
Systematic oligoaniline-based derivatives: ACQ–AIE conversion with a tunable insertion effect and quantitative fluorescence “turn-on” detection of BSA†
Hao
Lu
 ,
Kun
Wang
,
Beibei
Liu
,
Meng
Wang
,
Mingming
Huang
,
Yue
Zhang
and
Jiping
Yang
*
,
Kun
Wang
,
Beibei
Liu
,
Meng
Wang
,
Mingming
Huang
,
Yue
Zhang
and
Jiping
Yang
*
Key Laboratory of Aerospace Advanced Materials and Performance, Ministry of Education, School of Materials Science and Engineering, Beihang University, Beijing 100191, China. E-mail: jyang@buaa.edu.cn
First published on 18th December 2018
Abstract
The aggregation-induced emission (AIE) phenomenon has attracted persistent attention in recent years. Currently, the molecular design is mainly based on ring-shaped molecules. Hence, a novel strategy was put forward to achieve conversion from aggregation-caused quenching to AIE in derivatives of aniline oligomers using a new “chain-insertion” pattern. Studies of aniline oligomer derivatives indicate a tunable insertion effect on the AIE behaviors of the derivatives. Fully substituted oligoanilines exhibited typical AIE behavior, while partially substituted derivatives were AIE-inactive. In addition, luminescence wavelength and solid fluorescence in aniline derivatives can be effectively regulated by insertion and chain length. The resulting model will contribute to simplified and systematic research into oligomer-based AIEgens. Moreover, fully substituted anilines show a quantitative turn-on fluorescence response towards bovine serum albumin (BSA) with a detection limit in the order of micrograms per liter. Thus, fully substituted anilines show great potential for use as fluorescent probes in BSA sensors.
Introduction
Currently, the design of new molecular systems with aggregation-induced emission (AIE) is an active area of research in organic luminescent materials.1 Conceptually presented and thoroughly investigated by Prof. Tang's group, the restriction of intramolecular motion is considered to be the main factor in overcoming the notorious aggregation-caused quenching (ACQ) phenomenon.2 Accordingly, the modification of normal ACQ luminogens is becoming an attractive strategy to convert ACQ to AIE, which is mostly achieved with a twisted conformation and suppressed π–π stacking.3 Various AIE systems with decorated aromatic rings, flexible chains and intramolecular rings on the periphery have been successfully investigated, including derivatives of coumarin, anthracene, naphthalene diimide, pyrrole, porphyrin, pyridine and diketopyrrolopyrrole.4However, from the perspective of linkage patterns between matrixes and modified moieties, all the aforementioned AIE systems are unitary in terms of their “core-side pattern” classification (Fig. 1a).5 Thus, there is a need to design new AIEgens with a new linkage pattern.
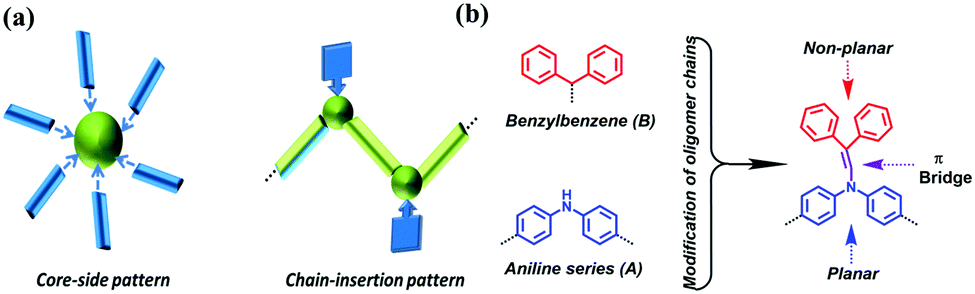 | ||
| Fig. 1 (a) Two patterns of fluorescent molecules with conversion from ACQ to AIE. (b) Guideline for the preparation of non-planar conjugated discrete oligoaniline derivatives. | ||
Long-chain organic molecules, which are as important as cyclic ones, are rarely explored in the AIE field. However, these molecules may provide another way to enrich the AIE family with a new linkage pattern called the “chain-insertion pattern” (Fig. 1a). Astoundingly, the ACQ compound 1,4-distyrylbenzene can turn into an AIE molecule when methyl groups are inserted into the ethylene chains.6 In fact, heteroatom oligomer systems play a key role in chain molecules. Helpfully, changes in chain length and the insertion of substituents into conjugated frameworks may lead to distinct optical behaviors related to color, efficiency and mechanism of emission.7
However, the elucidation of heteroatom oligomer analogues with different chain lengths and numbers of insertion units for ACQ–AIE conversion remains a challenging task. On one hand, extended conjugation through the empty pπ orbital in heteroatom oligomers is effective.8 On the other hand, differing from cyclic molecules, a small number of insertions can only partially avoid intermolecular stacking. Thus, it is of great importance to provide an effective strategy to show the effects of chain length and insertion groups on AIE heteroatom oligomer derivatives.
Oligoanilines are an important type of conjugated aromatic oligomers.9 Their planar π-conjugated aromatic rings are prone to forming strong intermolecular π–π stacking when aggregating, resulting in apparent ACQ phenomenon. Thus, achieving the definite and facile conversion from ACQ to AIE via a “chain-insertion” pattern using oligoaniline chains is directly relevant to the systematic expansion of heteroatom chain AIE compounds.
In this study, we proposed to avoid strong intermolecular π–π interactions by inserting nonplanar building blocks into the oligomer structures. For the expected oligoaniline derivatives, the following unknowns remain to be discussed: (1) the appropriate groups to combine with planar frameworks under facile synthetic conditions; (2) a detailed ACQ–AIE conversion study revealing how the introduction of nonplanar blocks affects the electronic distributions and the stereoscopic structures of oligoaniline derivatives; and (3) the effective influence of extended p–π conjugation on the AIE effect after introducing side conjugation of various types. Herein, we present the design and synthesis of oligoaniline derivatives with various chain lengths and insertion numbers. We also make efforts to explore the above issues to obtain an applicable ACQ–AIE conversion strategy for conjugated aromatic oligomers.
Results and discussion
Based on the aforementioned considerations, neutral aromatic rings should be an appropriate choice. Single benzene is excluded because the formed triphenylamine-type molecules have planar geometries;10 hence, non-planar benzylbenzene is chosen instead (Fig. 1b). Combining benzylbenzene with aniline oligomers can be realized through joint enamines, which is known as Stork enamine alkylation.11 Aniline monomer (A1), dimer (A2), trimer (A3) and tetramer (A4) were used to successfully synthesize a series of oligoaniline derivatives that differed only in the aniline chain length and the number of diphenyl enamine substituents (Scheme 1). Moreover, based on Buchwald–Hartwig amination,12 the incontrovertible di-substituted tetramer derivative was also obtained for comparison (Scheme 1).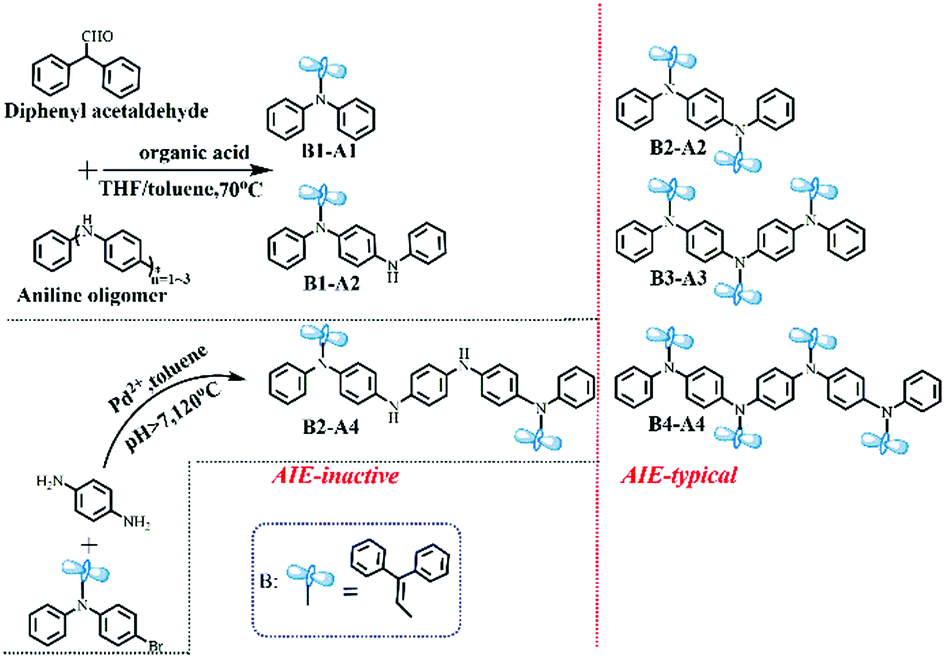 | ||
| Scheme 1 Synthesis of oligoaniline derivatives substituted with diphenyl enamines. | ||
Specifically, diphenyl enamine singly or partially substituted aniline oligomer derivatives (i.e., B1-A1, B1-A2 and B2-A4) and fully substituted aniline oligomer derivatives (i.e., B2-A2, B3-A3 and B4-A4) were prepared in good yields. All compounds were characterized using mass spectroscopy and spectroscopic methods (Fig. S1–S8, ESI†).
Photo-physical properties
The UV-vis absorption and PL emission spectra were obtained to study the optical properties of the derivatives. Their normalized absorption and emission spectra in THF solution (10 µM) are shown in Fig. 2. The basic spectroscopic parameters are summarized in Table S1 (ESI†).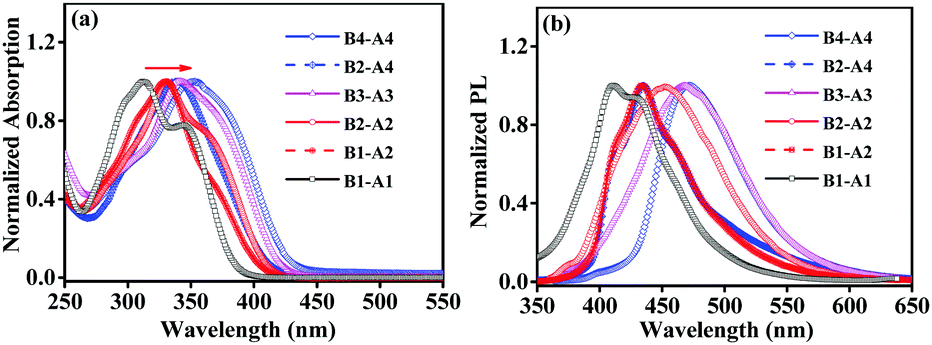 | ||
| Fig. 2 Normalized absorption (a) and emission (b) spectra of B1-A1, B1-A2, B2-A2, B3-A3, B2-A4 and B4-A4 in THF (concentration: 10 µM; excitation wavelengths: 344 nm for B1-A1, 329 nm for B1-A2, 335 nm for B2-A2, 340 nm for B3-A3, 338 nm for B2-A4 and 353 nm for B4-A4; EX slit: 5 nm; EM slit: 10 nm). | ||
As shown in Fig. 2a, four classes of absorption bands were observed between 310 and 360 nm. All aniline derivatives (B1-A1, B1-A2, B2-A2, B3-A3, B2-A4 and B4-A4) exhibited strong absorption capacities with molar extinction coefficients of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 550, 35900, 45900, 53667, 34300 and 31600 cm−1 mol−1 L, respectively. The monomer, dimer, trimer and tetramer derivatives showed different absorption bands with peaks at 312, 330/332, 342 and 339/353 nm, respectively. Meanwhile, some shoulder absorptions appeared at approximately 350 nm for B1-A1 and 365 nm for B2-A2; these bands were assigned to mild intramolecular charge transfer (CT) from the electron-rich aniline moiety to the relatively electron-deficient inserted benzylbenzene group. The CT absorptions may be partially merged with the red-shifted absorption band of the π–π* transition of the backbone when the aniline units and insertions are sufficient.13 Compared to their original oligomers, the absorption peaks of all compounds are red-shifted, indicating that the electron clouds of the introduced groups can spread to the oligomer chains through the ethylene–N bridges. In addition, the absorption peaks of derivatives A1–A4 gradually become bathochromic by about 10–20 nm as the number of chain units increases. The slightly enhanced extent of conjugation was attributed to moderately extended p–π conjugation. However, for serial derivatives with specific oligomer chains, the effect of insertion number was not progressive like that of chain length.
550, 35900, 45900, 53667, 34300 and 31600 cm−1 mol−1 L, respectively. The monomer, dimer, trimer and tetramer derivatives showed different absorption bands with peaks at 312, 330/332, 342 and 339/353 nm, respectively. Meanwhile, some shoulder absorptions appeared at approximately 350 nm for B1-A1 and 365 nm for B2-A2; these bands were assigned to mild intramolecular charge transfer (CT) from the electron-rich aniline moiety to the relatively electron-deficient inserted benzylbenzene group. The CT absorptions may be partially merged with the red-shifted absorption band of the π–π* transition of the backbone when the aniline units and insertions are sufficient.13 Compared to their original oligomers, the absorption peaks of all compounds are red-shifted, indicating that the electron clouds of the introduced groups can spread to the oligomer chains through the ethylene–N bridges. In addition, the absorption peaks of derivatives A1–A4 gradually become bathochromic by about 10–20 nm as the number of chain units increases. The slightly enhanced extent of conjugation was attributed to moderately extended p–π conjugation. However, for serial derivatives with specific oligomer chains, the effect of insertion number was not progressive like that of chain length.
As the number of naked NH moieties was more than one, the maximal absorption wavelength of B2-A4 is approximately 10 nm different from that of the fully substituted tetraaniline B4-A4. Meanwhile, the maximum absorption wavelength of aniline derivative B1-A2, which has just one naked NH moiety, was similar to that of the fully substituted aniline dimer B2-A2. The non-progressive effect of insertion number may be because the effective conjugation content of these molecules was achieved by introducing the proper quantity of conjugated moieties.14 These results can help provide a methodology for the molecular design of AIE compounds for future applications from the viewpoint of positional relationships between matrices and modifying factors.
The emission spectra of these compounds were recorded in THF (10 µM), as shown in Fig. 2b. The emission spectra of all aniline derivatives showed an emission peak in the range of 410–480 nm with different Stokes shifts ranging from 7450 to 9010 cm−1. The PL emission peaks of B2-A2, B3-A3 and B4-A4 were at 451, 467, and 472 nm, respectively, demonstrating smaller Stokes shifts than the corresponding partially substituted aniline derivatives. Owing to the absence of conspicuous D–A structures, the difference in Stokes shift was not large, and all compounds exhibited moderate Stokes shifts of Δλ = 90–150 nm.
Furthermore, the emissions of B1-A1, B1-A2, B2-A2, B3-A3, B2-A4 and B4-A4 in the solid powder state were also measured, revealing maximum emission peaks at 455, 469, 473, 481, 488 and 480 nm, respectively (Fig. S9, ESI†). The majority of the aniline derivatives exhibited obvious red shifts in the solid state comparing to in the solution state. When the degree of aggregation was enhanced, well-organized and flatter conformations formed by necessity, resulting in red shifts of the emission peaks. As B2-A4 and B4-A4 were already bulky, their emission wavelengths did not show marked differences between the solid and solution states.
Aggregation-induced emission characteristics
The AIE properties of the aniline derivatives were explored. The derivatives all dissolved in miscible solvents consisting of water and THF with water contents ranging from 0 to 90 vol%. As shown in Fig. 3a, for B2-A2, a tiny enhancement in emission was observed when the content of water was less than 60 vol%. In contrast, when the content of water was over 60 vol%, the luminescence increased dramatically. Usually, in an AIE test, the point of mutation indicates the formation of nano-aggregates at that time.15 The luminescence intensity of B2-A2 solution containing 90 vol% water in THF increased by six times compared to that of B2-A2 solution in pure THF, showing typical AIE properties.16 The fluorescent images shown in the inset of Fig. 3a visually demonstrate the difference in emission of B2-A2 in THF/water mixtures containing 0 and 90 vol% water. More specifically, obvious aggregation did not occur when the content of water was below 70 vol%; thus, the PL intensity was nearly unchanged. When the content of water reached 70 vol%, aggregation occurred, resulting in a fast increase in PL intensity based on the RIM mechanism. Aggregation was convincingly demonstrated by the Tyndall phenomena and Mie effect in the UV absorption curve of B2-A2 at various proportions of THF and water (Fig. S10, ESI†).17 When the content of water reached 90 vol%, heavy aggregation prevented vast non-radiative transitions. On the other hand, the absorption capacity distinctly increased. These factors resulted in a huge increase in PL intensity. In addition, the locations of the emission bands for different water contents were similar (∼451 nm), indicating that B2-A2 was totally neutral, and no strong intermolecular interactions occurred during aggregation.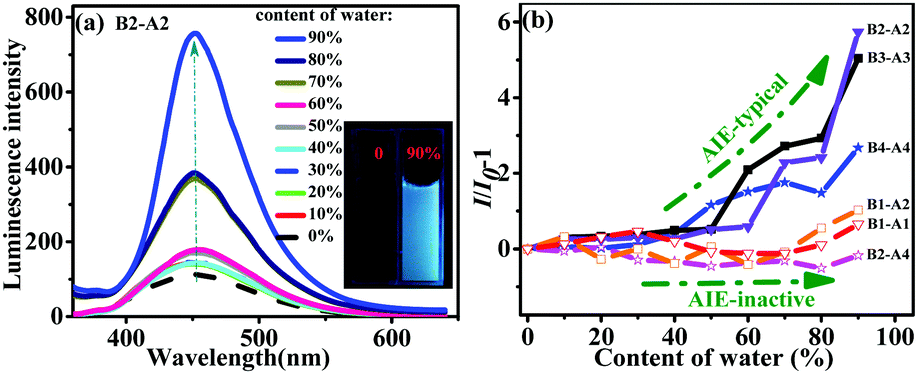 | ||
| Fig. 3 (a) Emission spectra of B2-A2 in THF/water mixtures under excitation at 335 nm (10 µM; EX slit: 5 nm; EM slit: 10 nm). Inset: Fluorescent images of B2-A2 in THF/water mixtures containing 0 and 90% water. (b) Plots of (I/I0) − 1 vs. water content for six aniline derivatives, where I0 is the luminescence intensity in pure THF. | ||
The emissions of B3-A3 and B4-A4 were similar to that of B2-A2, indicating typical AIE behaviors (Fig. S11d and e, ESI†). The turning points of B3-A3 and B4-A4 were observed at water contents of 50% and 40%, respectively, consistent with the increased molecular sizes of these derivatives. For B3-A3 and B4-A4, the variation in emission band locations at different water contents was small, with all bands located at approximately 466 and 476 nm. The slight change was mainly attributed to the lack of the sole naked NH moiety.
In contrast to emission phenomena of B1-A1, B1-A2 and B2-A4 showed mild fluorescence changes with increasing water percentage in THF (Fig. S11a–c, ESI†). The luminescence intensities of these derivatives in 90% water were almost identical to those in pure THF, indicating that AIE was inactive. Based on the change in peak wavelength, B1-A1 was a neutral molecule. In contrast, B1-A2 and B2-A4 exhibited huge variations in amplitude reaching 37 and 49 nm, respectively. Considering the AIE behaviors of the fully substituted anilines, we deduced that a higher density of naked NH moieties in aniline derivatives facilitates intermolecular interactions during aggregation, resulting in shifts in emission wavelengths and harmful effects to their AIE properties.
The fluorescent lifetimes and quantum yields of all aniline derivatives in both the solution and solid states were also tested and are presented in Fig. S12 and Table S2 (ESI†). For AIE-typical aniline derivatives (B2-A2, B3-A3 and B4-A4), their fluorescent lifetimes and quantum yields were both remarkably higher in the solid state compared to in the solution state, which is beneficial for real applications. For the AIE-inactive aniline derivatives (B1-A1, B1-A2 and B2-A4), due to the absence of adequate insertions to avoid nonradiative decay channels, their fluorescent lifetimes were distinctly lower in the solid state compared to in the solution state. The quantum yields of B1-A1, B1-A2 and B2-A4 were low in both the solution and solid states, suggesting AIE inactivity. These results indicate that the insertion number and chain length have notable effects on AIE.
As suggested by the AIE mechanism based on restricted intramolecular motion,18 in derivative B1-A1, the rotations of the four phenyl groups dissipate the excitation energy, resulting in weak emission in dilute solution. Free rotations cannot be prevented during aggregation due to the small molecular dimension and long ethylene–N principal chain, resulting in AIE inactivity. For B1-A2 and B2-A4, which have only unsaturated diphenyl moieties attached to the aniline chains, the number of insertions is too low to provide sufficient steric hindrance to restrain intramolecular motion and interaction during aggregation; thus, the channel for consuming the excitation energy cannot effectively be switched out. As a result, B1-A2 and B2-A4 show subdued emission in both the solution and aggregated states. For B2-A2, B3-A3 and B4-A4, the larger molecular sizes and sufficient number of insertions suppress free motion and intermolecular interactions, and the characteristic AIE phenomenon appears.
The AIE behaviors of all six aniline derivatives are summarized in Fig. 3b. Intuitively, multi-substituted aniline derivatives exhibited typical AIE characteristics, whereas mono-substituted aniline derivatives as well as B2-A4 were AIE-inactive. As the aniline oligomers (dimer, trimer and tetramer) are expressly ACQ molecules, we can conclude that increased diphenyl enamine substituents and successful ACQ–AIE conversion can be achieved with adequate insertions. In brief, these results revealed the notable effect of diphenyl enamine groups on the optical properties of aniline derivatives. The findings provide guidance for the modification of undesirable ACQ compounds with heteroatom chains.
Systematic study of aniline chain derivatives
Given the variety in insertion number and chain length, it is difficult to study the effects of individual factors on the ACQ–AIE process. Hence, based on monomers, dimers, trimers and tetramers, we developed a systematic plot to describe the effects of insertions for all aniline oligomers. Following the “chain-insertion” pattern, the effects of insertion on the luminescence behavior (fluorescence wavelength) and solid luminescence ability (fluorescence quantum yield) are generally illustrated in an X–Y scheme in Fig. 4. The performances of single-, partially and fully substituted aniline derivatives are shown by the orange (circle), purple (square) and blue (star) symbols, respectively.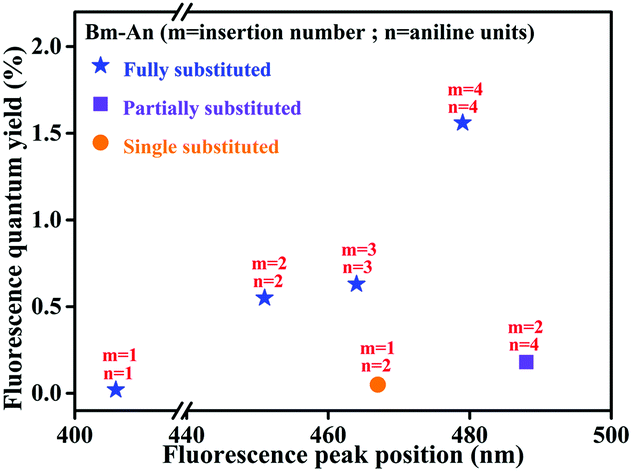 | ||
| Fig. 4 Systematic luminescence characteristics of all aniline derivatives (Bm-An: m = insertion number; n = aniline units). | ||
The AIE ability of the aniline derivative becomes stronger as the number of insertions increases. In other words, for fully substituted aniline derivatives, the fluorescence quantum yield increased from 0% to approximately 1.5% with increasing insertion number. In our linear system, more insertions corresponds to a more twisted conformation, which prevents π–π stacking and increases the potential for ACQ–AIE conversion. On the other hand, twisted conformations may be harmful to the effective conjugation length, especially in the aggregated state, which mainly determines the luminescent behaviors. In fact, for molecules with heteroatom chains, the extended conjugation lengths are easily saturated and disturbed as the chain length and number of insertions increase.19 The emission is blue-shifted with increasing number of insertions, which may be attributed to a decrease in planarity. Thus, insertion has opposite effects on solid luminescence ability and luminescence behavior during aggregation. Precisely controlling the degree of insertion may be an efficient way to achieve specific AIE behaviors in linear aniline systems.
Single crystal structural analysis
To better understand the photo-physical properties of aniline derivatives in the aggregated state, single-crystalline B2-A2 was obtained by the slow diffusion of its n-hexane/THF (2:1 v/v) solution. The crystallographic data can be obtained free of charge from the Cambridge Crystallographic Data Centre with serial number CCDC 1560810.† The single-crystal structure in Fig. 5a indicates an S-shaped chain with two π-connective diphenyl enamines. In this symmetric B2-A2 crystal, the torsion angle between adjacent aniline moieties is 67.2°, indicating a strong twisted conformation that restrains the free rotation of the phenyl and diphenyl enamine segments during aggregation.20 Moreover, some C–H⋯π hydrogen bonds exist between the phenyl rings and protons on the vinyl groups with distances of 3.4–3.7 Å. These bonds improve the stiffness of the entire molecule and partially inhibit free intramolecular rotation.21
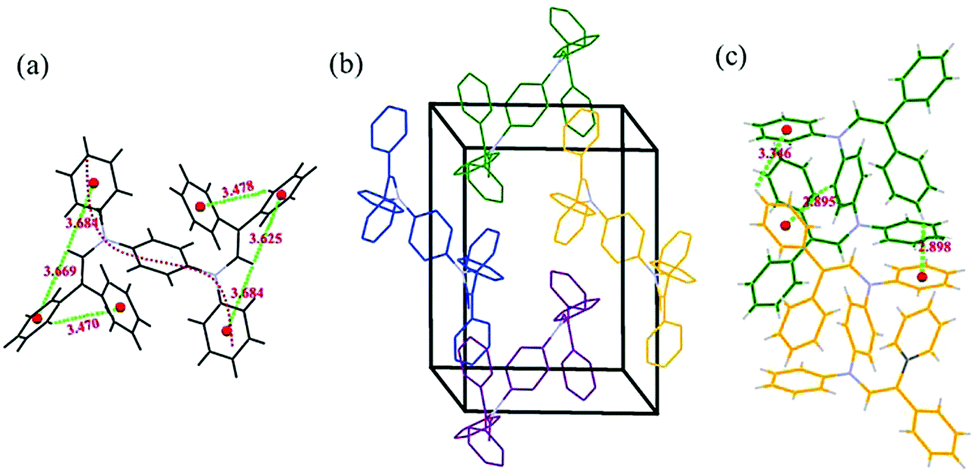 | ||
| Fig. 5 Intramolecular C–H⋯π interactions (a), packing structure (hydrogen atoms are deleted for clarity) (b), and intermolecular C–H⋯π interactions (c) of B2-A2. | ||
The packing structure of B2-A2 is given in Fig. 5b. Due to the loose molecular arrangement in B2-A2, planar intermolecular interactions are hampered, and high congestion results from the multiple diphenyl enamine modifications. In addition, C–H⋯π hydrogen bonds with lengths of 2.8–3.4 Å are formed between neighboring aniline derivative molecules (Fig. 5c). These weak forces further assist in restraining molecular motion, which reduces the non-radiative deactivation of excitons and contributes to the AIE effect.22 These results suggest that the twisting of main chains plays a key role in ACQ–AIE conversion, and the presence of suitable interpositions that prevent the planar stacking of the main chain may strengthen the AIE effect.
DFT calculations
To further explore the effect of insertion on ACQ–AIE conversion, we studied the orbital energy levels and optimum configurations of the aniline derivatives (Fig. S13 (ESI†) and Fig. 6) using density functional theory (DFT) at the B3LYP/6-31G(d) level in the Gaussian 09 package.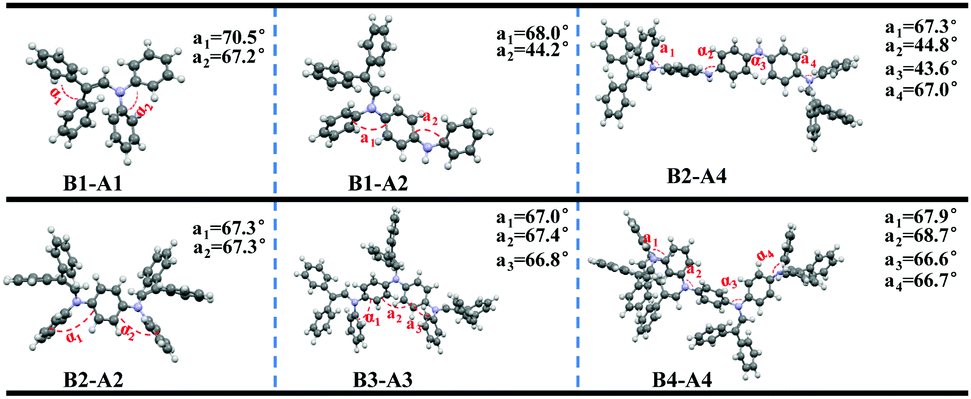 | ||
| Fig. 6 Optimal structures of the six diphenyl enamine-substituted aniline derivatives (B1-A1, B1-A2, B2-A2, B3-A3, B2-A4 and B4-A4; α1, α2, α3 and α4: dihedral angles between adjacent benzene rings in the aniline chain). | ||
The electronic densities of B1-A1 in both the HOMO and LUMO levels are equally distributed, suggesting that fully effective conjugation has been achieved in short-length aniline. A twisted structure formed, and the torsion angle between adjacent rings along the aniline chain reached 70.5°; however, the axis orientation has actually resulted in a flexible carbon and nitrogen chain owing to the comparable side groups. In fact, the steric effect is not adequate for the flexible carbon and nitrogen chain, resulting in AIE inactivity.
The electron clouds of B1-A2 and B2-A2 in the HOMO level are both spread over the entire molecules, whereas those in the LUMO level are predominantly localized at the introduced diphenyl enamine parts, indicating a certain degree of charge transition. Furthermore, the conformation of B2-A2 is crowded, preventing adverse intermolecular functions and contributing to the AIE effect. However, some aniline rings still exist nakedly in B1-A2 and tend to interact with the π planes of other nearby molecules, resulting in more non-radiative pathways. Thus, the steric effect of B1-A2 is insufficient for substantial changes to the ACQ properties, resulting in AIE activity.
We then studied the diphenyl enamine-substituted aniline trimer derivative B3-A3. As expected, the conformation of B3-A3 is strongly twisted; α1, α2 and α3 all reach 67°, resulting in strong AIE. In contrast to the A1 and A2 derivatives, the electron clouds of B3-A3 are not able to spread across the entire segments, and only approximately two diphenyl enamine groups work in both the HOMO and LUMO levels. This explains the slight change in aniline derivatives with increased chain length and insertion number, due to the existence of conjugate validity, which is totally different from the AIEgens as a result of a “core-side pattern.”
Due to the existence of continuous NH moieties, the structure of the tetramer derivative B2-A4 is relatively planar, and the peripheral steric hindrance may not be sufficient to reverse intrinsic planar interactions. In addition, the electron clouds of the NH moieties in both the HOMO and LUMO are totally separated, leading to distinct TICT phenomena. These results agree with the large bathochromic shift (49 nm) observed in the AIE test. The structure of B4-A4 is sufficiently twisty to ensure AIE behavior. Moreover, as for B3-A3, the electron clouds of B4-A4 cannot spread across the entire molecule in both the HOMO and LUMO. Considering the electron clouds B2-A2 and B3-A3, the large number of introduced diphenyl enamine groups are not able to drastically increase the conjugacy of the molecules. The effective conjugate lengths play an important role, as indicated by the absorption peaks of B2-A2, B3-A3 and B4-A4 at 332, 342 and 353 nm, respectively.
We compared the structures of aniline derivatives in the geometrical and excited states using B1-A1, B1-A2 and B2-A2 as representative molecules (Fig. S14, ESI†). For B1-A1, the angles between adjacent benzene rings are smaller in the excited state than in the ground state. The planarity of the entire B1-A1 molecule benefits from intermolecular π–π stacking and strengthened non-radiative transition, which is harmful for AIE. For B1-A2, the optimized structures in both the ground state and excited state were similar, with some distinctions in angles between adjacent benzene rings. The naked NH moiety was detrimental to the AIE effect in B1-A2. For B2-A2, two benzylbenzene insertions spread over the same side of aniline chain in the ground state. In contrast, in the excited state, the two benzylbenzene insertions were distributed bilaterally in the aniline chain. The helical configuration was favorable for impeding non-radiative energy dissipation, resulting in typical AIE behavior. These results indicate that insertions significantly affect the structures of aniline derivatives, resulting in different emission behaviors.
BSA detection
The level of serum albumin (SA) is a pivotal parameter of human health. As a homologous protein of SA, bovine serum albumin (BSA) is comprehensively researched as a model protein. Hence, efficient methods of BSA detection and quantification are particularly important. Unlike the undesirable ACQ phenomenon, the aggregation of AIE dyes improves fluorescence, which expands the effective range of a probe.23 Therefore, the typical AIE molecules B2-A2 and B3-A3 were applied for BSA detection and quantification.The selectivity of B2-A2 towards BSA was evaluated in a solution of PBS buffer (pH = 7.4) and THF (1/9, v/v). The standard BSA solution was prepared with a concentration of 670 mg ml−1, and 5 µl BSA solution was added in each of 14 experimental rounds. Thus, BSA was detected after the addition of volumes ranging from 5 to 70 µl (concentrations ranging from approximately 1.68–23.5 mg ml−1). With increasing BSA concentration together with the slightly decreasing polarity of the solvent mixture, the fluorescent intensity of the B2-A2 solution increased gradually. The enhancement factor reached approximately 79 in the presence of BSA at a concentration of 23.5 mg ml−1. This enhancement was attributed to mutual interactions between oleophilic B2-A2 and the hydrophobic cavities of BSA.24 These reactions hindered the intramolecular rotations of B2-A2, which in turn hindered non-radiative transitions and further enhanced fluorescence. Compared to the blank control, apparent aggregation was observed when 50 µl of BSA solution was added to the B2-A2 solution (inset of Fig. 7). Meanwhile, an apparent enhancement in fluorescence was observed by the naked eye under 365 nm ultraviolet light (inset of Fig. 7).
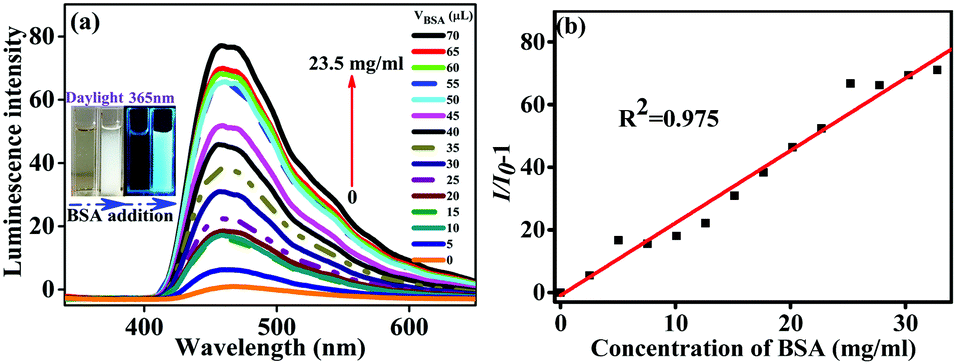 | ||
| Fig. 7 (a) Emission spectra of B2-A2 solutions (10 µM; excitation wavelength: 335 nm; EX slit: 5 nm; EM slit: 10 nm) containing various amounts of BSA. (b) Fitted linear curve of luminescence changes in response to different amounts of BSA. | ||
Noticeably, in many previous studies, the fluorescence peaks changed significantly during BSAS detection.25 In this study, the aggregation of B2-A2 and BSA did not result in peak displacement. The lack of covalent interactions and electrostatic interactions due to the neutrality of B2-A2 may explain this difference; this explanation was further confirmed by the lack of response to changes in pH and ions. In other words, the detection of BSA by aniline derivatives is not disturbed by micro-molecules and the environmental conditions. Moreover, based on the least-squares method, a large number of data points were fitted. A linear relationship between the fluorescence enhancement (X) and BSA solution concentration (Y) was observed: Y = 3.48X − 0.90. In consideration of the slight influence of increased water content, the linear coefficient R2 of 0.975 was sufficiently high. The detection limit for B2-A2 was measured to be approximately 3.56 µg l−1 (3δ per slope) for a precise turn-on bioprobe for the quantitative detection of BSA.
Furthermore, using a similar procedure, solutions of B3-A3 were also applied to detect BSA. As shown in Fig. S15 (ESI†), B3-A3 also exhibited favorable detection properties for BSA. The linear regression coefficient R2 reached 0.962, and the detection limit was approximately 4.78 µg l−1. These results reveal that aniline derivatives with typical AIE properties are preeminent detection molecules with high sensitivity and strong linear relationships.
We also measured the responses of AIE-inactive compounds (B1-A1, B1-A2 and B2-A4) towards BSA (Fig. S16, ESI†). Although the fluorescence intensities of the AIE-inactive compounds (B1-A1, B1-A2 and B2-A4) first increased with the addition of BSA, the emission responses were irregular. When the concentration of BSA reached certain values (1.85, 8.38 and 11.73 mg ml−1 for B1-A1, B1-A2 and B2-A4, respectively), the fluorescence intensities decreased. These results further demonstrate the advantages of B2-A2 and B3-A3 for the detection of BSA.
The time-dependent fluorescence spectra of B2-A2 and B3-A3 were monitored in response to different BSA concentrations. Due to the limited manipulation time, the time gap between adjacent tests was at least 6 s, and the entire measurement lasted 48 s. As shown in Fig. S17 (ESI†), as soon as BSA was added, the maximum emission intensity was immediately reached and maintained over an extended period. In contrast to a typical colorimetric method with a rapid detection time of approximately 10–55 s,26 this reaction was completed within 10 s, showing extremely fast response. This fast detection of BSA can be applied to some emergency treatments, such as hemorrhea and burns.
Selecting cholesterol, carbamide, glucose, L-arginine and γ-globulin as interfering components,27 the specificity of B2-A2 and B3-A3 for BSA in a simulated serum environment was further studied. As shown in Fig. S18 (ESI†), all micro-molecules did not interact with B2-A2 and B3-A3, as indicated by the negligible variation in the maximum emission intensities. In addition, the fluorescent intensities of B2-A2 and B3-A3, which were enhanced by BSA, were not affected by the interfering micro-molecules. As soon as γ-globulin was added into the B2-A2 and B3-A3 solutions, large precipitates appeared and quickly settled at the bottom of the cell. Thus, the B2-A2 and B3-A3 solutions cannot be used to detect γ-globulin. When all compounds were mixed, despite the effect of γ-globulin precipitation, the fluorescent intensities of B2-A2 and B3-A3 were still enhanced by 50%. These results indicate that B2-A2 and B3-A3 have the ability to detect BSA in complex environments. Further research on protein detection using fully substituted anilines is underway.
Conclusions
In summary, a series of aniline derivatives substituted with diphenyl enamine were successfully synthesized based on aniline monomer, dimer, trimer and tetramer examples. Meticulous control of insertions in the aniline oligomer chains can achieve successful ACQ–AIE conversion with varying numbers of nonplanar diphenyl enamine groups. Fully substituted oligoanilines exhibited typical AIE characteristics, while partially substituted derivatives were AIE-inactive. UV-vis and fluorescence studies along with DFT calculations offered detailed understandings of the effects of insertion on the intrinsic photo-physical properties of the derivatives. The luminescence wavelength and solid fluorescence of the aniline derivatives could be effectively regulated by chain length and insertion. The results are important for understanding the effects of insertion and chain length on the AIE characteristics of linear heteroatom chains, which can easily be comprehended macroscopically by applying the oligomer approach. This study will hence encourage further work to understand the effects of conjugated blocks on the AIE behaviors of heteroatom chain molecules and to design additional AIE systems. Moreover, fully substituted anilines show a quantitative turn-on fluorescence response towards BSA under physiological conditions. As probes, these aniline derivatives have detection limits in the order of micrograms per liter. Thus, these molecules show great potential for use as fluorescent probes in BSA sensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21574003) for financial support.Notes and references
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (c) H. Wang, D. Chen, Y. Zhang, P. Liu, J. Shi, X. Feng, B. Tong and Y. Dong, J. Mater. Chem. C, 2015, 3, 7621 RSC; (d) Z. Zhao, S. Chen, J. W. Y. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221 RSC; (e) H. Nie, K. Hu, Y. Cai, Q. Peng, Z. Zhao, R. Hu, J. Chen, S. J. Su, A. Qin and B. Z. Tang, Mater. Chem. Front., 2017, 1, 1125 RSC; (f) X. Q. Zhang, X. Y. Zhang, B. Yang and Y. Wei, Chin. J. Polym. Sci., 2014, 32, 871 CrossRef CAS; (g) S. Xu, Y. Yuan, X. Cai, C. J. Zhang, F. Hu, J. Liang, G. Zhang, D. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824 RSC; (h) X. Zhang, K. Wang, M. Liu, X. Zhang, L. Tao, Y. Chen and Y. Wei, Nanoscale, 2015, 7, 11486 RSC; (i) K. Wang, H. Lu, B. B. Liu and J. Yang, Eur. Polym. J., 2018, 101, 225 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740 RSC; (b) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 126, 2151 CrossRef; (c) J. Yang, Y. Gao, T. Jiang, W. Liu, C. Liu, N. Lu, B. Li, J. Mei, Q. Peng and J. Hua, Mater. Chem. Front., 2017, 1, 1396 RSC; (d) G. F. Zhang, T. Chen, Z. Q. Chen, M. P. Aldred, X. Meng and M. Q. Zhu, Chin. J. Chem., 2015, 33, 939 CrossRef CAS; (e) T. Yu, D. Ou, Z. Yang, Q. Huang, Z. Mao, J. Chen, Y. Zhang, S. Liu, J. Xu, M. R. Bryce and Z. Chi, Chem. Sci., 2017, 8, 1163 RSC; (f) K. Wang, J. Yang, C. Gong and H. Lu, Faraday Discuss., 2017, 196, 43 RSC.
- (a) H. Lu, Y. Zheng, X. Zhao, L. Wang, S. Ma, X. Han, B. Xu, W. Tian and H. Gao, Angew. Chem., Int. Ed., 2016, 128, 163 CrossRef; (b) S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797 CrossRef CAS PubMed; (c) Z. Zhao, P. Lu, J. W. Y. Lam, Z. Wang, C. Y. K. Chan, H. H. Y. Sung, I. D. Williams, Y. Ma and B. Z. Tang, Chem. Sci., 2011, 2, 672 RSC.
- (a) F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 127, 14700 CrossRef; (b) Z. Peng, Z. Wang, B. Tong, Y. Ji, J. Shi, J. Zhi and Y. Dong, Chin. J. Chem., 2016, 34, 1071 CrossRef CAS; (c) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084 CrossRef CAS PubMed; (d) L. Zong, Y. Xie, C. Wang, J. R. Li, Q. Li and Z. Li, Chem. Commun., 2016, 52, 11496 RSC; (e) L. Dong, G. Shang, J. Shi, J. Zhi, B. Tong and Y. Dong, J. Phys. Chem. C, 2017, 121, 11658 CrossRef CAS; (f) B. Guo, X. Cai, S. Xu, S. M. A. Fateminia, J. Liu, J. Liang, G. Feng, W. Wu and B. Liu, J. Mater. Chem. B, 2016, 4, 4690 RSC; (g) M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485 RSC; (h) X. Y. Shen, Y. J. Wang, H. Zhang, A. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2014, 50, 8747 RSC; (i) Z. Peng, Y. Ji, Z. Huang, B. Tong and J. Shi, Mater. Chem. Front., 2018, 2, 1175 RSC.
- S. Kim, B. Kim, J. Lee, H. Shin, Y.-I. Park and J. Park, Mater. Sci. Eng., R, 2016, 99, 1 CrossRef.
- (a) C. J. Bhongale, C. W. Chang, C. S. Lee, E. W. G. Diau and C. S. Hsu, J. Phys. Chem. B, 2005, 109, 13472 CrossRef CAS PubMed; (b) B. K. An, S. H. Gihm, J. W. Chung, C. R. Park, S. K. Kwon and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 3950 CrossRef CAS PubMed.
- (a) W. Hu, Q. Yan and D. Zhao, Chem. – Eur. J., 2011, 17, 7087 CrossRef CAS PubMed; (b) Y. Liu, X. Chen, Y. Lv, S. Chen, J. W. Y. Lam, F. Mahtab, H. S. Kwok, X. Tao and B. Z. Tang, Chem. – Eur. J., 2012, 18, 9929 CrossRef CAS PubMed.
- P. Chen, R. A. Lalancette and J. Frieder, J. Am. Chem. Soc., 2011, 133, 8802 CrossRef CAS PubMed.
- (a) L. Cao, C. Gong and J. Yang, Electrochim. Acta, 2016, 192, 422 CrossRef CAS; (b) Q. Chen, X. Yu, Z. Pei, Y. Yang, Y. Wei and Y. Ji, Chem. Sci., 2016, 8, 724 RSC; (c) Y. Yan, N. Sun, F. Li, X. Jia, C. Wang and D. Chao, ACS Appl. Mater. Interfaces, 2017, 9, 6497 CrossRef CAS PubMed; (d) C. Gong, J. Yang, L. Cao and Y. Gao, ChemElectroChem, 2017, 4, 521 CrossRef CAS.
- Y. Wang, H. D. Tran, L. Liao, X. Duan and R. B. Kaner, J. Am. Chem. Soc., 2010, 132, 10365 CrossRef CAS PubMed.
- R. Paspirgelyte, J. V. Grazulevicius, S. Grigalevicius and V. Jankauskas, Des. Monomers Polym., 2009, 12, 579 CrossRef CAS.
- S. Urgaonkar, J. H. Xu and J. G. Verkade, J. Org. Chem., 2003, 68, 8416 CrossRef CAS PubMed.
- L. Chen, C. Zhang, G. Lin, H. Nie, W. Luo, Z. Zhuang, S. Ding, R. Hu, S. J. Su, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2775 RSC.
- M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834 CrossRef CAS PubMed.
- Y. Zhang, J. H. Wang, J. Zheng and D. Li, Chem. Commun., 2015, 51, 6350 RSC.
- (a) L. Li, M. Chen, H. Zhang, H. Nie, J. Z. Sun, A. Qin and B. Z. Tang, Chem. Commun., 2015, 51, 4830 RSC; (b) Y. X. Li, X. F. Yang, J. L. Miao and G. X. Sun, J. Phys. Chem. C, 2016, 120, 21722 CrossRef CAS.
- (a) W. Fan, B. Sun, J. Ma, X. Li, H. Tan and L. Xu, Chem. – Eur. J., 2015, 21, 12947 CrossRef CAS PubMed; (b) Y. Qian, S. Li, Q. Wang, X. Sheng, S. Wu, S. Wang, J. Li and G. Yang, Soft Matter, 2012, 8, 757 RSC.
- (a) N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349 CrossRef CAS PubMed; (b) Q. Zhao, X. A. Zhang, Q. Wei, J. Wang, X. Y. Shen, A. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2012, 48, 11671 RSC; (c) C. F. A. Gomez-Duran, R. Hu, G. Feng, T. Li, F. Bu, M. Arseneault, B. Liu, E. Pena-Cabrera and B. Z. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 15168 CrossRef CAS PubMed.
- J. Li, A. Terec, Y. Wang, H. Joshi, Y. Lu, H. Sun and M. C. Stuparu, J. Am. Chem. Soc., 2017, 139, 3089 CrossRef CAS PubMed.
- Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, L. Ji, Z. Zhu, C. K. W. Jim, G. Yu, Q. Li, Z. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70 RSC.
- B. Chen, H. Nie, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 7541 RSC.
- W. Yang, C. Li, M. Zhang, W. Zhou, R. Xue, H. Liu and Y. Li, Phys. Chem. Chem. Phys., 2016, 18, 28052 RSC.
- S. Samanta, S. Halder and G. Das, Anal. Chem., 2018, 90, 7561 CrossRef CAS PubMed.
- L. Wang, L. Yang and D. Cao, Sens. Actuators, B, 2015, 221, 155 CrossRef CAS.
- J. Tong, T. Hu, A. Qin, J. Z. Sun and B. Z. Tang, Faraday Discuss., 2017, 196, 285 RSC.
- B. Deng, Y. Wang, P. Zhu, X. Xu and X. Ning, Anal. Chim. Acta, 2010, 683, 58 CrossRef CAS PubMed.
- N. Kang, S. Kasemsumran, Y. A. Woo, H. J. Kim and Y. Ozaki, Chemom. Intell. Lab. Syst., 2006, 82, 90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and spectral details are provided. X-ray data have been deposited in the CCDC 1560810. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c8qm00543e |
| This journal is © the Partner Organisations 2019 |