
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiparameter antibiotic resistance detection based on hydrodynamic trapping of individual E. coli†
Giampaolo
Pitruzzello
 *a,
Stephen
Thorpe
b,
Steven
Johnson
c,
Adrian
Evans
d,
Hermes
Gadêlha
e and
Thomas F.
Krauss
a
*a,
Stephen
Thorpe
b,
Steven
Johnson
c,
Adrian
Evans
d,
Hermes
Gadêlha
e and
Thomas F.
Krauss
a
aDepartment of Physics, University of York, YO105DD, York, UK. E-mail: gp744@york.ac.uk
bDepartment of Biology, University of York, YO105DD, York, UK
cDepartment of Electronic Engineering, University of York, YO105DD, York, UK
dYork General Hospital, Wigginton Road, YO318HE, York, UK
eDepartment of Mathematics, University of York, YO105DD, York, UK
First published on 14th March 2019
Abstract
There is an urgent need to develop novel methods for assessing the response of bacteria to antibiotics in a timely manner. Antibiotics are traditionally assessed via their effect on bacteria in a culture medium, which takes 24–48 h and exploits only a single parameter, i.e. growth. Here, we present a multiparameter approach at the single-cell level that takes approximately an hour from spiking the culture to correctly classify susceptible and resistant strains. By hydrodynamically trapping hundreds of bacteria, we simultaneously monitor the evolution of motility and morphology of individual bacteria upon drug administration. We show how this combined detection method provides insights into the activity of antimicrobials at the onset of their action which single parameter and traditional tests cannot offer. Our observations complement the current growth-based methods and highlight the need for future antimicrobial susceptibility tests to take multiple parameters into account.
Introduction
The excessive, inappropriate and often unnecessary use of antibiotics has dramatically accelerated the development of bacterial resistance.1,2 A key reason for this misuse, especially for the overuse of broad-spectrum antibiotics, is the lack of rapid and precise tests available to pinpoint specific antibiotics for a given infection. Most antimicrobial susceptibility tests (ASTs) are currently based on defining susceptibility as the inhibition of bacterial growth in the presence of antibiotics. The main limitation of these techniques is the need for detecting division at the community level, given that a minimum concentration of bacteria is needed to produce a detectable signal. For example, the traditional and still most commonly used clinical AST is based on the agar diffusion test or the broth dilution technique, both of which take 24–48 h, which is too slow for informing the correct choice of antibiotic in the early and often critical stages of an infection.3,4 Furthermore, culturing often favours certain bacteria over others, so is biased.5 Genotyping can also lead to biases, as the absence of specific resistance genes or mutations does not guarantee susceptibility to a specific antibiotic.6Since the majority of ASTs measures susceptibility of large bacterial populations, they average over billions of organisms. Thus, the response of individual bacteria is lost, which potentially contains very relevant information. Collecting single-cell statistically-rich data is crucial for unveiling underlying distributions within a bacterial population; indeed, there is growing evidence for heterogeneity and differentiation into sub-populations that can adopt specific strategies for developing resistance, such as acting dormant or modifying their growth rate.7 Hence, there is a need for new ways of defining and characterising susceptibility that consider the heterogeneity of bacterial populations and that offer rapid analysis. Phenotyping susceptibility6,8 at the single-cell level is emerging as a possible candidate to meet these requirements.7,9,10
The interest towards single bacteria analysis has recently gained significant attention, especially in the context of antimicrobial resistance.7,8 An important requirement for single-cell phenotyping is the availability of an immobilization technique that allows localising a significant number of single bacteria at specific locations over time. One of the first platforms proposed for single bacteria trapping and analysis is the so-called mother machine.11 Here, a single bacterium (the pole mother cell) is localised in a narrow dead-end microfluidic channel which constricts growth along a single direction in order to follow multiple generations. This configuration is advantageous for studying cell aging and proliferation over extended periods of time.12–14 However, the accumulation of secreted products and the increased shear along the channel might affect bacteria phenotype and division time.15 Notably, a slight variant of this platform has recently been applied to the study of antimicrobial resistance by measuring the differential growth rate of E. coli upon exposure to antibiotics in multiple dead-end channels.16
Single bacteria localisation can also be achieved through a variety of other techniques, such as optical17,18 or acoustic19 trapping, hydrogel or agarose immobilisation assays,4 droplet microfluidics,20–22 dielectrophoresis23,24 or by antibodies tethered on the sensor surface and binding to proteins on the bacteria membrane.25,26
In order to achieve the immobilisation of hundreds of single bacteria over time, here we employ hydrodynamic trapping9,10 within micro wells because it is label-free, it can readily be integrated into a microfluidic assay and it is highly scalable, thereby enabling the creation of high-density trap arrays. Compared to the previously mentioned trapping methods, hydrodynamic trapping affords single cell mechanical immobilisation over time at specific, regular and known locations. On one hand, the trapping principle itself (i.e. mechanically impeding bacteria swimming), eliminates the need for molecular recognition elements (i.e. antibodies which are costly and may not be available for a particular strain), the generation of droplets or external equipment such as a laser, for either trapping or droplets readout, or acoustic transducers. On the other hand, arraying single cells at regular positions facilitates automation in the detection and data analysis, which is not straightforward to control with antibodies tethering or agarose immobilisation, for example. Furthemore, this method offers the possibility of continuous medium recirculation, which ensures that the trapped cells are exposed to a constant environment over time, unlike the mother-machine inspired devices. Finally, daughter cells following division are removed from the trapping areas, thus ensuring truly single cell assay. However, hydrodynamic trapping suffers from relatively poor trapping efficiency, as we will detail later on. The method has been pioneered for and widely applied to the study of eukaryotic cells, exploiting either micro-wells structures acting as mechanical barriers9,10,27 or the difference in hydraulic resistance between different fluidic paths to drive cells towards small openings.28–30 However, it has not been applied as extensively to the study of single bacterial cells, with only a few examples available in the relevant literature.31,32
For most of the aforementioned bacteria trapping platforms, growth is employed as a signature of susceptibility to antibiotics, either by direct imaging4,16 or by indirect measurements such as fluorescence21,22 or Raman analysis.23 Recent studies have instead focused on motility as the signature of bacterial susceptibility.33–35 Motility is a fundamental property of many types of bacteria which plays a crucial role in the early stages of infection.36,37 In contrast to a growth-based assay, motility could represent a more rapid way of assessing bacterial viability upon drug administration and to classify resistant strains. Highly sensitive methods for detecting motility have included the use of an AFM cantilever to record motility-induced vibrations33,34,38 and fluctuations of the voltage drop across a micro channel.35 However, these techniques usually interrogate hundreds of bacteria in parallel, which makes them unable to follow the critical dynamics response of individual organisms. The effect of antibiotics on the motility at the single-cell level, has so far only been studied using antibody tethering in conjunction with surface plasmon resonance imaging (SPRi).26 All of these tests use motility as the only signature to determine susceptibility, while the response of bacteria to an antibiotic is a multiparameter problem. To this purpose, we also exploit bacterial morphology as a second indicator of response to antibiotics, which has already been shown to be a suitable parameter to detect susceptibility.4,39 However, to the best of our knowledge, the combined analysis has never been performed at the single-cell level before.
Our assay consists of arrays of cup-shaped structures acting as mechanical barriers to trap live bacteria flowing in a microfluidic channel. The trap dimensions are designed to fit a single E. coli per trap in most cases, as illustrated in Fig. 1. The hydrodynamic interaction between the bacteria and such traps is shown to carry rich information about bacterial behaviour, including sensitivity to changes in cell phenotype in response to environmental challenges, including antibiotic action. In particular, we have identified three signatures of the bacterial response to such challenges: the fraction of bacterial population entering the traps (hydrodynamic trapping efficiency), the amplitude of the movement of each bacterium within the trap, and the bacterial morphology (length of the bacterium). We demonstrate that these signatures, which are dependent on both motility and morphology, can be used to successfully classify resistant and susceptible strains of E. coli to different antibiotics in about an hour from spiking the culture. The method also provides useful insights into the antibiotic mode of actions and their effect of bacterial viability over time, right at the onset of their action. Our results are then correlated with a traditional microdilution assay in order to directly link our phenotypic signatures to a standard method of detecting susceptibility.
 | ||
| Fig. 1 (a) Sketch of the hydrodynamic trap array. Traps (in green) are fabricated in SU8, an electron beam negative resist, on a glass slide, and are sealed with a PDMS channel to allow injection of the bacterial solution. (b) Phase contrast image of a section of a microfluidic channel and trap array, some of which are populated with bacteria. (c) Atomic force microscopy (AFM) image of a single trap on a microscope slide. | ||
Results
A preliminary set of experiments is conducted as a control to prove the ability of the assay to discern motility at the single bacteria level. In particular, we have studied the behaviour of two different strains of E. coli in different viability conditions. We have used wild type E. coli MG1655 (colour-coded in green) as a motile peritrichous strain, meaning that is has multiple flagella (four on average) arranged randomly over the cell. In addition, we ran trapping assays using a non-motile E. coli strain, BW25113, which has a reduced expression of flagella genes and is thus unable to swim40 (colour-coded in orange). BW25113 is then exposed to heat to kill the cells and provide a control group.Intensity traces and evaluation of bacterial motility
Videos of the trapping experiments are recorded and subsequently post processed by using a custom MATLAB script. In particular, the average pixel intensity (or luminance) over each trap area is traced over time (see movie S1† for an illustration of the operation principle). Typical traces illustrating the time variation are shown in Fig. 2(a) and (b). The presence of bacteria causes an abrupt change in the intensity across the trap area, while motion results in increased fluctuations due to oscillations of constricted bacteria. We quantify this degree of motility by normalising the standard deviation of the trapped bacterium to the background noise calculated for the empty trap (bacterial motility = σtrapped/σempty). For example, a motility of 10 means that the standard deviation produced by the trapped bacterium is ten times greater than the background noise. Fig. 2(a) shows a typical example of a trapped motile E. coli MG1655, while 2(b) illustrates a typical non-motile E. coli BW25113. The difference in the trace noise is very apparent. Fig. 2(c) and (d) show micrographs of an empty and filled trap respectively.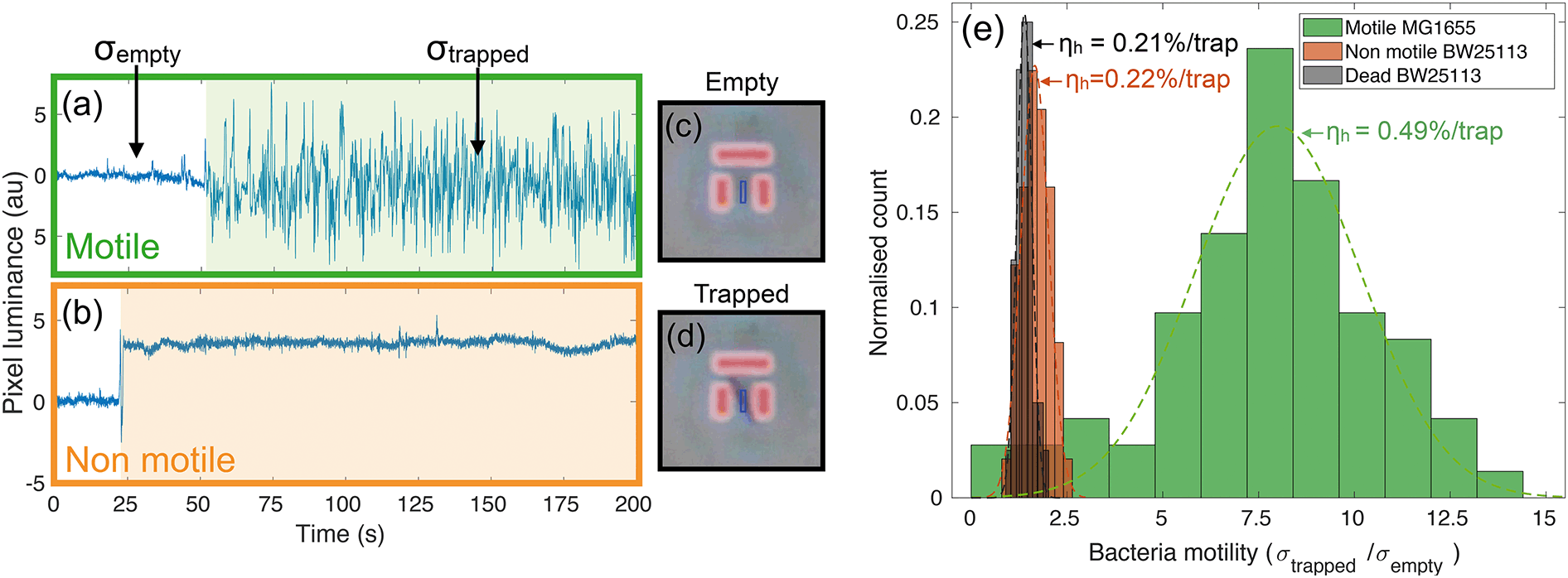 | ||
| Fig. 2 Typical traces from motile (a) and non-motile (b) strains together with phase contrast micrographs of an empty (c) and a filled (d) trap. The change in brightness (or luminance) is a signature of bacterial trapping, while variance is indicative of motion inside the trap. The light shaded areas in (a) and (b) highlight the length of time that bacteria remain in the trap. The standard deviation σtrapped is calculated over this time frame, while σempty is referred to the empty trap. The blue boxes in panels (c) and (d) represent the area over which the average pixel intensity is calculated. Panel (e) shows the distributions of motility for motile (MG1655), non-motile (BW25113) and dead bacteria strains along with the measured trapping efficiencies per trap. | ||
We measured bacterial motility for both strains in independent trapping experiments. In particular, three different experiments were conducted with the motile E. coli MG1655 strain and two with the non-motile BW25113, each consisting of three 5-minute-long videos being recorded and analysed. Fig. 2(e) shows the distributions of motility values for the different conditions, along with the measured values of trapping efficiency ηh (see next paragraph). The distributions are fitted to gaussian curves in order to retrieve an average motility and the relative spread. Motile E. coli MG1655 (green data in Fig. 2(e)) showed an average motility of (8.0 ± 0.4), while the non-motile BW25113 (orange data in Fig. 2(e)) showed a motility of (1.7 ± 0.1). We then repeated the trapping experiment with dead bacteria that had been exposed to a heat treatment of 60 °C for 2 hours. The relative distribution is represented by the black data in Fig. 2(e), which produced an average motility for dead bacteria of (1.4 ± 0.1). This shows that a consistent measurement of motility can be obtained with our trapping assay, which allows us to discriminate motile from non-motile cells. Interestingly, we can also distinguish the alive, non-motile BW25113 from the same strain after heat treatment. The former shows a slightly higher and statistically relevant signal than the dead case, which is likely due to motility mechanisms that do not require the active use of flagella, such as gliding or twitching.41,42
Hydrodynamic trapping efficiency
The design principle of such hydrodynamic traps is optimised to maximise the fluid flow through them.9,31,32 However, the fluidic resistance is always much higher and the fluid velocity much lower inside the traps compared to the surrounding regions.31 The majority of fluidic streamlines thus diverts from the interior regions of the trap because of the non-slip boundary condition at each interface.43 This effect is qualitatively illustrated in the insets of Fig. 3, which show the fluid velocity with the associated streamlines obtained from a 2D FEM simulation in COMSOL Multiphysics. Thus, the trapping efficiency of such assays is relatively low compared to differential-pressure hydrodynamic trapping.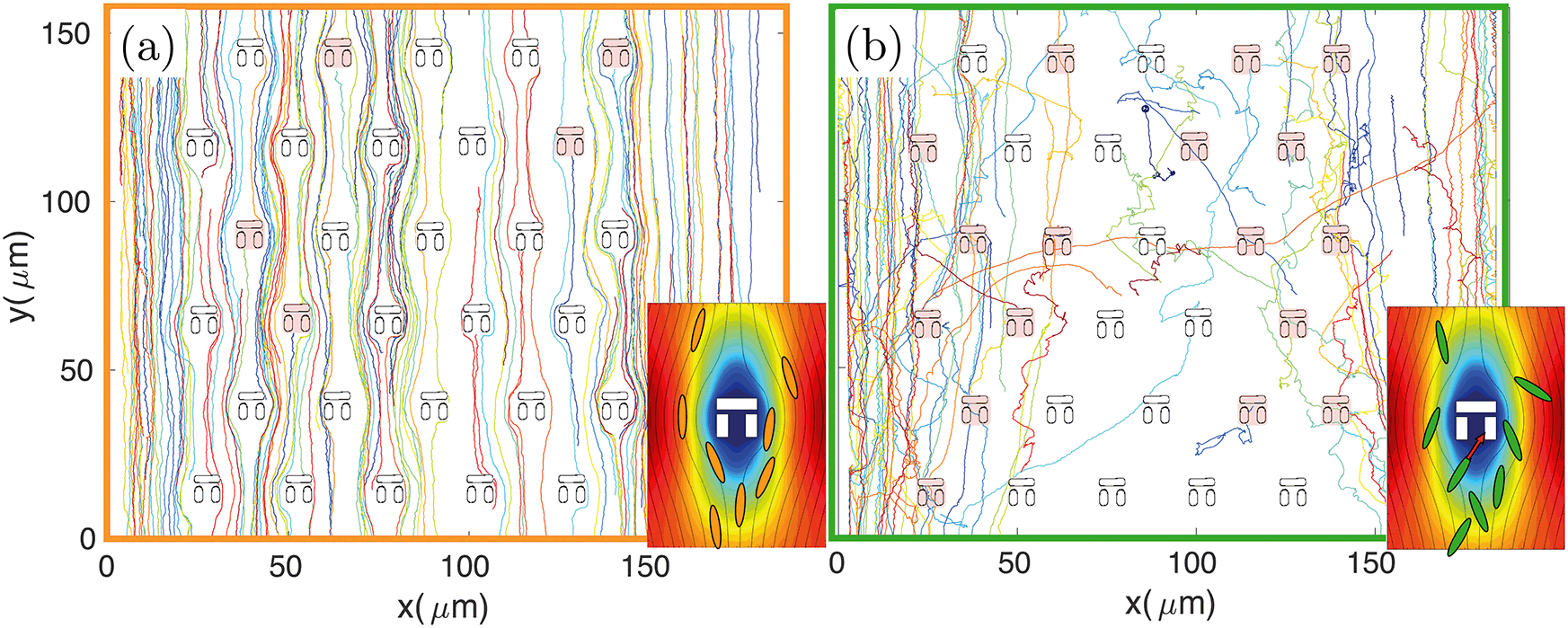 | ||
| Fig. 3 Typical trajectories for a non-motile (a) and a motile strain (b) obtained by tracking E. coli BW25133 and MG1655, respectively. The trap layout is superimposed and those traps that have been filled by one or more bacteria are shaded in red. Non-motile trajectories follow the streamlines of the fluid flow, which tend to avoid the stagnation regions, while trajectories of motile bacteria are more segmented, allowing more traps to be filled. The insets show the fluid velocity field and associated streamlines obtained by a FEM simulation in COMSOL Multiphysics, illustrating the streamlines diverting from the inner regions of the traps. | ||
For these reasons the number of trapped bacteria is strictly linked to bacterial motility. A passive object in the flow is less likely to enter any of the traps as it will simply follow the fluidic streamlines. The only chance for a non-motile organism to get trapped is to happen to flow along one of the few streamlines going through a trap. In contrast, a swimming organism has an increased probability of getting trapped because it can actively move across streamlines, due to its rheotactic ability44 as well as because swimming naturally increases the frequency of encountering a trap. This behaviour is qualitatively illustrated in Fig. 3 where bacteria are tracked over time. For the non-motile strain BW25113 (panel (a) and inset), bacterial trajectories follow the streamlines of the underlying flow, thus tending to avoid the traps. In the motile case of strain MG1655 (panel (b) and relative inset), however, the trajectories are more segmented and bacteria actively cross streamlines enabling them to enter the stagnant regions within the traps.
In order to quantify the efficiency of this process, we consider the number of bacteria, N0, that swim through the field of view in a given time period (typically 5 minutes, corresponding to the length of the recorded videos for each experiment). Of these, Nh will enter a trap. The fraction of bacteria can be used to define the hydrodynamic efficiency ηh = Nh/N0. This quantity is then normalised to the number of available traps in the field of view (typically ranging from 25 to 33). We have measured the efficiency from the same set of experiments described in the previous paragraph and have obtained values of (0.49 ± 0.02) %/trap for the motile E. coli strain MG1655, (0.23 ± 0.01) %/trap for the non-motile E. coli strain BW25113 and (0.21 ± 0.01) %/trap for dead bacteria. Values are reported next to the corresponding motility distributions in Fig. 2(e). These numbers correspond respectively to about (16.2 ± 0.7) %, (7.6 ± 0.3) % and (6.9 ± 0.3) % absolute trapping efficiencies for 33 traps over 5 minutes. The remaining probability of trapping for dead organisms is expected because of the design of the traps and because dead organisms still experience Brownian motion associated with any object suspended in a fluid. Notably, at the bacteria concentration we used (107 cfu ml−1), nearly all the traps in the field of view were filled within the 5 minute long analysis. It is clear that motile bacteria are approximately twice as likely to swim into a trap than non-motile or dead ones. This result correlates well with the motility analysis, supporting the aforementioned connection between bacteria motility, ability to cross streamlines and, consequently, trapping efficiency.
Clearly, the absolute values of the trapping efficiency depend on the fluid velocity relative to the bacteria swimming speed. We can identify a lower and an upper bound for the flow rate: a) if it is too small compared to the bacteria swimming speed, bacteria are easily able to migrate across streamlines and enter a trap, but less likely to stay trapped for long since they can swim against the (weak) flow; b) conversely, if the fluid is too fast, the trapping efficiency drops because cells are preferentially dragged along the channel. The optimal flow rate balancing these two effects has been determined empirically to be between 10 and 15 nl min−1. Therefore, a rate of 10 nl min−1 is employed in subsequent experiments. Further details are provided in Fig. S1.†
Morphology selectivity
The physical geometry of the traps intrinsically selects bacteria according to their size and shape. In order to illustrate this point, we have studied the trapping process for bacteria with different morphology by exploiting the natural size distribution within the same population of E. coli MG1655 and for two different trap geometries. Fig. 4(a) and (b) show the corresponding length and eccentricity distributions as determined from the residence time in the traps for the same type of trap. Blue data represents bacteria that have escaped within seconds of being trapped, while the red distributions refer to those that were trapped for at least 3 minutes.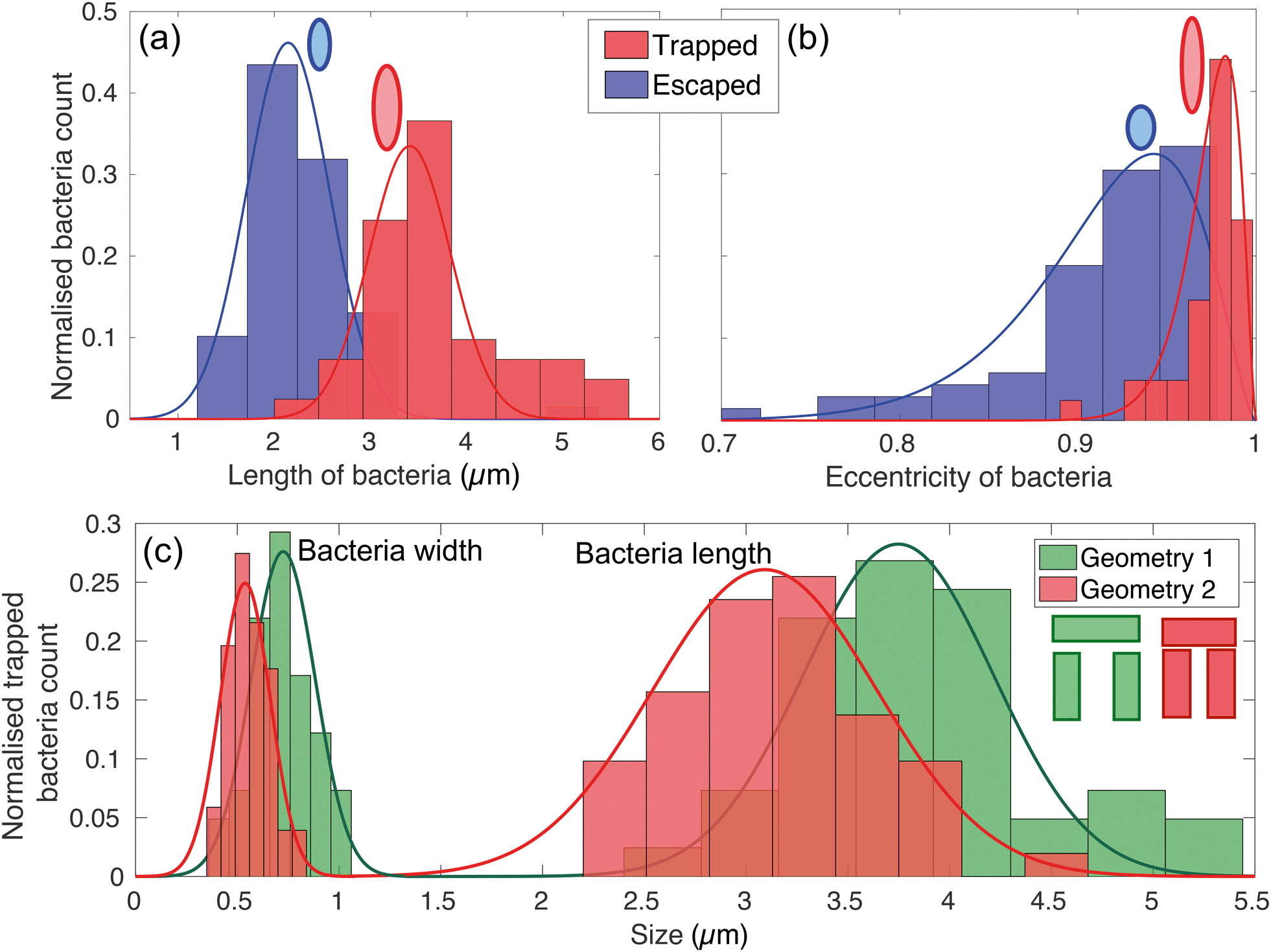 | ||
| Fig. 4 Length (a) and eccentricity (b) distributions of bacteria that have entered a trap. The eccentricity is calculated by modelling the rod-shaped E. coli as an ellipsoid. The blue curves represent bacteria that have escaped within a few seconds, while the red curves refer to bacteria that have been trapped for at least 3 minutes. The lengths are fitted to Gaussian distributions, while the eccentricities with an asymmetric beta distribution. (c) Shows the distribution of width and length of trapped bacteria for the two different geometries sketched in the inset. | ||
We note a clear difference in the trapping duration between different shapes of bacteria. Elongated bacteria, characterised by a major axis a = (3.41 ± 0.51) μm and an eccentricity e of 0.97 ± 0.01, exhibit extended residence time within the trap. Shorter bacteria ((2.14 ± 0.46) μm long and with e = 0.91 ± 0.03) escape the traps immediately after having entered. The reason for this marked difference is the fact that the shorter and more rounded bacteria are able to rotate more easily within the trap and thereby gain access to the vertical gap between the vertical and horizontal trap walls. Conversely, the motion of elongated bacteria is physically restricted, so they remain trapped for longer. See movies S2 and S3† for an illustration of this mechanism.
This trapping effect is also explicitly demonstrated in Fig. 4(c), where the distributions of width and length of trapped bacteria only are shown for the two different geometries sketched in the inset (geometry 1 and 2 colour-coded in green and red respectively). The key differences between the two are the width of the trap (1.83 μm and 1.66 μm for geometry 1 and 2 respectively) and the vertical gap (455 nm and 315 nm, respectively). The larger these dimensions, the larger the bacterium needs to be to remain trapped. In particular, the size of the vertical gap controls the minimum width of the bacteria to be trapped, while the width of the trap determines the minimum length of bacteria able to rotate within the trapping area and thus escape.
Antimicrobial susceptibility assay
Having established that the hydrodynamic traps are sensitive to both bacterial motility and shape, we now proceed to show how these signatures can be used to provide a measure of antibiotic susceptibility by classifying resistant and susceptible strains. The wild type motile E. coli MG1655 was used as a susceptible strain. Resistant colonies were grown from the same strain being modified by bacterial transformation with specific plasmids encoding resistance to three different antibiotics, namely trimethoprim, ampicillin and kanamycin. The susceptibility of the wild type and the minimum inhibitory concentration (MIC) values were determined by a standard resazurin microdilution assay. We found MIC values of 1–2 μg ml−1 for trimethoprim, 4–8 μg ml−1 for ampicillin and 8–16 μg ml−1 for kanamycin. The same method was then used to verify that the transformed strains were resistant to >512 μg ml−1 of the three drugs (see the methods section and Fig. S2 and S3† for more details). Furthermore, the viability of the transformed colonies was proven by measuring standard growth curves (see Fig. S4†).We thus chose to expose the different strains in our trapping experiments to a concentration of 10 μg ml−1 of each drug, which corresponds to a few times the MIC for trimethoprim and ampicillin, and a near-MIC value for kanamycin. The reason for selecting these three antibiotics lies in their different modes of action, which allows us to demonstrate the generality of our assay. Specifically, the bacteriostatic antibiotic trimethoprim inhibits the synthesis of folic acid,45 while the bactericidal ampicillin and kanamycin inhibit the synthesis of the cell wall46 and proteins,47 respectively.
Fig. 5 reports the results for the three different drugs administered to both the susceptible and resistant strains. Rows report different antibiotics (ampicillin (AMP, panels (a–c)), kanamycin (KAN, panels (d–f)) and trimethoprim (TRI, panels (g–i)), respectively), while columns correspond to the three quantities being evaluated as signatures (motility, trapping efficiency and length, respectively). Red curves with circle markers refer to the susceptible wild type E. coli MG1655, while blue curves with square markers are obtained for the resistant strains. Each data point is calculated from data extracted from a 5 minutes long video that is post-processed to provide measures of total cell count, number of trapped bacteria, intensity traces over the trap areas and cell morphology. Values of motility and trapping efficiency are normalised in each case in order to provide a homogeneous measure between 0 and 1 and to facilitate comparison. Negative times serve as no-drug control measurements. The bacterial samples were subsequently spiked with antibiotics at time t = 0 and injected into the same chip (as indicated by the vertical dashed lines). The only exception is represented by the black dot-dash line in each graph, which is obtained from a control experiment with the wild type E. coli MG1655 with no antibiotics being administered at any time point. This control results serve to confirm that our phenotypical signatures are not significantly affected by the microfluidic environment. In fact, bacteria maintain constant motility and show only a slight decrease in length over the entire duration of the experiment, likely due to the pressure they experience within the thin microfluidic channel. Therefore, any subsequent change in these signatures can be ascribed to the action of the antibiotics.
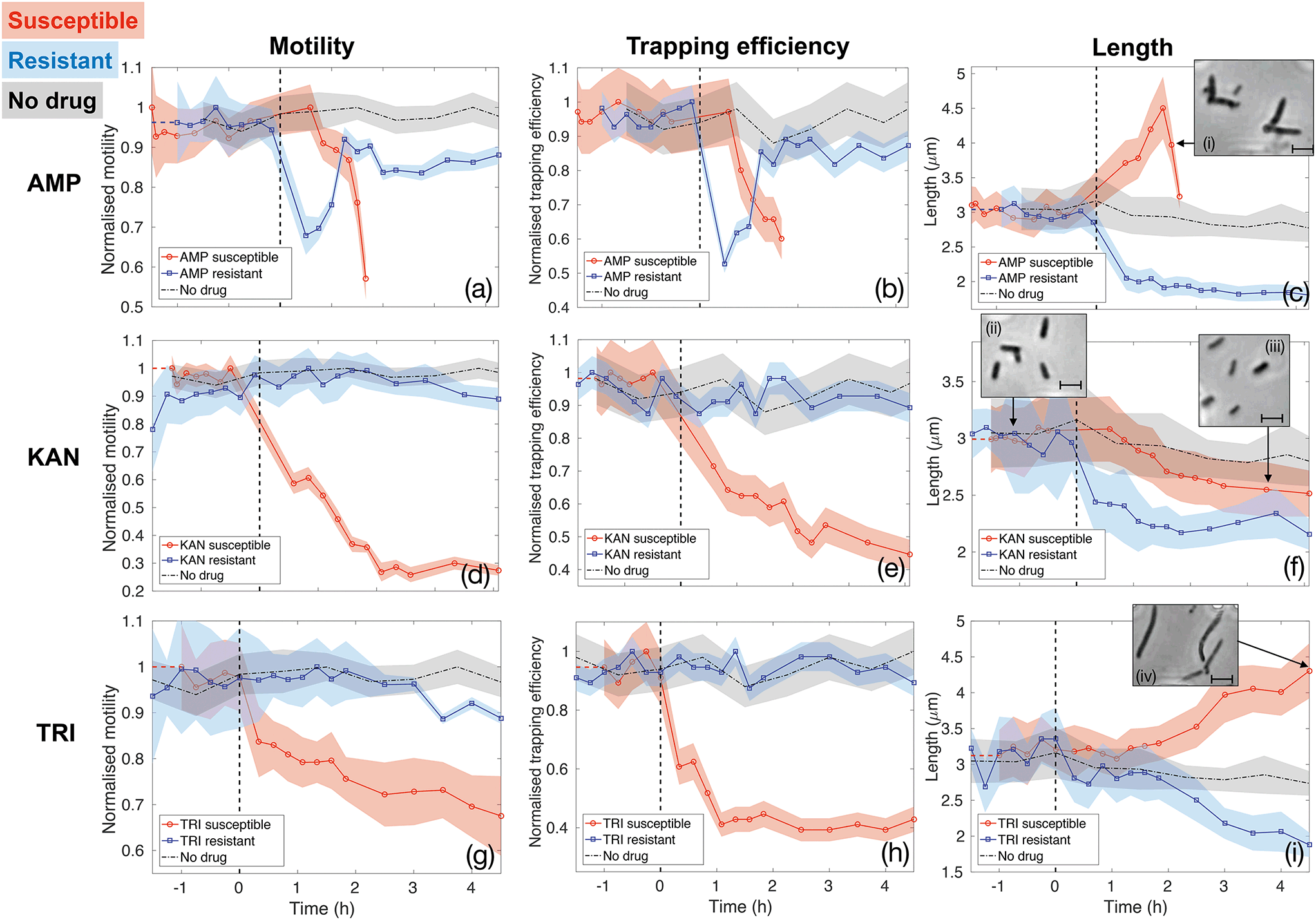 | ||
| Fig. 5 Motility, trapping efficiency and length of susceptible (red curves with circle markers) and resistant (blue curves with square markers) E. coli MG1655 exposed to ampicillin (AMP, panels (a–c)), kanamycin (KAN, panels (d–f)) and trimethoprim (TRI, panels (g–i)). A common decreasing motility and trapping efficiency trend is observed for susceptible strains exposed to all three antibiotics (red curves in panels a, b, d, e, g and h). Motility and trapping efficiency of resistant strains are not affected by KAN and TRI (blue curves in panels d, e, g and h), while a temporary loss is measured during the first 1.5 hours of exposure to AMP (blue curves in panels a and b). The third column (panels c, f and i) illustrates a variety of length responses to the antibiotics. Insets of these panels show single frames illustrating typical bacterial morphologies at different stages. Scale bars correspond to 3 μm. The time at which the samples were spiked with antibiotics (t = 0) is indicated by the vertical black dashed lines in each panel. The dot-dashed black curve in each graph refers to a no-drug control over the entire duration of the experiments, thus showing that bacteria motility and morphology are not significantly affected by the microfluidic environment. | ||
The first column (panels (a), (d) and (g)) shows how the bacterial motility varies over time following exposure of E. coli to each of the three antibiotics. A clear and common decreasing trend is observed for the susceptible strain (red curves with circle markers) for all three antibiotics, which directly corresponds to the similarly decreasing trapping efficiency, as illustrated in the second column (red curves in panels b, e and h). On the other hand, resistant strains (blue curves with square markers) behave differently. Specifically, ampicillin causes a rapid loss of motility also in the resistant case. However, motility is almost totally recovered after 1.5 h of continuous exposure to the drug (see panel (a)). The same behaviour is reflected in the trapping efficiency (panel (b)). Kanamycin and trimethoprim do not affect motility of resistant strains, which remains constant upon exposure to the drugs (blue curves in panels (d) and (g)) and, consequently, trapping efficiency is not influenced either (blue curves in panels (e) and (h)). These are already clear signatures that enable to detect resistance by differentiating the two strains.
Although the decreasing trends for the susceptible cases are common to the three antibiotics, the dynamics and time scales differ. These differences are confirmed by the morphological investigation over time reported in the third column (panels (c), (f) and (i)). The combination of the motility and the morphology information allows to probe the mechanisms of antibiotic action, to explain the observed differences between the three drugs and to increase the confidence and decrease the time needed for the classification of resistance and susceptibility.
In particular, ampicillin caused a significant decrease in motility to the susceptible strain after about 1 hour from spiking the culture (red curve in panel(a)). This reduction in motility can be explained by two mechanisms. Firstly, ampicillin is known to interfere with transcription of genes required for flagella synthesis and activation.48 Secondly, ampicillin is a β-lactam antibiotic that binds to the penicillin binding protein (PBP) and inhibits cross linking of the peptidoglycan layer in the cell wall, which is crucial for maintaining cell morphology and balancing turgor pressure, leading ultimately to cell lysis. Critically, the process of cell lysis is preceded by an elongation phase and the formation of a bulge structure,46 which make the swimming action less efficient.49 This significant change in morphology can be considered an early signature of susceptibility that occurs before cell lysis. It can be observed using our assay as a drop in trapping efficiency and motility and confirmed by the length analysis (see Fig. 5(c) and insets (i), illustrating the formation of the bulge structure). Similarly, the resistant strain also experiences a rapid loss of motility immediately after the exposure to ampicillin (blue curve in Fig. 5(a)). This could produce ambiguity until the resistant strain recovers motility, about 1.5 hours from the administration or until cell lysis is observed in the susceptible case. Instead, the pronounced filamentation process of the susceptible strain, allows to clearly distinguish it from the resistant one, as the latter shows a rapid decrease in length upon ampicillin exposure (see panel (c) and relative inset (i)). This proves how the simultaneous assessment of multiple parameters could be advantageous in the classification process and provide a richness of information that would be otherwise lost in a single-parameter assay.
The opposite situation is instead observed for kanamycin (second row of Fig. 5). The morphological signature shows rather similar behaviour for the susceptible and resistant colonies (Fig. 5(f)), as they both shrink upon the action of kanamycin, thus not providing notable insights. Conversely, a significant loss of motility, and consequently of trapping efficiency, is observed only 40 minutes after the administration of kanamycin to the susceptible strain (red curves in Fig. 5(d) and (e)). Contrarily, motility and trapping efficiency of resistant strains are almost not affected, remaining constant over time (blue curves in Fig. 5(d) and (e)). This antibiotic is an aminoglycoside compound which binds to the 30S ribosomal subunit causing misreading of the mRNA instructions and synthesis of non-functional and toxic peptides.47 This rapid loss of motility revealed here for susceptible bacteria is likely due to lesser translation of motility genes.
Finally, trimethoprim, which indirectly affects DNA synthesis by targeting the production of tetrahydrofolic acid (THF),45 shows the least pronounced trend for the susceptible bacteria (Fig. 5(g) and (h)), likely because the lack of THF does not have a strong and direct effect on flagella activation and motion in general. The reason for the decreased motility is instead a filamentary phenotypical change, which started after around 1.5 hours of exposure to trimethoprim and steadily increased over time (see Fig. 5(i) and the inset (iv)). Filamentation is a defence mechanism adopted by the bacteria which results in growth without division. The increased length is also known to hamper the bacteria rotational diffusion and swimming ability, so affecting both morphology and motility.49 However, the motility signature can still be considered the most valuable, as the length of the susceptible and resistant strain are not clearly differentiated until about 2 hours of exposure to trimethoprim (Fig. 5(i)). Conversely, the motility of resistant E. coli remains approximately constant upon the action of trimethoprim (Fig. 5(g)). Experiments with trimethoprim have been conducted in Mueller-Hinton (MH) broth because the Luria-Bertani (LB) used elsewhere contains traces of folic acid which might be taken up by bacteria, thus partly counteracting the action of the drug.
Comparison to growth-based assays
In order to validate our results, two different controls have been conducted to correlate our phenotypical signatures with the standard definition of susceptibility based on growth and bacterial division. The first evidence is represented by a bacterial count being performed on the same experiments described above. The total number of bacteria swimming through the channel is monitored over time. The results are reported in Fig. S5.† The resistant strains continued to divide even in the presence of the antibiotics following an expected logistic model of growth. On the other hand, the number of susceptible bacteria decreases in the presence of ampicillin, given its bactericidal nature causing cell lysis, while trimethoprim and kanamycin prevent division, thus maintaining the number of bacteria approximately constant over time.The second validation is a standard resazurin-based microdilution assay. Wild type and transformed E. coli MG1655 are grown in a standard 96-well plate in a serial dilution of antibiotics in order to verify their viability after an overnight incubation. The conclusion from this experiment is twofold: on one hand, we can confirm that we are indeed operating in the susceptibility range of E. coli MG1655, while on the other hand, it is clear that the transformation of the bacteria has been successful since the strains have acquired resistance to >512 μg ml−1 of the respective antibiotics (see results in Fig. S2 and S3† for further details).
In conclusion, these final experiments enabled us to confirm the classification provided by our microfluidic platform by linking our motility-morphology phenotypical signatures to growth-based techniques, which represent the gold standard in the definition of susceptibility and resistance.
Discussion
The platform presented here enables multi-parameter susceptibility analysis by linking motility and morphology in response to drug exposure at the single-cell level. Our study provides three major insights.Firstly, we demonstrate that the combined assessment of motility and morphology at the single-cell level could guide the design of novel susceptibility tests and, more generally, to study bacterial behaviour upon antibiotics administration. While the two parameters have been used separately as indicators of susceptibility,4,26,34,35,39 their combination at the single cell level is presented here for the first time. In particular, we have shown the possibility to correctly classify susceptible and resistant strains in less than two hours (from spiking the culture) for three drugs of a very different nature, i.e. drugs that are either bacteriostatic (trimethoprim) or use different modes of bactericidal action (ampicillin and kanamycin). This combination of information from the two domains enabled us to confirm the different modes of action of the antibiotics and to increase the confidence in the classification by selecting the best indicator for each drug. Our phenotypical signatures are correlated to the standard growth-based definition of susceptibility by quantifying bacterial proliferation within the microchannel and by a standard microdilution assay.
Secondly, the trapping assay, by operating at the level of individual cells, is able to respond on short timescales. In contrast, traditional bulk tests based on detecting bacterial growth cannot achieve such a short time resolution, because of their intrinsic averaging procedure, which occurs over billions of cells.9,16 Our approach directly analyses a significant number of individual cells in parallel and averages only after the actual readout. This feature has also been exploited in other single bacteria works, even if a truly single cell level is either not achieved34,35,38 or the throughput capability is lower.17,18,26 Here, changes in motility and morphology are recorded on the timescale of the antibiotic action, thereby not only providing a rapid assessment of susceptibility but also information regarding the kinetics and time evolution of antibiotic action.
Finally, the assay can be tuned to be selective. Even though we do not use molecular recognition methods such as antibodies, we can separate bacteria according to size, shape and motility, as shown in Fig. 2 and 4(a) and (b). Our method also allows to choose the size of the bacteria to be trapped by modifying the trap geometry (Fig. 4(c)). This feature can be advantageous for tuning the assay to be selective to certain bacteria, potentially extending its employability to cocci organisms and to polymicrobial cases. We also note that the size-selectivity could be used to filter bacteria in terms of size or shape in a microfluidic circuit. The geometry of the traps could be adjusted along the same microfluidic channel in order to trap progressively longer/larger bacteria and to study them at different stages of their life cycle or in different conditions.
The main limitation of our approach is that it works for motile strains. Even though non-motile bacteria can be trapped, their trapping efficiency is rather low, which limits the multi-parametric capability of the assay. Nevertheless, motile strains are responsible for a large variety of infections and motility plays a fundamental role in early-colonisation, chemotaxis and in the growth and proliferation of biofilms.36,37
In the context of antimicrobial resistance (AMR), one could envisage a stand-alone chip with several channels in order to test multiple antibiotics in parallel, potentially at different concentrations, and provide insights into the antibiotic activity at the onset of its action. We have already observed significant changes on a timescale of a few hours from spiking the culture. More generally, the assay offers a unique platform that is able to localize and confine hundreds of individual bacteria enabling them to be studied in parallel as well as allowing for high-throughput time-dependent analysis. Furthermore, localisation via hydrodynamic trapping does not require expensive consumables or complex instrumentation, such as those required by antibody-based capture or optical traps. This simplicity is a great advantage for monitoring cell behaviour over time, while probing the heterogeneity that is typical of bacterial populations.
Methods
Traps and microfluidic fabrication
Traps are fabricated in the negative electron beam resist SU8 on standard microscope slides. The access gap is typically 1.7 μm wide, the vertical bars are 4 μm long, the horizontal gap is typically 300 nm wide and all the blocks are 2.1 μm tall. The microfluidic channel is 1.5 μm deep, 170 μm wide and several mm long. It is made out of PDMS by mixing the silicon elastomer and the curing agent (DowCorning) in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The mixture is poured onto the SU8 mould on a silicon substrate featuring 8 parallel channels, cured for at least 12 h at 60 °C and peeled off. The PDMS stamp is permanently bonded to the microscope slide with the traps by oxygen plasma treatment (22sccm of oxygen for 25 seconds at a DC voltage of about 180 V).
:1 ratio. The mixture is poured onto the SU8 mould on a silicon substrate featuring 8 parallel channels, cured for at least 12 h at 60 °C and peeled off. The PDMS stamp is permanently bonded to the microscope slide with the traps by oxygen plasma treatment (22sccm of oxygen for 25 seconds at a DC voltage of about 180 V).
Bacteria culturing, transformation and experiments
E. coli MG1655 and BW25113 were grown overnight at 37 °C to saturation phase in Luria-Bertani (LB) or Muller-Hinton (MH) broth, before being diluted in LB or MH to a concentration of 107 cfu ml−1. Bacterial transformation was achieved through electroporation in order to facilitate plasmid uptake. Details about the plasmid selection and the transformation process can be found in the ESI.† Before starting every experiment, the microfluidic chips were flushed with a 1% solution of bovine serum albumin (BSA) in phosphate buffer saline (PBS) solution for 30 minutes by using a syringe pump at 1 μl min−1 to prevent attachment of bacteria to surfaces. The bacterial sample is then injected at a flow rate of 10 nl min−1. HD videos were recorded with a DSLR camera (Nikon D3300) at 50 fps connected to an inverted phase contrast microscope (Leica DM IRB) equipped with a 60× magnification objective.Microdilution resazurin assay
The microdilution assay has been performed on standard 96-well plates by exposing wild type and transformed E. coli MG1655 to a serial dilution of ampicillin, kanamycin and trimethoprim in order to determine their MIC and to verify that the transformed strains have acquired resistance to the antibiotics. Resazurin is employed as a reporter dye to indicate the viability status of the cells. Detailed procedures are reported in the ESI† and results in Fig. S2 and S3.†Growth curve assay
The growth curve assay has been carried out by measuring the optical density (OD) of the growing colonies with a time resolution of 20 minutes. Growth curves are obtained for the wild type E. coli MG1655 and for the three transformed strains. Detailed procedures and resulting curves are included in the supplementary information (Fig. S4†).Data analysis
Videos were post-processed with a custom-written MATLAB script which converts them into grayscale and subtracts the median frame calculated over the entire duration of the videos. A region of interest (ROI) was centred on each trap to define the area over which the average pixel intensity is calculated. Typically, 33 traps are present in each field of view and videos are 5 minutes long. Intensity traces were obtained for each trap over time and were used to calculate the standard deviation corresponding to the trapping events and for counting the bacteria. The total number of cells swimming through the field of view in each video is of the order of several hundreds (up to 2000 in some cases). The number of trapped bacteria is of the order of several tens of individuals, up to 150 in some cases. Tracking was performed on one-minute long videos with the TrackMate extension of ImageJ.50 Cell detection and length measurements were achieved with the MATLAB regionprop function after being manually calibrated for around 200 cells. This calibration procedure is required because of the typical halos present in phase contrast micrographs which cause misjudgement of the cell boundaries. We explicitly verified that the correction factor was constant within a ∼5% error across four independent experiments, so that it was not necessary to repeat the calibration for each experiment.Author contributions
G. P., S. J., H. G. and T. F. K. contributed to the conceptualisation and methodology of the research. G. P. contributed to the software aspect and to data acquisition, analysis, curation and visualisation. S. T. and A. E. provided biological resources and clinical knowledge. G. P. and T. F. K. wrote and edited the manuscript. T. F. K. supervised the project. S. J. and T. F. K acquired funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the EPSRC Programme Grant EP/P030017/1 “Resonant and shaped photonics” and the EPSRC grant EP/M027538/1 “Targeted”. T. F. K. acknowledges the Royal Society Wolfson Research Merit Award Scheme. G. P., H. G. and T. F. K. also wish to thank Stevens Paz Sánchez from the University of São Paulo for help with data analysis.References
- J. O'Neill, Indep. Rev. AMR, 2015, pp. 1–36 Search PubMed.
- I. Roca, M. Akova, F. Baquero, J. Carlet, M. Cavaleri, S. Coenen, J. Cohen, D. Findlay, I. Gyssens, O. E. Heure, G. Kahlmeter, H. Kruse, R. Laxminarayan, E. Liébana, L. López-Cerero, A. MacGowan, M. Martins, J. Rodríguez-Baño, J. M. Rolain, C. Segovia, B. Sigauque, E. Taconelli, E. Wellington and J. Vila, New Microbes New Infect., 2015, 6, 22–29 CrossRef CAS PubMed.
- M. Balouiri, S. Moulay and K. I. Saad, J. Pharm. Anal., 2016, 6, 71–79 CrossRef PubMed.
- J. Choi, J. Yoo, M. Lee, E.-G. Kim, J. S. Lee, S. Lee, S. Joo, S. H. Song, E.-C. Kim, J. C. Lee, H. C. Kim, Y.-G. Jung and S. Kwon, Sci. Transl. Med., 2014, 6, 267ra174 CrossRef PubMed.
- T. K. Price, T. Dune, E. E. Hilt, K. J. Thomas-White, S. Kliethermes, C. Brincat, L. Brubaker, A. J. Wolfe, E. R. Muller and P. C. Schreckenberger, J. Clin. Microbiol., 2016, 54, 1216–1222 CrossRef CAS PubMed.
- M. Davenport, K. E. Mach, L. M. D. Shortliffe, N. Banaei, T. H. Wang and J. C. Liao, Nat. Rev. Urol., 2017, 14, 298–310 Search PubMed.
- K. Kogermann, M. Putrinš and T. Tenson, Eur. J. Pharm. Sci., 2016, 95, 2–16 CrossRef CAS PubMed.
- K. Syal, M. Mo, H. Yu, R. Iriya, W. Jing, S. Guodong, S. Wang, T. E. Grys, S. E. Haydel and N. Tao, Theranostics, 2017, 7, 1795–1805 CrossRef CAS PubMed.
- D. Di Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- D. Di Carlo, N. Aghdam and L. P. Lee, Anal. Chem., 2006, 78, 4925–4930 CrossRef CAS PubMed.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- Z. Long, E. Nugent, A. Javer, P. Cicuta, B. Sclavi, M. Cosentino Lagomarsino and K. D. Dorfman, Lab Chip, 2013, 13, 947–954 RSC.
- A. Amir, F. Babaeipour, D. B. McIntosh, D. R. Nelson and S. Jun, Proc. Natl. Acad. Sci. U. S. A., 2013, 111, 5778–5783 CrossRef PubMed.
- S. Taheri-Araghi, S. Bradde, J. T. Sauls, N. S. Hill, P. A. Levin, J. Paulsson, M. Vergassola and S. Jun, Curr. Biol., 2015, 25, 385–391 CrossRef CAS PubMed.
- D. Yang, A. D. Jennings, E. Borrego, S. T. Retterer and J. Männik, Front. Microbiol., 2018, 9, 1–12 CrossRef PubMed.
- Ö. Baltekin, A. Boucharin, E. Tano, D. I. Andersson and J. Elf, Proc. Natl. Acad. Sci. U. S. A., 2017, 201708558 Search PubMed.
- T. van Leest and J. Caro, Lab Chip, 2013, 13, 4358 RSC.
- H. Xin, Q. Liu and B. Li, Sci. Rep., 2014, 3, 1–8 Search PubMed.
- B. Hammarström, T. Laurell and J. Nilsson, Lab Chip, 2012, 12, 4296–4304 RSC.
- J. Q. Boedicker, L. Li, T. R. Kline and R. F. Ismagilov, Lab Chip, 2008, 8, 1265–1272 RSC.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- W. Postek, P. Gargulinski, O. Scheler, T. S. Kaminski and P. Garstecki, Lab Chip, 2018, 18–21 Search PubMed.
- D. S. Liao, J. Raveendran, S. Golchi and A. Docoslis, Sens Biosensing Res, 2015, 6, 59–66 CrossRef.
- C. Páez-Avilés, E. Juanola-Feliu, J. Punter-Villagrasa, B. Del Moral Zamora, A. Homs-Corbera, J. Colomer-Farrarons, P. L. Miribel-Català and J. Samitier, Sensors, 2016, 16, 1541 CrossRef PubMed.
- E. Rostova, C. Ben Adiba, G. Dietler and S. K. Sekatskii, Biosensors, 2016, 6, 52 CrossRef PubMed.
- K. Syal, R. Iriya, Y. Yang, H. Yu, S. Wang, S. E. Haydel, H. Y. Chen and N. Tao, ACS Nano, 2016, 10, 845–852 CrossRef CAS PubMed.
- D. Wlodkowic, S. Faley, M. Zagnoni, J. P. Wikswo and J. M. Cooper, Anal. Chem., 2009, 81, 5517–5523 CrossRef CAS PubMed.
- L. Bell, A. Seshia, D. Lando, E. Laue, M. Palayret, S. F. Lee and D. Klenerman, Sens. Actuators, B, 2014, 192, 36–41 CrossRef CAS PubMed.
- R. J. Kimmerling, G. Lee Szeto, J. W. Li, A. S. Genshaft, S. W. Kazer, K. R. Payer, J. de Riba Borrajo, P. C. Blainey, D. J. Irvine, A. K. Shalek and S. R. Manalis, Nat. Commun., 2016, 7, 10220 CrossRef CAS PubMed.
- E. L. Jackson-Holmes, T. C. Mcdevitt and H. Lu, Lab Chip, 2017, 17, 3634–3642 RSC.
- C. Probst, A. Grünberger, W. Wiechert and D. Kohlheyer, Micromachines, 2013, 4, 357–369 CrossRef.
- M.-C. Kim, B. C. Isenberg, J. Sutin, A. Meller, J. Y. Wong and C. M. Klapperich, Lab Chip, 2011, 11, 1089 RSC.
- S. Kasas, F. S. Ruggeri, C. Benadiba, C. Maillard, P. Stupar, H. Tournu, G. Dietler and G. Longo, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 378–381 CrossRef CAS PubMed.
- G. Longo, L. Alonso-Sarduy, L. M. Rio, A. Bizzini, A. Trampuz, J. Notz, G. Dietler and S. Kasas, Nat. Nanotechnol., 2013, 8, 522–526 CrossRef CAS PubMed.
- V. Kara, C. Duan, K. Gupta, S. Kurosawa, D. J. Stearns-Kurosawa and K. Ekinci, Lab Chip, 2018, 18, 743–753 RSC.
- C. Josenhans and S. Suerbaum, Int. J. Med. Microbiol., 2002, 291, 605–614 CrossRef CAS PubMed.
- Q. Duan, M. Zhou, L. Zhu and G. Zhu, J. Basic Microbiol., 2013, 53, 1–8 CrossRef PubMed.
- H. Etayash, M. F. Khan, K. Kaur and T. Thundat, Nat. Commun., 2016, 7, 12947 CrossRef CAS PubMed.
- Y. Matsumoto, S. Sakakihara, A. Grushnikov, K. Kikuchi, H. Noji, A. Yamaguchi, R. Iino, Y. Yagi and K. Nishino, PLoS One, 2016, 11, 1–17 Search PubMed.
- T. K. Wood, A. F. González Barrios, M. Herzberg and J. Lee, Appl. Microbiol. Biotechnol., 2006, 72, 361–367 CrossRef CAS PubMed.
- R. M. Harshey, Annu. Rev. Microbiol., 2003, 57, 249–273 CrossRef CAS PubMed.
- K. Son, D. R. Brumley and R. Stocker, Nat. Rev. Microbiol., 2015, 13, 761–775 CrossRef CAS PubMed.
- J. S. Guasto, R. Rusconi and R. Stocker, Annu. Rev. Fluid Mech., 2012, 44, 373–400 CrossRef.
- H. C. Fu Marcos, T. R. Powers and R. Stocker, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4780–4785 CrossRef PubMed.
- R. N. Brogden, A. A. Carmine, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1982, 23, 405–430 CrossRef CAS PubMed.
- Z. Yao, D. Kahne and R. Kishony, Mol. Cell, 2012, 48, 705–712 CrossRef CAS PubMed.
- L. P. Kotra, J. Haddad and S. Mobashery, Antimicrob. Agents Chemother., 2000, 44, 3249–3256 CrossRef CAS PubMed.
- N. Kaldalu, R. Mei and K. Lewis, Antimicrob. Agents Chemother., 2004, 48, 890–896 CrossRef CAS PubMed.
- Ò. Guadayol, K. L. Thornton and S. Humphries, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- J. Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, Methods, 2017, 115, 80–90 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01397g |
| This journal is © The Royal Society of Chemistry 2019 |