
Reversible insertion of CO into an aluminium–carbon bond†
Richard Y.
Kong
 and
Mark R.
Crimmin
*
and
Mark R.
Crimmin
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, 80 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 2nd May 2019
Abstract
A [2.2.1] aluminium metallobicycle is capable of reversibly inserting CO to form a [2.2.2] metallobicycle at 100 °C. Computational studies reveal a highly asynchronous, but concerted, transition state for CO insertion. The coordination of CO to aluminium precedes C–C bond formation. The reversible migratory insertion of CO at aluminium thus mimics well-established transition-metal reactivity.
Migratory insertion, and its microscopic reverse elimination, are fundamental transformations in transition metal chemistry. In a migratory insertion reaction, two ligands on the transition metal combine to form one with no changes in formal oxidation state. Perhaps the most widely studied and applied variant of this reactivity involves the 1,1-insertion of CO into metal–alkyl bonds. This reaction requires a cis-relation of the ligands and can be thought of in terms of an intramolecular nucleophilic attack of the alkyl group on CO to generate a new acyl ligand.1 The coordination site vacated during the reaction may be trapped by external ligand. The reactivity underpins some of the most widely applied homogeneously catalysed industrial processes including the Cativa acetic acid synthesis2 and hydroformylation. In 2008, it was estimated that hydroformylation accounted for the production of 10.4 million metric tonnes of carbonylated hydrocarbons.3 The reverse reaction elimination has been applied in organic synthesis and used to decarbonylate carbonyl functional groups.4
Over the last decade there has been a concerted effort from both academia and industry to alleviate concerns surrounding transition metal toxicity, scarcity, and cost by replacing transition metals with more abundant main group elements.5 Reports on the insertion of CO into main-group–element bonds are rare. In the case of aluminium, the most abundant metal in the Earth's crust, they are limited to three examples: the insertion of CO into the Al–C bonds of a trialkylaluminium,6 aluminacyclopropene,7 and an aluminium-bound carbon chain.8 In contrast to the well-established transition-metal systems, however, these insertion reactions have been reported to be non-reversible. Thus, while the reversible activation of dihydrogen,9–11 carbon dioxide,12 and alkenes13–17 has been documented at main-group centres,18,19 reports on reversible processes involving CO are hitherto unknown.
Herein we report the reversible insertion of CO into the Al–C bond of an aluminium-based metallobicycle. The reversibility is demonstrated through the elimination of CO from the product under a N2 atmosphere and isotopic exchange with 13CO. Through a combination of DFT calculations and kinetics, CO insertion within our system has been shown to proceed by an asynchronous but concerted mechanism. To the best of our knowledge, our findings represent the first reversible insertion chemistry of CO at a main group metal and a new variant of migratory insertion reactivity.
The reaction of the low-valent aluminium complex 120 with 1,3-cyclohexadiene furnishes 2, a [4+1] cycloaddition product that contains a [2.2.1] metallobicycle (Scheme 1a). The reaction is analogous to the recently reported reduction of 1,3,5,7-cyclooctatetraene by an aluminyl anion performed by Coles and co-workers.21 We have previously reported that 1 reacts reversibly with a series of terminal and strained alkenes to form metallocyclopropanes as the [2+1] cycloaddition product.17 In this case, the formation of metallobicycle 2 is a non-reversible process.
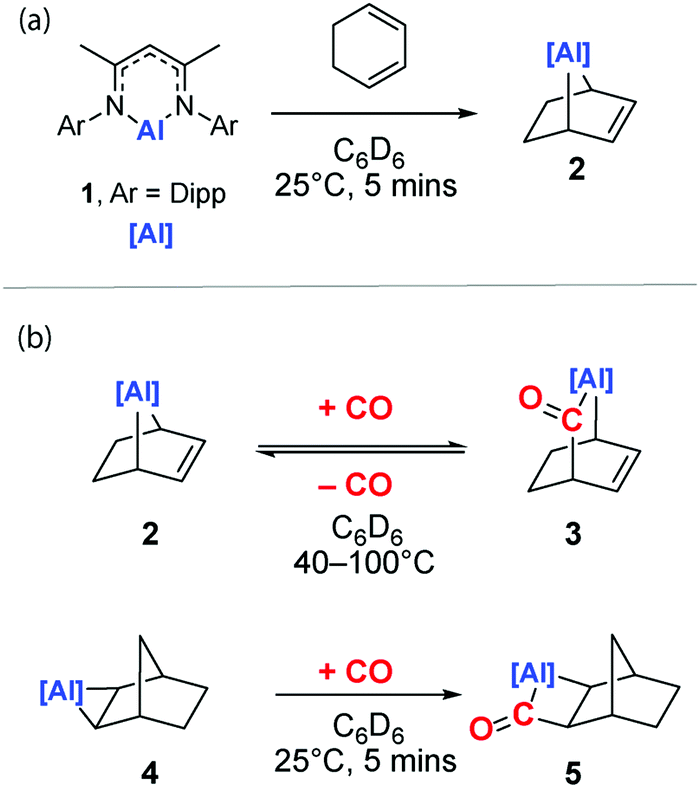 | ||
| Scheme 1 Synthesis of (a) [2.2.1] metallobicycle 2 and (b) reversible CO insertion. | ||
Compound 2 forms single crystals that contain a unit cell which possesses a C2 symmetry axis bisecting the N–Al–N angle of the β-diketiminate ligand. The entirety of the cyclohexene ring fragment is disordered over the mirror plane precluding unequivocal measurement of carbon–carbon bond lengths within the fragment. The shortening of the Al–N bond distance to 1.9095(15) Å and expansion of the N–Al–N angle to 95.96° in 2 compared to the metrics reported for 1 (Al–N: 1.957(2) Å, N–Al–N: 89.86(8)°)20 are consistent with the formation of an Al(III) centre in 2. The 1H NMR spectrum of 2 in C6D6 shows a diagnostic alkene proton resonance at δH = 6.02 ppm along with the bridgehead methine positions at δH = 2.04 ppm. The latter protons are shielded due to the proximity of aluminium and so resonate further upfield relative to its hydrocarbyl homologue norbornene (δH = 2.74). The 1H NMR data in combination with the structural constraints imposed by Bredt's rule on the location of the alkene moiety allow the unequivocal assignment of 2.
Exposure of a C6D6 solution of 2 to 1 atm. of CO results in its slow conversion to 3 at 25 °C (Scheme 1). 3 is the result of insertion of CO into the Al–C bond of 2. The reaction converts an alkyl ligand into an acyl ligand and results in a ring-expansion to form a [2.2.2] metallobicycle. Despite multiple attempts at crystallisation, single crystals of 3 suitable for X-ray diffraction were not obtained due to the instability of this compound. Nevertheless, the structure of 3 can be determined through multinuclear NMR spectrometry. The desymmetrisation of the metallacycle is clear in the now two unique alkene environments which resonate at δH = 5.56 and 6.61 ppm δC = 121.7 and 139.4 ppm in the respective spectra. The newly incorporated acyl group is identifiable in the 13C NMR spectrum as a broad singlet at δC = 255.3 ppm consistent with our previous reports of aluminium acyls.8 A correlation between the acyl carbon and the bridgehead methine (δH = 2.77) is identifiable in the 1H–13C HMBC spectrum. The incorporation of an acyl group is further confirmed by assignment of the acyl stretch in the infrared spectrum at 1620 cm−1. While prolonged heating at 40 °C (>48 h) allows for the formation 3 as the major product, at higher temperatures (100 °C) equilibration to an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2:3 is observed. The data suggest that insertion of CO into the Al–C bond of 2 is reversible.
:1 mixture of 2:3 is observed. The data suggest that insertion of CO into the Al–C bond of 2 is reversible.
Reversible CO insertion was confirmed in two separate experiments. Heating samples of 3 in C6D6 at 100 °C under an atmosphere of N2 results in quantitative reformation of 2. Furthermore, heating a solution of 3 in C6D6 at 100 °C for 3 h under 1 atm. of 13CO results in the exchange of the 13C isotopic label into the 12C position of the acyl group. This degenerate exchange reaction is evident by the change in multiplicity of the bridgehead methine resonance in the 1H NMR spectrum due to a 2J13C–1H coupling of 5.5 Hz as well as the signal intensity enhancement quaternary carbonyl resonance in the 13C NMR spectrum (see the ESI†).
Reports of the insertion of CO into Al–C bonds remain rare. In the absence of crystallographic characterization of 3, the insertion of CO into a related heterocycle, 4, was studied to improve the confidence of our structural assignment. Reaction of 4 with CO generates the insertion product 5 within five minutes at 25 °C (Scheme 1c). Like 2, 5 demonstrates a diagnostic 13C NMR resonance for the carbonyl moiety δC = 292.3 ppm. In the solid state, 5 possesses a Cs symmetry plane bisecting the N–Al–N angle and passing through the atoms Al1 and C11 (Fig. 1b). Despite disorder of the norbornyl fragment over this plane of symmetry, the insertion of CO to the Al–C bond of 4 to form a metallocyclobutanone is clear. The sum of internal angles (∑∠ = 358.66°) of the metallocycle imply a near planar geometry of the four-membered ring. While the formation of 5 is supported by solid-state and solution-state characterisation (see ESI†), 5 decomposes at 25 °C over 12 h to an intractable mixture of products.
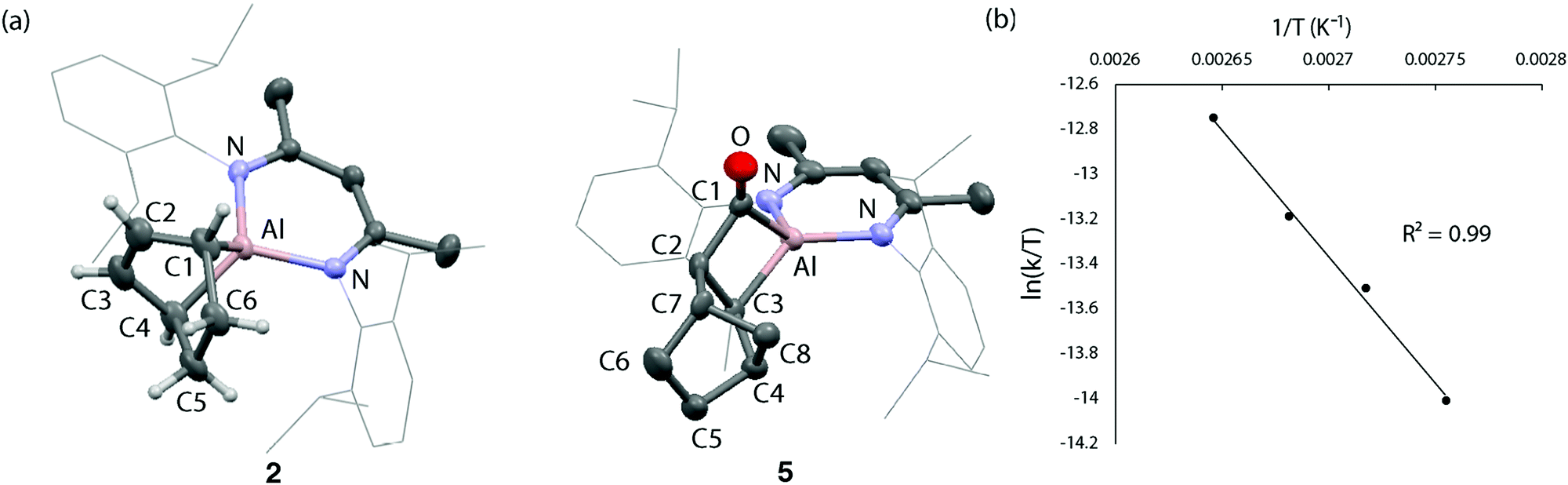 | ||
| Fig. 1 (a) Solid state structure of 2 and 5. (b) Eyring analysis on the elimination of CO from 3 to form 2. | ||
In order to quantify the reversible behaviour of 2 + CO ↔ 3, kinetics measurements were made by 1H NMR spectroscopy. Rates were measured using a low concentration of 3 (2 mM) in toluene-d8 a J Youngs NMR tube under a N2 atmosphere with a modest headspace, as such the liberated CO is at high-dilution and not expected to participate in the forward reaction (see the ESI†). Under these conditions, the elimination of CO was found to fit first order kinetics with respect to the concentration of 3. Eyring analysis using this approach over a 90–105 °C temperature range gave activation parameters for the back reaction of ΔH‡ = 23.5 ± 1.7 kcal mol−1 and ΔS‡ = −10.3 ± 4.5 cal K−1 mol−1 (Fig. 1c). The negative entropy of activation is consistent with a highly ordered transition state. The associated Gibbs activation energy is ΔG‡298K = 26.6 ± 3.0 kcal mol−1.
To provide further insight into the reactivity, density functional theory (DFT) calculations were performed (Fig. 2). The formation of 3 by insertion of CO into the Al–C bond of 2 was found to proceed viaTS-1 corresponding to a calculated activation barrier of ΔG‡298K = 20.0 kcal mol−1 for the forward-reaction, ΔG‡298K = 28.7 kcal mol−1 for the back-reaction, within error of the experimentally determined activation parameters. The conversion of 4 to 5 proceeds through TS-2, calculated to have an activation barrier of ΔG‡298K = 7.8 kcal mol−1. The transformation of 4 to 5 is calculated to be more exergonic than 2 to 3, 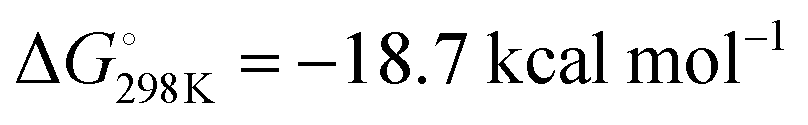 but the low activation barrier means that the back reaction should also be feasible, ΔG‡298K = 26.5 kcal mol−1. Experimentally the decomposition of 5 occurs before the elimination of CO can be observed.
but the low activation barrier means that the back reaction should also be feasible, ΔG‡298K = 26.5 kcal mol−1. Experimentally the decomposition of 5 occurs before the elimination of CO can be observed.
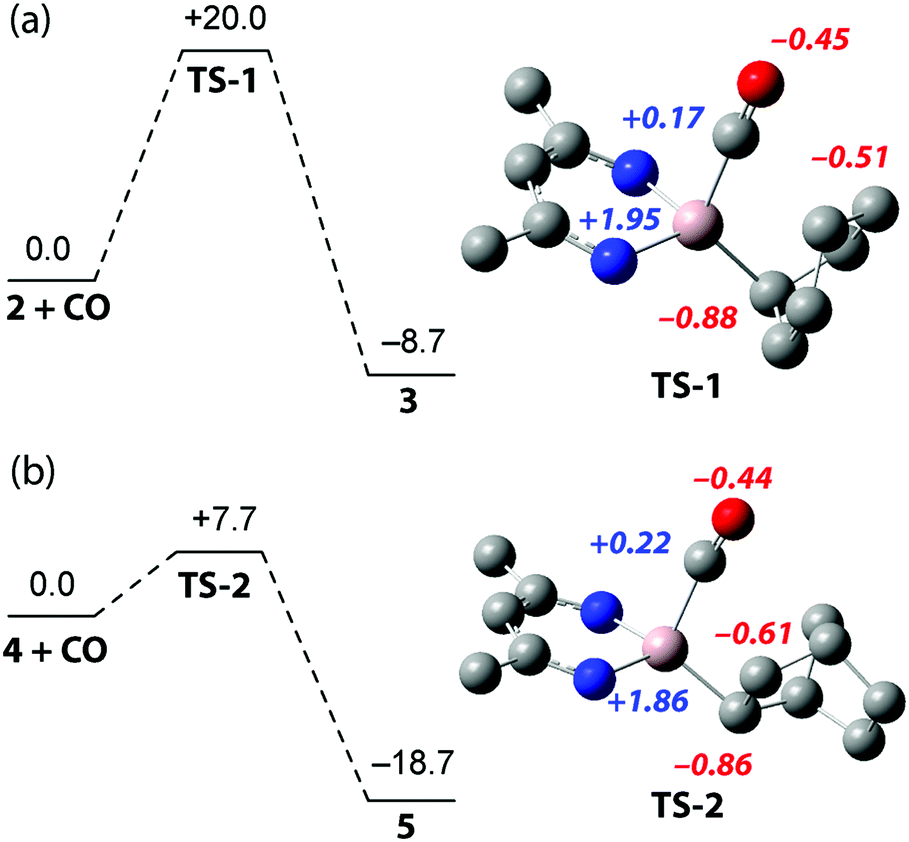 | ||
| Fig. 2 Calculated pathway and associated transition states for the formation of (a) 3 and (b) 5. Gibbs energies in kcal mol−1. Representations of TS-1 and TS-2 have been truncated for clarity and annotated with calculated NPA charges. | ||
Both TS-1 and TS-2 directly connect reactants and products and involve an asynchronous concerted Al–C and C–C bond forming process. In contrast to more established migratory insertion reactions of transition metal complexes and a previously calculated pathway for CO insertion into a trialkylaluminium complex,22 there is no intermediate in these pathways. Despite this, there is a clear parallel between the asynchronicity of these transition states and the stepwise coordination/insertion processes documented for transition metal mediated carbonylation. NBO analysis (Wiberg bond indices) allows the characterisation a strong Al–CO interaction in the transition states relative the products (TS-1, 0.45; 3, 0.48; TS-2, 0.50; 5, 0.53). Incipient C⋯C bond formation is also clear from these data (TS-1, 0.46; 3, 0.92; TS-2, 0.41; 5, 0.95). The notable disparity in values between the transition-state and product suggest that C–C bond formation occurs after Al–C bond formation and that CO binding to Al is an important consideration. Analysis of the NPA charges (Fig. 2) indicates that C–C bond formation occurs via a nucleophilic attack on the bound CO by the electronegative aluminium alkyl. The role of electrophilic aluminium centres as Lewis acids in migratory insertion reactions of transition metal carbonyls has been established before.23,24 It has been shown that, through coordination of the CO moiety, aluminium additives such as AlBr3 can not only lower the activation barrier of migratory insertion in transition metal carbonyls but also stabilise the resulting acyl product.
In summary, we report the reversible insertion of CO into an Al–C bond that results in equilibration between aluminium alkyl and acyl complexes. To the best of our knowledge, this is the first example of such reversible behaviour in main group systems and a long overdue parallel of the migratory insertion reaction established for transition metal chemistry.
We are grateful to the Royal Society for provision of a University Research Fellowship (MRC) and the ERC (FluoroFix: 677367) for generous funding. We thank Imperial College London for the award of a President's Scholarship (RYK). Dr Clare Bakewell is thanked for running preliminary experiments with compound 4.
Conflicts of interest
There are no conflicts of interest.Notes and references
- J. F. Hartwig, Organotransition metal chemistry: from bonding to catalysis, University Science Books, 2010, pp. 349–365 Search PubMed.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- K. Ohno and J. Tsuji, J. Am. Chem. Soc., 1968, 90, 99–107 CrossRef CAS.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- M. R. Mason, B. Song and K. Kirschbaum, J. Am. Chem. Soc., 2004, 126, 11812–11813 CrossRef CAS PubMed.
- X. Li, C. Ni, H. Song and C. Cui, Chem. Commun., 2006, 1763–1765 RSC.
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- M. Ullrich, A. J. Lough and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 52–53 CrossRef CAS PubMed.
- S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590–593 CrossRef CAS PubMed.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed.
- Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef CAS PubMed.
- F. Lips, J. C. Fettinger, A. Mansikkamäki, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2014, 136, 634–637 CrossRef CAS PubMed.
- D. Wu, R. Ganguly, Y. Li, S. N. Hoo, H. Hirao and R. Kinjo, Chem. Sci., 2015, 6, 7150–7155 RSC.
- A. J. Boutland, A. Carroll, C. Alvarez Lamsfus, A. Stasch, L. Maron and C. Jones, J. Am. Chem. Soc., 2017, 139, 18190–18193 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452–2458 RSC.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- M. R. Mason, B. Song, Y. Han and X. Hu, Inorg. Chim. Acta, 2008, 361, 3332–3337 CrossRef CAS.
- S. B. Butts, S. H. Strauss, E. M. Holt, R. E. Stimson, N. W. Alcock and D. F. Shriver, J. Am. Chem. Soc., 1980, 102, 5093–5100 CrossRef CAS.
- T. G. Richmond, F. Basolo and D. F. Shriver, Inorg. Chem., 1982, 21, 1272–1273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic and crystallographic data, the latter in.cif format. CCDC 1905522 and 1905523. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02818h |
| This journal is © The Royal Society of Chemistry 2019 |