution to the lower energy absorption. Un-conjugated bridging ppy-Pt-(S,S)Cy shows a similar transition (423 nm, fOSC = 0.0533, HOMO → LUMO, 88%) (Fig. 2 and Fig. S6, ESI†) to ppy-Pt-3-Cl-Ph, except that the pyridine rings and un-conjugated bridging cyclohexane have and doesn’t have a contribution to the lower energy absorption, respectively.
Table 1 Photophysical data of the Pt(II) complexes at room temperature. A sample without emission data means that it is non-emissive
|
|
Medium |
λ
abs/nm (ε/dm3 mol−1 cm−1) |
λ
em/nm |
Stokes shift/nm |
Φ
|
f/% |
|
ppy-Pt-Ph
|
THF |
333(2.00 × 104); 401(1.33 × 104) |
|
|
|
|
| THF/water |
274; 403 |
598 |
195 |
0.001 |
96 |
| Solid |
|
584 |
|
0.057 |
|
|
ppy-Pt-3-Cl-Ph
|
THF |
393(1.08 × 104); 445(5.10 × 103) |
|
|
|
|
| THF/water |
280; 334 |
597 |
263 |
0.002 |
94 |
| Solid |
|
580 |
|
0.16 |
|
|
ppy-Pt-NaPh-Ph
|
THF |
365(2.33 × 104) 403(1.53 × 104); 458(8.90 × 103) |
|
|
|
|
| THF/water |
359 |
602 |
243 |
0.001 |
96 |
| Solid |
|
604 |
|
0.017 |
|
|
bhq-Pt-Ph
|
THF |
334(2.63 × 104); 407(1.23 × 104) |
|
|
|
|
| THF/water |
280; 350; 410 |
592 |
182 |
0.001 |
92 |
| Solid |
|
576 |
|
0.047 |
|
|
bhq-Pt-NaPh-Ph
|
THF |
369(1.79 × 104); 406(1.14 × 104); 460(6.70 × 103) |
|
|
|
|
| THF/water |
378 |
604 |
226 |
0.001 |
96 |
| Solid |
|
605 |
|
0.047 |
|
|
ppy-Pt-(R,R)Cy
|
THF |
289(2.38 × 104); 372(1.33 × 104); 400(1.14 × 104) |
|
|
|
|
| THF/water |
253; 297; 371; 402 |
569 |
168 |
0.006 |
96 |
| Solid |
|
592 |
|
0.15 |
|
|
ppy-Pt-(S,S)Cy
|
THF |
288(3.49 × 104); 371(1.49 × 104); 399(1.28 × 104) |
|
|
|
|
| THF/water |
253; 297; 372; 404 |
569 |
168 |
0.006 |
96 |
| Solid |
|
593 |
|
0.15 |
|
|
ppy-Pt-3-F-(R,R)Cy
|
THF |
287(3.32 × 104); 369(1.36 × 104); 398(1.12 × 104) |
|
|
|
|
| THF/water |
252; 298; 377; 402 |
595 |
194 |
0.010 |
96 |
| Solid |
|
596 |
|
0.17 |
|
|
ppy-Pt-3-F-(S,S)Cy
|
THF |
287(3.45 × 104); 370(1.62 × 104); 398(1.21 × 104) |
|
|
|
|
| THF/water |
253; 297; 378; 403 |
594 |
191 |
0.009 |
96 |
| Solid |
|
597 |
|
0.18 |
|
|
ppy-Pt-3-Cl-(R,R)Cy
|
THF |
282(5.00 × 104); 370(2.07 × 104); 395(1.43 × 104) |
|
|
|
|
| THF/water |
253; 298; 380; 402 |
595 |
193 |
0.016 |
96 |
| Solid |
|
599 |
|
0.20 |
|
|
ppy-Pt-3-Cl-(S,S)Cy
|
THF |
286(3.82 × 104); 372(2.11 × 104); 398(1.40 × 104) |
|
|
|
|
| THF/water |
252; 299; 380; 402 |
595 |
193 |
0.016 |
96 |
| Solid |
|
598 |
|
0.21 |
|
|
ppy-Pt-3-Cl-(R,R)diPh
|
THF |
287(3.57 × 104); 377(1.56 × 104); 455(4.50 × 103) |
|
|
|
|
| THF/water |
254; 299; 377; 459 |
595 |
136 |
0.012 |
96 |
| Solid |
|
587 |
|
0.33 |
|
|
ppy-Pt-3-Cl-(S,S)diPh
|
THF |
288(3.22 × 104); 378(1.62 × 104); 456(4.30 × 103) |
|
|
|
|
| THF/water |
254; 299; 378; 459 |
595 |
136 |
0.012 |
96 |
| Solid |
|
586 |
|
0.35 |
|
|
ppy-Pt-3,5-Cl-(R,R)diPh
|
THF |
292(2.29 × 104); 376(1.81 × 104); 457(4.90 × 103) |
|
|
|
|
| THF/water |
297; 381; 454 |
606 |
152 |
0.012 |
94 |
| Solid |
|
610 |
|
0.21 |
|
|
ppy-Pt-3,5-Cl-(S,S)diPh
|
THF |
292(2.43 × 104); 375(1.83 × 104); 456(5.30 × 103) |
|
|
|
|
| THF/water |
298; 380; 454 |
605 |
151 |
0.014 |
94 |
| Solid |
|
607 |
|
0.20 |
|
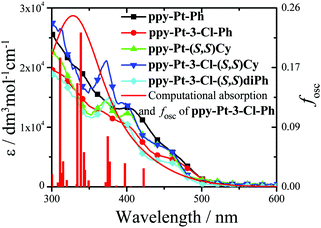 |
| | Fig. 1 Computational and experimental (in THF) absorption spectra of some Pt(II) complexes. | |
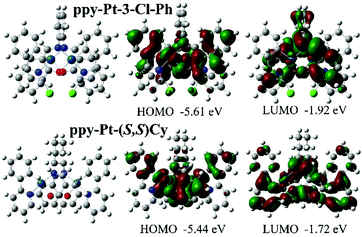 |
| | Fig. 2 Computational energy level diagram and frontier molecular orbitals of ppy-Pt-3-Cl-Ph and ppy-Pt-(S,S)Cy (in THF). | |
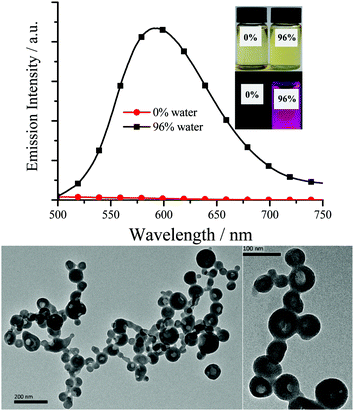 |
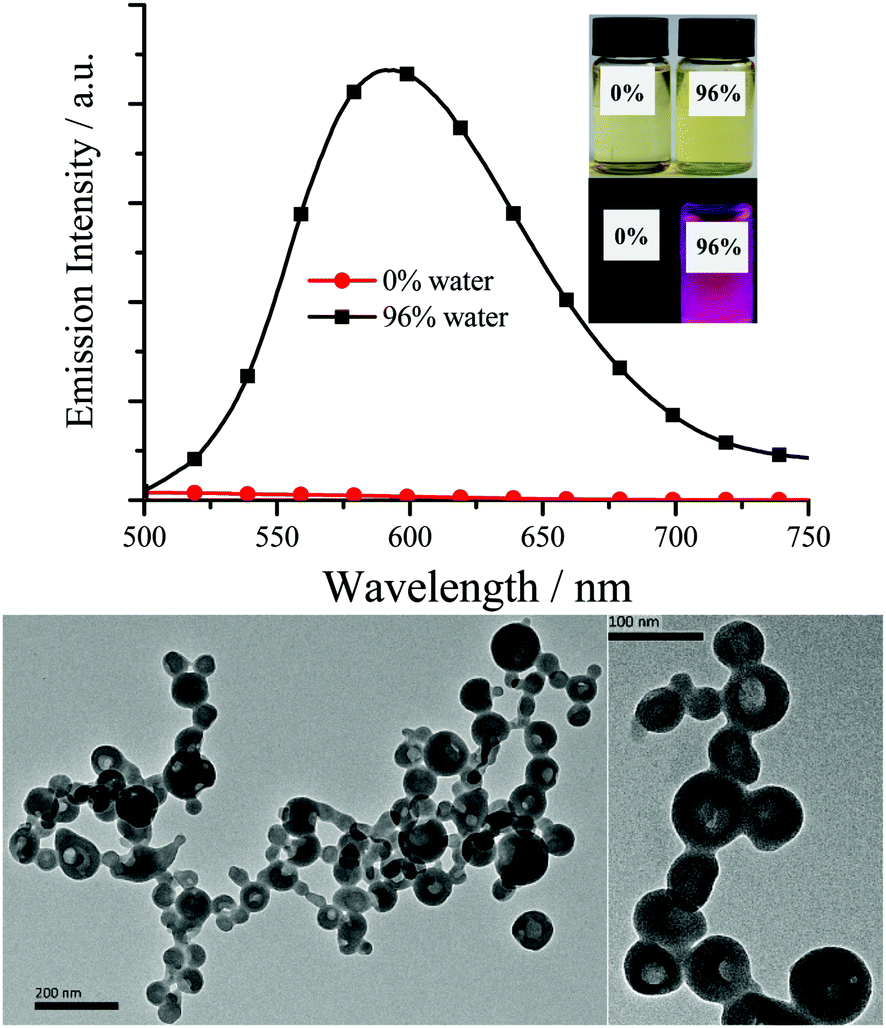
| | Fig. 3 Top: Emission spectra of ppy-Pt-3-Cl-(S,S)Cy in THF and THF/water (2.0 × 10−5 mol dm−3). Inset: Their photographs under room light and 360 nm UV light. Bottom: Transmission electron microscopy of ppy-Pt-3-Cl-(S,S)Cy (THF/water, 96% water) on carbon-coated copper grids after solvent evaporation. | |
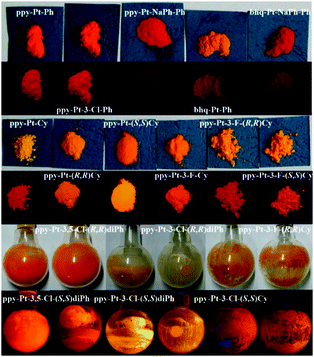 |
| | Fig. 4 Photographs of the Pt(II) complexes under room light and 360 nm UV light. | |
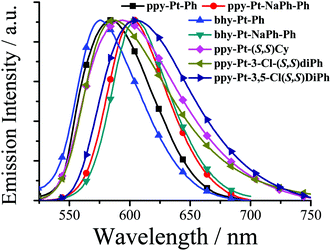 |
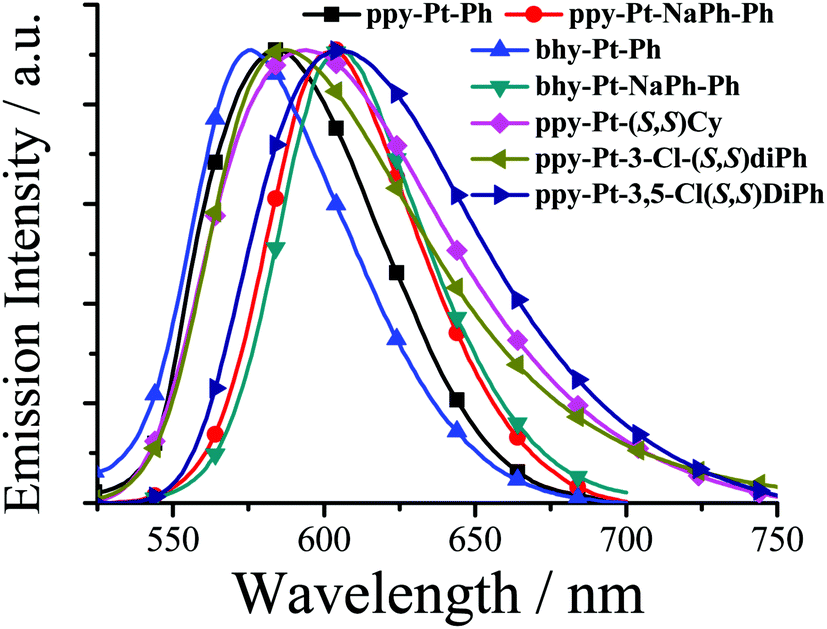
| | Fig. 5 Normalized emission spectra of some Pt(II) complexes in the solid state. | |
As expected, all the Pt(II) complexes are non-emissive (Table 1) in pure dilute CH2Cl2, MeCN, benzene, DMSO or THF, as the intramolecular rotations (IVs) of the bridging Salen ligand provide a possible way to non-radiatively annihilate their excited states (Scheme 1c). Nonetheless, their aggregates either in THF/water or the solid state (Fig. 3–5 and Fig. S7–S11, ESI†) exhibit strong yellow–red emission (λem = 576–610 nm; Φ up to 0.35) with relative long decay lifetimes (1.68, 2.34, 1.72, and 4.02 μs for ppy-Pt-(R,R)Cy, ppy-Pt-(S,S)Cy, ppy-Pt-3-Cl-(R,R)diPh, and ppy-Pt-3-Cl-(S,S)diPh, respectively, Fig. S12, ESI†), revealing their AIP nature (water is an insoluble solvent). The absorption spectrum of ppy-Pt-3-Cl-(S,S)Cy in THF/water is similar to that in the organic solution of THF (Fig. S2, ESI†), except that there is an obvious tail in the visible region, indicating the formation of aggregated hollow nanoparticles (Fig. 3). Moreover, their absorption and excitation spectra are similar (Fig. S2, ESI†), suggesting they have a common excitation pathway, which would not be the case for a different (e.g., dimer) species.
Single crystal structures
The molecular structures and arrangements play a key role in AIE.65–68 Non-covalent interactions are also frequently involved to assist the molecules to orient and assemble. In order to enhance AIE, molecules should stack closely with not only strong non-covalent intermolecular interactions but also weak intermolecular face-to-face π–π* interactions. The former can ensure elimination of IVs; and the latter would prevent the formation of excimers. We tried many different ways to grow single crystals of these Pt(II) complexes. After dozens of attempts, we obtained suitable single crystals of ppy-Pt-3-Cl-Ph (CCDC: 1589703), ppy-Pt-NaPh-Ph (CCDC: 1589704), racemic ppy-Pt-Cy (CCDC: 1589683), ppy-Pt-(S,S)Cy (CCDC: 1589684), and ppy-Pt-(R,R)Cy (CCDC: 1589685) for X-ray structure determination by slow diffusion/evaporation of a CH2Cl2/hexane or CHCl3/hexane solution.† The X-ray single crystals of racemic ppy-Pt-Cy were obtained from a mixture of enantiopure complexes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in solution. In general, strong intermolecular interactions in the crystal are in the range 1.9–2.3 Å and 2.2–3.0 Å for hydrogen bonds and non-hydrogen bonds, respectively.18,36,38,58,63,69
:1) in solution. In general, strong intermolecular interactions in the crystal are in the range 1.9–2.3 Å and 2.2–3.0 Å for hydrogen bonds and non-hydrogen bonds, respectively.18,36,38,58,63,69
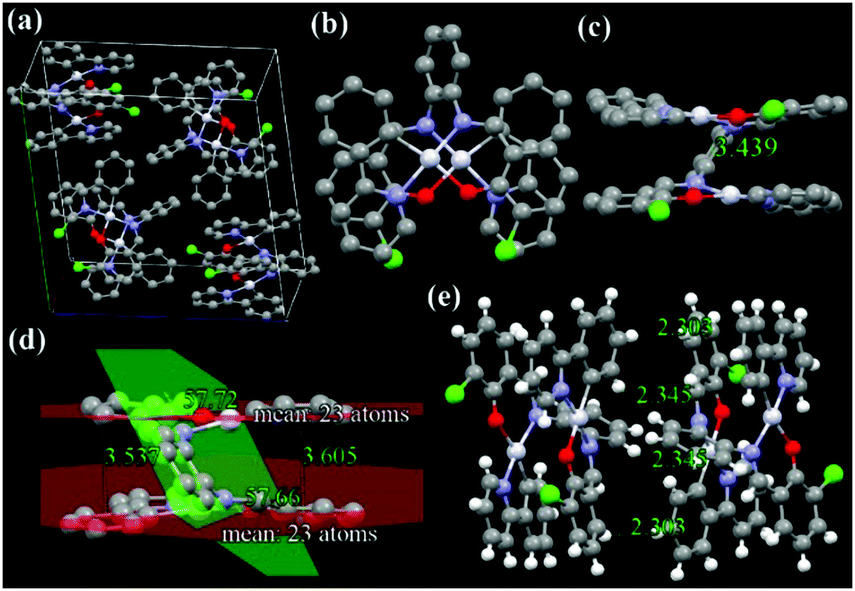
The X-ray single-crystal structures and arrangements of ppy-Pt-3-Cl-Ph are shown in Fig. 6 and Fig. S13 (ESI†). ppy-Pt-3-Cl-Ph molecules are stacked to exhibit lamellar packing with a distance of 7.092 Å. Unlike the square-planar mononuclear Pt(Salen) complexes (Scheme 1a), the I-type binuclear ppy-Pt-3-Cl-Ph complex bears two planes of Pt(II) coordination systems that are linked by a central bridge of a benzene ring. The four coordination atoms of C^N^O^N and central PtII are almost located in one plane and the Pt–N, Pt–O, and Pt–C bonds are of similar lengths (2.006–2.068 Å) to what was found in normal Pt(Salen) complexes,54 which indicates that the ppy-Pt-3-Cl-Ph complex has a suitable square-planar coordination environment for chelating the PtII cation. Since the dihedral angle between these two Pt(II) coordination systems and the central benzene bridge is 57.72° and 57.66°, respectively, the two Pt(II) coordination systems are almost parallel with an interplanar distance (d) of 3.537–3.605 Å, which reveals that there are some weak intramolecular π–π stacking interactions in the ppy-Pt-3-Cl-Ph complex. Some modest intramolecular PtII–PtII interactions (3.439 Å) are observed as well. Due to its staggered arrangement, there are no strong intermolecular π–π stacking interactions and PtII–PtII interactions (7.390 Å) between two neighboring ppy-Pt-3-Cl-Ph molecules, which would be beneficial in preventing the formation of excimers. On the other hand, many strong intermolecular noncovalent interactions including not only normal H–H (2.303–2.851 Å), C–H (2.793–2.797 Å), and O–H (2.650 Å) but also Cl–H (2.911 Å, halogen bonds) exist between the two neighboring ppy-Pt-3-Cl-Ph molecules. These strong intermolecular interactions could restrict the IVs of the bridging ligand, and thus the phosphorescence is enhanced finally.
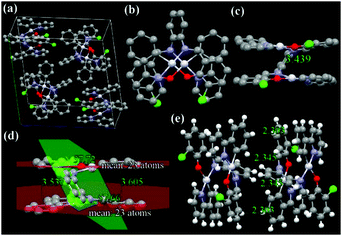 |
| | Fig. 6 X-ray single crystal structures and packing of ppy-Pt-3-Cl-Ph molecules (a: molecule packing in one crystal cell; b: top view; c and d: side view; e: intermolecular interactions of the two closest molecules). H atoms are omitted in a–d. | |
With π-extended conjugation in the bridging ligand, ppy-Pt-NaPh-Ph molecules (Fig. 7 and Fig. S14, ESI†) exhibit a similar lamellar packing to ppy-Pt-3-Cl-Ph molecules. The dihedral angle between the two Pt(II) coordination systems and the central benzene bridge is 51.38° and 50.55°, respectively. Their interplanar distances are in the range of 3.376–3.456 Å, indicating that intramolecular π–π stacking interactions in ppy-Pt-NaPh-Ph are stronger than those in ppy-Pt-3-Cl-Ph. Moreover, stronger intramolecular PtII–PtII interactions (3.370 Å) and weaker intermolecular H–H (2.430–2.945 Å) and C–H (2.842 Å) interactions are observed for ppy-Pt-NaPh-Ph molecules. All the above factors lead to solid-state ppy-Pt-NaPh-Ph (λem = 605 nm; Φ = 0.017) emitting red-shifted emission along with a much lower Φ than solid-state ppy-Pt-3-Cl-Ph (λem = 580 nm; Φ = 0.16).
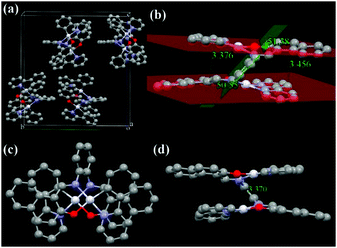 |
| | Fig. 7 X-ray single crystal structures and packing of ppy-Pt-NaPh-Ph molecules (a: molecule packing in one crystal cell; b and d: side view; c: top view). H atoms and CHCl3 solvent molecules are omitted. | |
Linked with an un-conjugated Cy ligand, ppy-Pt-(S,S)Cy and ppy-Pt-(R,R)Cy molecules (Fig. 8 and Fig. S15, ESI†) exhibit lamellar packing as well. The molecular structures and arrangements of the ppy-Pt-(S,S)Cy and ppy-Pt-(R,R)Cy enantiomers are similar and mirrored, which might contribute to their similar absorption and AIP properties.38,63,69 In this section, ppy-Pt-(S,S)Cy was discussed as an example. Since the un-conjugated Cy bridging ligand is flexible, the two Pt(II) coordination systems are not parallel with a dihedral angle of 14.12°. There are no intramolecular π–π stacking interactions and PtII–PtII interactions (4.481 Å) in one ppy-Pt-(S,S)Cy molecule. For two neighboring ppy-Pt-(S,S)Cy molecules, however, not only some strong intermolecular H–H (2.353–2.692 Å) and C–H (2.875 Å) interactions but also strong Pt–H (2.817 Å) interactions are observed. Therefore, solid-state ppy-Pt-(S,S)Cy (λem = 593 nm; Φ = 0.15) has a high Φ.
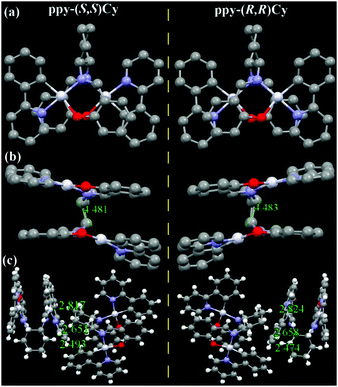 |
| | Fig. 8 X-ray single crystal structures and packing of ppy-Pt-(S,S)Cy and ppy-Pt-(R,R)Cy molecules (a: top view; b: side view; c: intermolecular interactions of the two neighboring molecules). Some H atoms and CH2Cl2 solvent molecules are omitted. | |
Racemic ppy-Pt-Cy (Fig. 9 and Fig. S16, ESI†) has a totally different molecular arrangement from enantiopure ppy-Pt-(S,S)Cy and ppy-Pt-(R,R)Cy, even though they have similar AIP properties. In the single crystals of ppy-Pt-Cy, one pair of (S,S) and (R,R) enantiomers (1:1) was found, and thus these (R)/(S)-chirality-induced interactions38,63 between the two enantiomers would help ppy-Pt-Cy molecules arrange in herringbone packing. Similar strong intermolecular H–H (2.390–2.876 Å), C–H (2.825 Å), and Pt–H (2.881 Å) interactions are observed in two neighboring ppy-Pt-Cy molecules. We failed to grow single crystals of the complexes bearing diPh bridging ligands. Our previous work38 reveals that diPh ligands have a 1,2-diphenylethane bridge but not a circular bridge of cyclohexane; hence, diPh ligands are more flexible than Cy ligands. We speculate that, compared with Cy-linked complexes, diPh-linked complexes would be more flexible as well and consequently pack more closely to enhance AIP. The Φ of solid-state ppy-Pt-3-Cl-(S,S)diPh is up to 0.35, which is unexpected for binuclear Pt(II) complexes70 and even higher than that of mononuclear Pt(Salen) complexes (Φ up to 0.27 in dilute MeCN).54 The presence of Cl and F atoms would aid in AIP through intermolecular halogen bonds (Fig. S13, ESI†).36–38
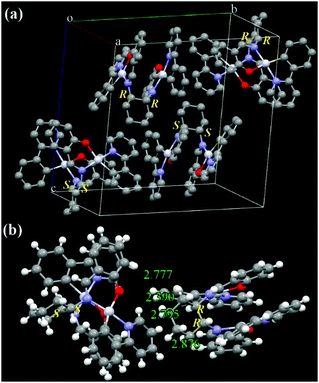 |
| | Fig. 9 X-ray single crystal structures and packing of racemic ppy-Pt-Cy molecules (a: molecule packing in one crystal cell; b: intermolecular interactions of the two neighboring molecules). Some H atoms are omitted. | |
It is interesting that similar cyclometallated [(ppy)Pt(pop)] (pop = 2-(5-phenyl-1,3,4-oxadiazol-2-yl)phenol) Pt(II) complexes71 are known to be emissive in solution. However, some similar cyclometallated [(C^N)Pt(N^O)] complexes57 and [(C^N)Pt(μ-SA)Pt(C^N)] complexes58 show strong AIP as well. This might be caused by the differences of their chemical and electronic structures.
Chirality properties
The un-conjugated Pt(II) complexes with Cy and diPh bridging ligands have two (S,S) or (R,R) chiral carbons and might be used in some special applications of enantioselective catalysis,42 chiral recognition,60,61 and CPL,62,63 and thus their chirality properties were studied by circular dichroism (CD) spectra and TD-DFT calculations (Fig. 10). As example, racemic ppy-Pt-Cy has no CD signals in dilute MeCN. Enantiopure ppy-Pt-(S,S)Cy exhibits three positive Cotton peaks at 390, 353, and 255 nm and two negative Cotton peaks at 448 and 300 nm. On the other hand, enantiopure ppy-Pt-(R,R)Cy shows exactly the mirror CD spectrum with two positive Cotton peaks at 488 and 300 nm and three negative Cotton peaks at 391, 352, and 255 nm, indicating the fact that ppy-Pt-(S,S)Cy and ppy-Pt-(R,R)Cy are a pair of enantiomers. The calculated CD spectrum of ppy-Pt-(S,S)Cy fits well with the experimental CD spectrum (Fig. 10), which further confirms its (S,S) chirality. ppy-Pt-3-F-(S,S)Cy, ppy-Pt-3-F(R,R)Cy, ppy-Pt-3-Cl-(S,S)diPh, and ppy-Pt-3-Cl-(R,R)diPh have similar chirality properties to ppy-Pt-(S,S)Cy and ppy-Pt-(R,R)Cy, which is consistent with their similar conjugation and chiral carbons. Our previous work38 demonstrated that the free bridging Cy and diPh ligands have identical CD signals to the corresponding complexes at high energy (<370 nm), and thus the high-energy CD signals can be assigned to the chiral bridging Salen ligands themselves (1ILCT) and the lower-energy CD signals (>370 nm) can be mainly assigned to 1LLCT and 1MLCT.
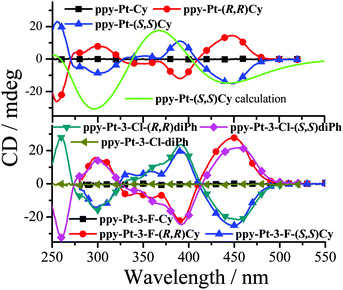 |
| | Fig. 10 Experimental CD spectra of ppy-Pt-(S,S)Cy, ppy-Pt-(R,R)Cy, racemic ppy-Pt-cy, ppy-Pt-3-F-(S,S)Cy, ppy-Pt-3-F(R,R)Cy, racemic ppy-Pt-3-F-Cy, ppy-Pt-3-Cl-(S,S)diPh, ppy-Pt-3-Cl-(R,R)diPh, and racemic ppy-Pt-3-Cl-diPh (5.0 × 10−5 mol dm−3 in MeCN). Computational CD spectra of ppy-Pt-(S,S)Cy (in THF). | |
Luminescence mechanism
DFT was used to calculate optimized singlet- and triplet-state structures of gas-state ppy-Pt-(S,S)Cy. As shown in Fig. 11, in the gas state or dilute solution, the optimized singlet- and triplet-state structures of ppy-Pt-(S,S)Cy are different from its single-crystal structure, which would indicate that the C–N single bonds of the bridging Salen ligand (Scheme 1c) may be rotatable and provide a possible way to non-radiatively annihilate the excited states consequently.63 The time-resolved emission spectra (Fig. S12, ESI†) reveal that the Pt(II) complexes only have one type of emission decay lifetime in the region of 1.68–4.02 μs. This indicates that the emission may be either phosphorescence or delayed fluorescence. For metal–organic complexes, delayed fluorescence is often observed for Cu(I), Ag(I) Au(I), and Sn(IV) complexes.72,73 For Pt(II) complexes, the emission is often phosphorescence rather than delayed fluorescence.1–8 Moreover, we used DFT to calculate the singlet–triplet energy gap ΔEST, defined as the gap between the lowest energy triplet state (T1) and the lowest energy singlet state (S1). When ΔEST is sufficiently small, taken usually as <0.1 eV, the upconversion from the triplet state to the singlet state by reverse intersystem-crossing becomes possible.74 The ΔEST of ppy-Pt-(S,S)Cy is 0.27 eV, which is much bigger than 0.1 eV. Therefore, the emission of Pt(II) complexes is phosphorescence rather than delayed fluorescence.
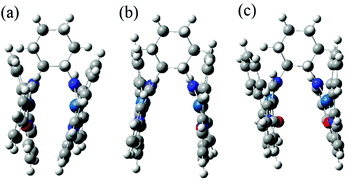 |
| | Fig. 11 Single-crystal structure (a) and optimized singlet- (b) and triplet-state (c) structures of gas-state ppy-Pt-(S,S)Cy. | |
It is surprising that the quantum yield (around 0.01) of Pt(II) complexes in the THF/water mixture is quite low. The solid powders of ppy-Pt-(S,S)Cy have intense and sharp X-ray powder diffraction peaks (Fig. S17, ESI†), which indicate that the solid powders are mainly microcrystalline and the emission might be caused by the aggregated crystallites. However, the cast film of ppy-Pt-3-F-(S,S)Cy-doped poly(methyl methacrylate) (PMMA) emits strong emission as well, revealing that the amorphous and aggregated Pt(II) complex in PMMA is emissive (Fig. S18, ESI†). Therefore, the low quantum yield in the THF/water mixture might be caused by self-assembly (Fig. 3). Of course, we can’t exclude the possibility of crystallization-induced emission.
Experimental
Materials and instrumentation
All reagents were purchased from commercial suppliers and used without further purification. All commercial chemicals were used without further purification unless otherwise stated. UV/visible absorption spectra were recorded using a UV 765 spectrophotometer with quartz cuvettes of 1 cm pathlength. Fluorescence spectra were obtained using an F-7000 fluorescence spectrophotometer (Hitachi) at room temperature. The slit width was 5.0 nm for both excitation and emission. The photon multiplier voltage was 700 V. Fluorescence lifetime data were determined in a 1 mm cell on a Hamamatsu FL920S instrument. The single-crystals were obtained by slow diffusion/evaporation of a CH2Cl2/CHCl3/hexane solution at room temperature during about one week.
1H and 13C NMR spectra were recorded on a Brüker Advance 400 spectrometer (1H: 400 MHz, 13C: 101 MHz). Chemical shifts (δ) for 1H and 13C NMR spectra are given in ppm relative to TMS. The residual solvent signals were used as references for 1H and 13C NMR spectra and the chemical shifts converted to the TMS scale (CDCl3: δH = 7.26 ppm, δC = 77.16 ppm). HR-MS were obtained with a Waters-Q-TOF-Premier (electrospray ionization mass spectrometry, ESI). X-Ray single-crystal diffraction data were collected on an Oxford Xcalibur E CCD area-detector diffractometer.
Measurement of fluorescence quantum yield (Φ)
The quantum yield of a solution sample was measured by the optically dilute method of Demas and Crosby with a standard of quinine sulfate (Φr = 0.55, quinine in 0.05 mol dm−3 sulfuric acid) calculated by: Φs = Φr(Br/Bs)(ns/nr)2(Ds/Dr), where the subscripts s and r refer to the sample and reference standard solution respectively; n is the refractive index of the solvents; and D is the integrated intensity. The excitation intensity B is calculated by: B = 1 – 10–AL, where A is the absorbance at the excitation wavelength and L is the optical path length (L = 1 cm in all cases). The refractive indices of the solvents at room temperature are taken from standard sources. Errors for the Φ values (±10%) are estimated. The quantum yield of a solid sample was measured by an integrating sphere.
Computational details
The program Gaussian 09 was employed to perform DFT and TD-DFT calculations based on the X-ray single-crystal structure of the Pt(II) complexes. The ground state geometry was optimized using DFT and excited states were predicted using the ground state geometry using TD-DFT, from which UV/vis absorption and CD spectra were predicted. Calculations were run at the PBE0/SDD/6-31G level (pcm method; 50 singlet–singlet transitions).64
General procedure for the preparation of the Pt(II) complexes
Bridging ligands were synthesized according to our previous work.38 The Pt(II) complexes were synthesized according to a procedure modified from that reported in the literature.58 Pt(II) dichloro-bridged dimers were synthesized from the starting materials of K2PtCl4 and 2-phenylpyridine according to a previous report.59 A solution of Pt(II) dichloro-bridged dimers, 0.5 equiv. of the bridging ligand and 5 equiv. of Na2CO3 in 2-ethoxyethanol was heated to reflux for 24 h under a N2 atmosphere. Then the reaction mixture was concentrated under reduced pressure. A few milliliters of dichloromethane was added gradually to give the crude product, which was purified by silica gel column chromatography using dichloromethane as the eluent to give pure products with moderate yields (10–28%).
ppy-Pt-Ph
.
(12% yield); HPLC m/z 1034.1566 [[M + Na]+] (calcd m/z 1034.1541).
ppy-Pt-3-Cl-Ph
.
(15% yield); HPLC m/z 1102.0755 [[M + Na]+] (calcd m/z 1102.0761).
ppy-Pt-NaPh-Ph
.
(10% yield); HPLC m/z 11134.1854 [[M + Na]+] (calcd m/z 1134.1841).
bhq-Pt-Ph
.
(11% yield); HPLC m/z 1082.1566 [[M + Na]+] (calcd m/z 1082.1541).
bhq-Pt-NaPh-Ph
.
(10% yield); HPLC m/z 1182.1883 [[M + Na]+] (calcd m/z 1182.1854).
ppy-Pt-(R,R)Cy
.
(25% yield); 1H NMR (400 MHz, CDCl3, δ): 9.14 (s, 2H), 8.14 (s, 2H), 7.66 (t, 2H), 7.40 (d, 2H), 7.33 (s, 2H), 7.13 (s, 2H), 7.00 (dd, 8H), 6.81 (d, 2H), 6.59 (d, 2H), 6.07 (t, 2H), 4.95 (d, 2H), 2.60 (d, 2H), 1.94 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, CDCl3, δ): 167.13, 165.16, 160.28, 146.51, 146.03, 143.54, 137.54, 134.09, 133.98, 133.51, 129.19, 123.10, 122.87, 122.22, 121.58, 120.36, 117.61, 113.81, 75.22, 33.95, 24.83; HPLC m/z 1041.2037 [[M + Na]+] (calcd m/z 1041.2031).
ppy-Pt-(S,S)Cy
.
(27% yield); 1H NMR (400 MHz, CDCl3, δ): 9.14 (s, 2H), 8.14 (s, 2H), 7.66 (t, 2H), 7.40 (d, 2H), 7.33 (s, 2H), 7.13 (s, 2H), 7.00 (dd, 8H), 6.81 (d, 2H), 6.59 (d, 2H), 6.07 (t, 2H), 4.95 (d, 2H), 2.60 (d, 2H), 1.94 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, CDCl3, δ): 167.13, 165.16, 160.28, 146.51, 146.03, 143.54, 137.54, 134.09, 133.98, 133.51, 129.19, 123.10, 122.87, 122.22, 121.58, 120.36, 117.61, 113.81, 75.22, 33.95, 24.83; HPLC m/z 1041.2037 [[M + Na]+] (calcd m/z 1041.2031).
ppy-Pt-3-F-(R,R)Cy
.
(28% yield); 1H NMR (400 MHz, CDCl3, δ): 9.23 (dd, 2H), 7.94 (d, 2H), 7.67 (td, 2H), 7.36–7.28 (m, 4H), 7.19–7.09 (m, 4H), 7.08–6.98 (m, 4H), 6.71 (ddd, 2H), 6.54 (d, J = 8.0 Hz, 2H), 5.97 (td, 2H), 4.96 (d, 2H), 2.53 (d, 2H), 2.00 (d, 4H), 1.60 (s, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.76, 159.61, 159.58, 155.63, 154.29, 154.15, 153.22, 146.73, 146.15, 143.09, 137.51, 133.86, 129.13, 128.39, 128.35, 124.88, 124.83, 122.97, 122.29, 120.73, 117.48, 117.38, 117.30, 111.99, 111.92, 76.07, 32.86, 24.69; HPLC m/z 1077.8883 [[M + Na]+] (calcd m/z 1077.8875).
ppy-Pt-3-F-(S,S)Cy
.
(26% yield); 1H NMR (400 MHz, CDCl3, δ): 9.23 (dd, 2H), 7.94 (d, 2H), 7.67 (td, 2H), 7.36–7.28 (m, 4H), 7.19–7.09 (m, 4H), 7.08–6.98 (m, 4H), 6.71 (ddd, 2H), 6.54 (d, 2H), 5.97 (td, 2H), 4.96 (d, 2H), 2.53 (d, 2H), 2.00 (d, 4H), 1.60 (s, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.76, 159.61, 159.58, 155.63, 154.29, 154.15, 153.22, 146.73, 146.15, 143.09, 137.51, 133.86, 129.13, 128.39, 128.35, 124.88, 124.83, 122.97, 122.29, 120.73, 117.48, 117.38, 117.30, 111.99, 111.92, 76.07, 32.86, 24.69; HPLC m/z 1077.8883 [[M + Na]+] (calcd m/z 1077.8875).
ppy-Pt-3-Cl-(R,R)Cy
.
(24% yield); 1H NMR (400 MHz, CDCl3, δ): 9.35 (s, 2H), 7.95 (s, 2H), 7.67 (td, 2H), 7.33 (dd, 4H), 7.23–6.88 (m, 10H), 6.68 (dd, 2H), 5.98 (t, 2H), 4.95 (d, 2H), 2.53 (d, 2H), 2.02 (d, 4H), 1.61 (t, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.50, 160.24, 159.26, 147.28, 146.33, 143.18, 137.43, 133.88, 132.89, 132.18, 129.09, 125.57, 123.68, 122.98, 122.22, 120.83, 117.27, 113.39, 76.18, 33.05, 24.46; HPLC m/z 1109.1252 [[M + Na]+] (calcd m/z 1109.1249).
ppy-Pt-3-Cl-(S,S)Cy
.
(22% yield); 1H NMR (400 MHz, CDCl3, δ): 9.35 (s, 2H), 7.95 (s, 2H), 7.67 (td, 2H), 7.33 (dd, 4H), 7.23–6.88 (m, 10H), 6.68 (dd, 2H), 5.98 (t, 2H), 4.95 (d, 2H), 2.53 (d, 2H), 2.02 (d, 4H), 1.61 (t, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.50, 160.24, 159.26, 147.28, 146.33, 143.18, 137.43, 133.88, 132.89, 132.18, 129.09, 125.57, 123.68, 122.98, 122.22, 120.83, 117.27, 113.39, 76.18, 33.05, 24.46; HPLC m/z 1109.1252 [[M + Na]+] (calcd m/z 1109.1249).
ppy-Pt-3-Cl-(R,R)diPh
.
(13% yield); 1H NMR (400 MHz, CDCl3, δ): 9.45 (d, 2H), 7.78–7.66 (m, 4H), 7.56–7.45 (m, 6H), 7.37–7.27 (m, 8H), 7.23–7.19 (m, 2H), 7.13 (ddd, 8H), 7.00 (d, 2H), 6.25 (dd, 2H), 5.95 (t, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.43, 165.06, 158.99, 147.11, 146.92, 140.93, 140.41, 137.57, 133.05, 132.80, 132.00, 129.24, 128.84, 128.51, 128.08, 125.37, 123.42, 122.59, 120.62, 117.51, 113.27, 73.08; HPLC m/z 1207.1412 [[M + Na]+] (calcd m/z 1207.1408).
ppy-Pt-3-Cl-(S,S)diPh
.
(15% yield); 1H NMR (400 MHz, CDCl3, δ): 9.45 (d, 2H), 7.78–7.66 (m, 4H), 7.56–7.45 (m, 6H), 7.37–7.27 (m, 8H), 7.23–7.19 (m, 2H), 7.13 (ddd, 8H), 7.00 (d, 2H), 6.25 (dd, 2H), 5.95 (t, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.43, 165.06, 158.99, 147.11, 146.92, 140.93, 140.41, 137.57, 133.05, 132.80, 132.00, 129.24, 128.84, 128.51, 128.08, 125.37, 123.42, 122.59, 120.62, 117.51, 113.27, 73.08; HPLC m/z 1207.1412 [[M + Na]+] (calcd m/z 1207.1408).
ppy-Pt-3,5-Cl-(R,R)diPh
.
(13% yield); 1H NMR (400 MHz, CDCl3, δ): 9.37 (d, 2H), 7.78–7.70 (m, 4H), 7.40 (ddd, 16H), 7.13 (ddd, 8H), 7.02 (d, 2H), 6.17 (d, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.00, 164.58, 157.50, 146.98, 146.64, 140.15, 137.89, 132.60, 131.91, 131.01, 129.10, 129.00, 128.85, 128.30, 126.27, 123.53, 123.33, 123.19, 120.60, 117.58, 116.77, 72.91; HPLC m/z 1275.0639 [[M + Na]+] (calcd m/z 1275.0629).
ppy-Pt-3,5-Cl-(S,S)diPh
.
(13% yield); 1H NMR (400 MHz, CDCl3, δ): 9.37 (d, 2H), 7.78–7.70 (m, 4H), 7.40 (ddd, 16H), 7.13 (ddd, 8H), 7.02 (d, 2H), 6.17 (d, 2H); 13C NMR (101 MHz, CDCl3, δ): 166.00, 164.58, 157.50, 146.98, 146.64, 140.15, 137.89, 132.60, 131.91, 131.01, 129.10, 129.00, 128.85, 128.30, 126.27, 123.53, 123.33, 123.19, 120.60, 117.58, 116.77, 72.91; HPLC m/z 1275.0639 [[M + Na]+] (calcd m/z 1275.0629).
Conclusions
In summary, we have developed a straightforward way to prepare a new class of binuclear cyclometalated Pt(II) complexes with well-known tetradentate Salen ligands as bridging ligands. These double-layer stacked complexes exhibit strong AIP through well-tuned intramolecular Pt–Pt interactions and strong multiple intermolecular interactions, such as H–H, C–H, O–H, Cl–H, and Pt–H interactions. Furthermore, chirality can be facile to introduce by the presence of chiral (S,S) and (R,R) cyclohexane and 1,2-diphenylethane in the bridging Salen ligands, which might provide a new paradigm in the design of AIP active dyes for developing phosphorescent materials, enantioselective catalysis, chiral recognition, CPL, and so on.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21871192) and Sichuan Science and Technology Program (no. 2018JY0559). We acknowledge the comprehensive training platform of the specialized laboratory of the College of Chemistry, Sichuan University, for material analysis. We would like to thank the Analytical & Testing Center of Sichuan University for CCD X-ray single crystal diffractometer work and circular dichroism CD spectrometer work. We are grateful to Daibing Luo and Yani Xie for help with the single crystal and circular dichroism measurements.
Notes and references
- C. W. Chan, L. K. Cheng and C. M. Che, Coord. Chem. Rev., 1994, 132, 87–97 CrossRef CAS.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819–1828 CrossRef CAS.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- L. Murphy and J. A. G. Williams, Top. Organomet. Chem., 2010, 28, 75–111 CrossRef CAS.
- Y. Feng, J. H. Cheng, L. Zhou, X. G. Zhou and H. F. Xiang, Analyst, 2012, 137, 4885–4901 RSC.
- H. F. Xiang, J. H. Cheng, X. F. Ma, X. G. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W. Y. Tong, W. H. Ang, W. L. Kwong and C. M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC.
- S. Campagna, F. Puntoriero and F. Nastasi, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- Y. Chi and P. T. Chou, Chem. Soc. Rev., 2007, 36, 1421–1431 RSC.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- A. Y. Y. Tam and V. W. W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- V. W. W. Yam, V. K. M. Au and S. Y. L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- X. F. Ma, R. Sun, J. H. Cheng, J. Y. Liu, F. Gou, H. F. Xiang and X. G. Zhou, J. Chem. Educ., 2016, 93, 345–350 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- L. Ravotto and P. Ceroni, Coord. Chem. Rev., 2017, 346, 62–76 CrossRef CAS.
- K. Saito, Y. Hamada, H. Takahashi, T. Koshiyama and M. Kato, Jpn. J. Appl. Phys., Part 2, 2005, 44, L500 CrossRef CAS.
- B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28–29 CrossRef CAS PubMed.
- B. Ma, P. I. Djurovich, S. Garon, B. Alleyne and M. E. Thompson, Adv. Funct. Mater., 2006, 16, 2438–2446 CrossRef CAS.
- A. A. Rachford and F. N. Castellano, Inorg.
Chem., 2009, 48, 10865–10867 CrossRef CAS PubMed.
- S. Jamali, R. Czerwieniec, R. Kia, Z. Jamshidi and M. Zabel, Dalton Trans., 2011, 40, 9123–9130 RSC.
- S. W. Lai, M. C. W. Chan, T. C. Cheung, S. M. Peng and C. M. Che, Inorg. Chem., 1999, 38, 4046–4055 CrossRef CAS.
- W. Lu, M. C. W. Chan, N. Zhu, C. M. Che, C. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS PubMed.
- C. K. Koo, B. Lam, S. K. Leung, M. H. W. Lam and W. Y. A. Wong, J. Am. Chem. Soc., 2006, 128, 16434–16435 CrossRef CAS PubMed.
- K. W. Wang, J. L. Chen, Y. M. Cheng, M. W. Chung, C. C. Hsieh, G. H. Lee, P. T. Chou, K. Chen and Y. Chi, Inorg. Chem., 2010, 49, 1372–1383 CrossRef CAS PubMed.
- R. P. L. Tang, K. M. C. Wong, N. Zhu and V. W. W. Yam, Dalton Trans., 2009, 3911–3922 RSC.
- H. Leopold, M. Tenne, A. Tronnier, S. Metz, I. Mgnster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2016, 55, 15779–15782 CrossRef CAS PubMed.
- P. Pinter, Y. Unger and T. Strassner, ChemPhotoChem, 2017, 1, 113–115 CrossRef CAS.
- L. Zhou, P. Y. Cai, Y. Feng, J. H. Cheng, H. F. Xiang, J. Liu, D. Wu and X. G. Zhou, Anal. Chim. Acta, 2012, 735, 96–106 CrossRef CAS PubMed.
- X. F. Ma, J. H. Cheng, J. Y. Liu, X. G. Zhou and H. F. Xiang, New J. Chem., 2015, 39, 492 RSC.
- J. H. Cheng, K. Y. Wei, X. F. Ma, X. G. Zhou and H. F. Xiang, J. Phys. Chem. C, 2013, 117, 16552–16563 CrossRef CAS.
- J. H. Cheng, Y. H. Zhang, X. F. Ma, X. G. Zhou and H. F. Xiang, Chem. Commun., 2013, 49, 11791–11793 RSC.
- J. H. Cheng, Y. X. Li, R. Sun, J. Y. Liu, F. Gou, X. G. Zhou, H. F. Xiang and J. Liu, J. Mater. Chem. C, 2015, 3, 11099–11110 RSC.
- X. H. Zhang, J. Shi, G. Y. Shen, F. Gou, J. H. Cheng, X. G. Zhou and H. F. Xiang, Mater. Chem. Front., 2017, 1, 1041–1050 RSC.
- G. Y. Shen, F. Gou, J. H. Cheng, X. H. Zhang, X. G. Zhou and H. F. Xiang, RSC Adv., 2017, 7, 40640–40649 RSC.
- J. H. Cheng, X. F. Ma, Y. H. Zhang, J. Y. Liu, X. G. Zhou and H. F. Xiang, Inorg. Chem., 2014, 53, 3210–3219 CrossRef CAS PubMed.
- J. H. Cheng, F. Gou, X. H. Zhang, G. Y. Shen, X. G. Zhou and H. F. Xiang, Inorg. Chem., 2016, 55, 9221–9229 CrossRef CAS PubMed.
- D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37–52 CrossRef CAS PubMed.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- E. Lamour, S. Routier, J. L. Bernier, J. P. Catteau, C. Bailly and H. Vezin, J. Am. Chem. Soc., 1999, 121, 1862–1869 CrossRef CAS.
- P. Wu, D. L. Ma, C. H. Leung, S. C. Yan, N. Y. Zhu, R. Abagyan and C. M. Che, Chem. – Eur. J., 2009, 15, 13008–13021 CrossRef CAS PubMed.
- H. Miyasaka, A. Saitoh and S. Abe, Coord. Chem. Rev., 2007, 251, 2622–2664 CrossRef CAS.
- M. Andruh, Chem. Commun., 2011, 47, 3025–3042 RSC.
- S. J. Wezenberg, E. C. Escudero-Adán, J. Benet-Buchholz and A. W. Kleij, Chem. – Eur. J., 2009, 15, 5695–5700 CrossRef CAS PubMed.
- G. Consiglio, S. Failla, P. Finocchiaro, I. P. Oliveri, R. Purrello and S. Di Bella, Inorg. Chem., 2010, 49, 5134–5142 CrossRef CAS PubMed.
- Y. Hai, J. J. Chen, P. Zhao, H. Lv, Y. Yu, P. Xu and J. L. Zhang, Chem. Commun., 2011, 47, 2435–2437 RSC.
- H. Y. Yin, J. Tang and J. L. Zhang, Eur. J. Inorg. Chem., 2017, 5085–5093 CrossRef CAS.
- P. F. Wang, Z. R. Hong, Z. Y. Xie, S. W. Tong, O. Y. Wong, C. S. Lee, N. B. Wong, L. S. Hung and S. T. Lee, Chem. Commun., 2003, 1664–1665 RSC.
- C. M. Che, S. C. Chan, H. F. Xiang, M. C. W. Chan, Y. Liu and Y. Wang, Chem. Commun., 2004, 1484–1845 RSC.
- H. F. Xiang, S. C. Chan, K. K. Y. Wu, C. M. Che and P. T. Lai, Chem. Commun., 2005, 1408–1410 RSC.
- C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Y. Zhu and H. Yersin, Chem. – Eur. J., 2010, 16, 233–247 CrossRef CAS PubMed.
- J. Zhang, F. C. Zhao, X. J. Zhu, W. K. Wong, D. G. Ma and W. Y. Wong, J. Mater. Chem., 2012, 22, 16448–16457 RSC.
- N. Komiya, T. Muraoka, M. Iida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054–16061 CrossRef CAS PubMed.
- S. J. Liu, H. B. Sun, Y. Ma, S. G. Ye, X. M. Liu, X. H. Zhou, X. Mou, L. H. Wang, Q. Zhao and W. Huang, J. Mater. Chem., 2012, 22, 22167–22173 RSC.
- F. Gou, J. H. Cheng, X. H. Zhang, G. Y. Shen, X. G. Zhou and H. F. Xiang, Eur. J. Inorg. Chem., 2016, 4862–4866 CrossRef CAS.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- L. Pu, Chem. Rev., 2004, 104, 1687–1716 CrossRef CAS PubMed.
- L. Pu, Acc. Chem. Res., 2017, 50, 1032–1040 CrossRef CAS PubMed.
- E. M. Sanchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.– Eur. J., 2015, 21, 13488–13500 CrossRef CAS PubMed.
- J. T. Song, M. Wang, X. G. Zhou and H. F. Xiang, Chem. – Eur. J., 2018, 24, 7128–7132 CrossRef CAS PubMed.
- C. Garino, A. Erenzi, G. Barone and L. Salassa, J. Chem. Educ., 2016, 93, 292–298 CrossRef CAS.
- J. S. Ni, H. X. Liu, J. K. Liu, M. J. Jiang, Z. Zhao, Y. C. Chen, R. T. K. Kwok, J. W. Y. Lam, Q. Peng and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1498–1507 RSC.
- G. X. Huang, R. S. Wen, Z. M. Wang, B. S. Li and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1884–1892 RSC.
- H. Nie, K. Hu, Y. J. Cai, Q. Peng, Z. J. Zhao, R. R. Hu, J. W. Chen, S. J. Su, A. J. Qin and B. Z. Tang, Mater. Chem. Front., 2017, 1, 1125–1129 RSC.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 1573–1579 RSC.
- M. Wang, C. Cheng, J. T. Song, J. Wang, X. G. Zhou, H. F. Xiang and J. Liu, Chin. J. Chem., 2018, 36, 698–707 CrossRef CAS.
- E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS.
- G. Z. Lu, Y. M. Jing, H. B. Han, Y. L. Fang and Y. X. Zheng, Organometallics, 2017, 36, 448–454 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. H. Li, R. F. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Z. Y. Yang, Z. Mao, Z. L. Xie, Y. Zhang, S. W. Liu, J. Zhao, J. R. Xu, Z. G. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General, materials, computational details, crystallographic information files (CIF), crystallographic data, absorption, and fluorescence emission data. CCDC 1589703, 1589704, 1589683, 1589684, and 1589685. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00105k |
|
| This journal is © the Partner Organisations 2019 |

 and
Haifeng
Xiang
and
Haifeng
Xiang