
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduction of 1,3,5,7-cyclooctatetraene by a molecular calcium hydride: an even electron polarised insertion/deprotonation mechanism†
Michael S.
Hill
 *a,
Mary F.
Mahon
a,
Andrew S. S.
Wilson
a,
Chiara
Dinoi
b,
Laurent
Maron
*b and
Emma
Richards
c
*a,
Mary F.
Mahon
a,
Andrew S. S.
Wilson
a,
Chiara
Dinoi
b,
Laurent
Maron
*b and
Emma
Richards
c
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
bUniversité de Toulouse et CNRS, INSA, UPS, UMR 5215, LPCNO, 135 Avenue de Rangueil, F-31077 Toulouse, France
cSchool of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, UK
First published on 17th April 2019
Abstract
Reaction of a dimeric β-diketiminato calcium hydride with 1,3,5,7-cyclooctatetraene enables two electron aromatisation of the [8]annulene to provide an inverse sandwich dicalcium cyclooctatetraenyl derivative. This reactivity does not proceed through sequential single electron transfer but via a consecutive polarised Ca–H/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C insertion and deprotonation pathway that occurs at the intact dimeric hydride reagent.
C insertion and deprotonation pathway that occurs at the intact dimeric hydride reagent.
Harder and co-workers’ β-diketiminato calcium hydride, [(BDI)Ca(THF)H]2 (1),1,2 (BDI = HC{(Me)CN-2,6-i-Pr2}2) initiated significant interest in molecular hydride derivatives of the heavier alkaline earth elements. Although a variety of dimeric and polynuclear structural types have now been described,3–14 even greater interest has arisen from their emergent utility as stoichiometric and catalytic reagents.15–18 Compound 1, for example, has been shown to act as a catalyst for the hydrogenation of activated alkene and diene substrates,19 a process which typifies the polarised metathetical and C
C insertion reactivity mediated by such redox-invariable 2+ metal centres. Our own research has highlighted that the highly nucleophilic character of the Ca–H bond of the THF-free variant of 1, [(BDI)CaH]2 (2), enables its direct reaction with a wide range of unactivated terminal alkenes to provide the corresponding dimeric n-alkyl species.20,21 Notably, these reactions occur via highly polarised Ca–H/CC insertion pathways and through the apparent retention of dimeric hydride and hydrido-alkyl intermediates (Scheme 1). The resultant alkyl derivatives are also sufficiently nucleophilic to effect the heterolytic cleavage of H2 or,22 even more remarkably, the direct nucleophilic displacement of hydride from benzene to provide the corresponding alkylated benzene derivatives.20,23
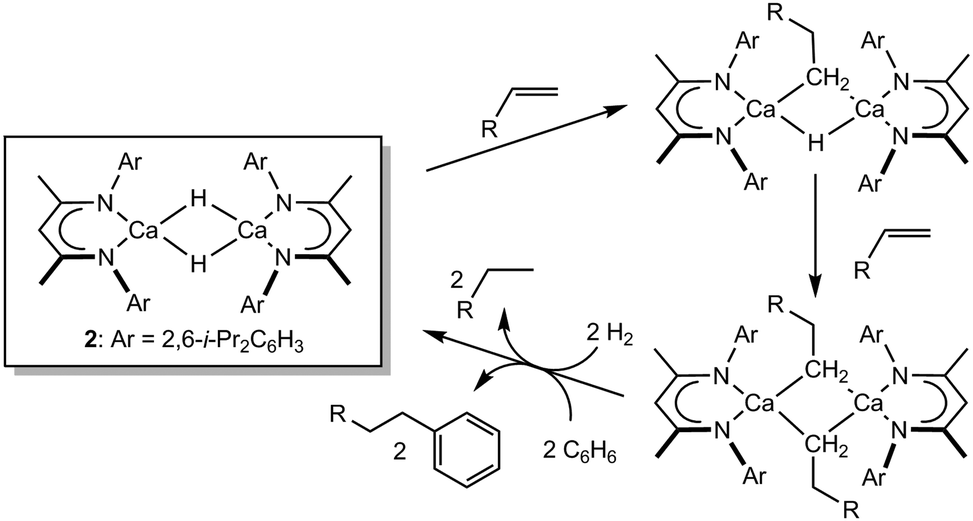 | ||
| Scheme 1 Reactivity of compound 2 with alkenes and that of the resultant calcium alkyls with H2 and benzene. | ||
With these observations in mind, our attention was drawn to Harder's recent demonstration of the potency of such hydride reagents toward the transfer hydrogenation of C–C multiple bonds.24 This chemistry utilised 1,4-cyclohexadiene (1,4-CHD) as the source of dihydrogen during which the cyclic diene is aromatised to benzene. Density functional theory (DFT) calculations demonstrated that this transformation ensues through a sequence of 1,4-CHD deprotonation and hydride elimination from the resultant cyclohexadienyl intermediate. This observation is particularly striking as it exemplifies a formal two electron oxidation of 1,4-CHD, but mediated by a hydridic reagent of a type that is more typically considered as a source of reducing electron equivalents. In this latter context, Evans has raised similar questions with regard to the potential of organolanthanide and actinide hydrides to provide reductive behaviour comparable to lower oxidation state species.25,26 A suitable test case was provided by the reduction of 1,3,5,7-cyclooctatetraene (1,3,5,7-C8H8) to the cyclooctatetraenyl dianion, [COT]2−, by [(C5Me5)2LnH]2 (Ln = Y, La and Sm),26 and the thorium hydride [(C5Me5)2ThH2]2.25 These transformations occur with retention of the oxidation state of the metal and through oxidation of the supporting ligands, eliminating either hydrogen gas or (C5Me5)2. Although no definitive mechanistic rationale has been presented, the authors’ comparison of the H2/2H− reduction potential (E° = −2.25 V) with the first and second reduction potentials of 1,3,5,7-C8H8 itself (E° = −1.83 and −1.99 V), invokes a tacit assumption of sequential single electron reduction steps analogous to those implicated in the direct reduction of 1,3,5,7-C8H8 by alkali metals (e.g. Na+/Na, E° = −2.71 V).27–34
Structurally characterised alkaline earth complexes comprising the cyclooctatetraenyl dianion are currently limited to examples in which the reducing equivalents of electrons are provided by the unambiguous oxidation of an s-block element centre. The heavier homoleptic alkaline earth complexes, [AeCOT] (Ae = Ca, Sr and Ba), for example, were synthesised by metal vapour co-condensation with 1,3,5,7-C8H8, whilst the magnesium derivative, [Mg(COT)(THF)2.5],35,36 was accessed through the direct combination of the [8]annulene and magnesium. In a process reminiscent of these ‘dissolving metal’ reductions, [{(BDI)Mg(THF)}2(COT)], has been prepared via the two-electron reduction of 1,3,5,7-C8H8 with the magnesium(I) dimer [(BDI)Mg]2.37 In contrast, a variety of heavier alkaline earth inverse sandwich derivatives, [(i-Pr4C5H)Ae(μ-COT)Ae(i-Pr4C5H)] (Ae = Ca and Ba)38 and [{(Me3Si)2N}{Ae(THF)x}(μ-COT)(THF)xAe{N(SiMe3)2}] (Ae = Ca, x = 1; Ae = Sr, x = 2),39 have been prepared by salt metathesis of the alkaline earth halide precursors, [{(C5Hi-Pr4)AeX(THF)2}2] (Ae = Ca, X = Cl; Ae = Ba, X = I) and AeI2 (Ae = Ca, Sr) with disodium cyclooctatetraenide or potassium hexamethyldisilazide and dipotassium cyclooctatetraenide, respectively. In all these latter cases, however, the [COT]2− dianions were generated by direct reduction of 1,3,5,7-C8H8 by the relevant group 1 metal.
Although alkaline earth hydride-mediated reactivity of 1,3,5,7-C8H8 is yet to be described, the reactions of diphenylacetylene with either compound 2 or a similarly dimeric amidinato calcium hydride have been shown to result in reduction to the stilbene dianion, [Ph(H)C–C(H)Ph]2−.21,40 Notably, the reductive reactivity of this latter species toward I2 and H2 led it to being posited as a potential Ca(I) synthon. With these diverse considerations in mind, we describe herein our observations of the ability of compound 2 to effect the generation of the [COT]2− dianion.
Addition of one equivalent of 1,3,5,7-C8H8 to a C6D6 solution of compound 2 was accompanied by the immediate vigorous evolution of gaseous hydrogen (Scheme 2). This observation was accompanied by the appearance of new BDI methine and [COT]2− signals at δ 4.49 and 5.60 ppm with a relative ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 in the resultant 1H NMR spectrum (Fig. S1, ESI†). A preparative scale reaction‡ allowed the solid state structure of the dicalcium cyclooctatetraenyl derivative, [(BDI)Ca(μ-COT)Ca(BDI)] (3), to be confirmed by a single crystal X-ray diffraction analysis of a crystal grown from a saturated hexane solution at −35 °C (Fig. 1).§
:8 in the resultant 1H NMR spectrum (Fig. S1, ESI†). A preparative scale reaction‡ allowed the solid state structure of the dicalcium cyclooctatetraenyl derivative, [(BDI)Ca(μ-COT)Ca(BDI)] (3), to be confirmed by a single crystal X-ray diffraction analysis of a crystal grown from a saturated hexane solution at −35 °C (Fig. 1).§
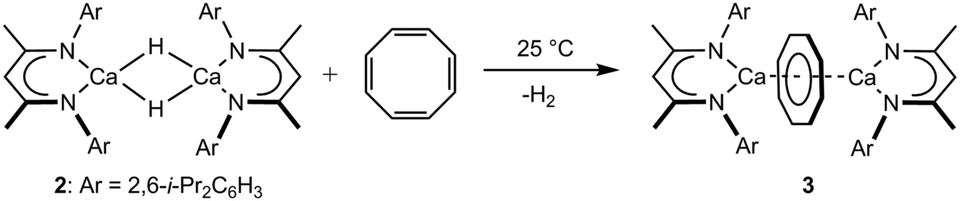 | ||
| Scheme 2 Synthesis of compound 3. | ||
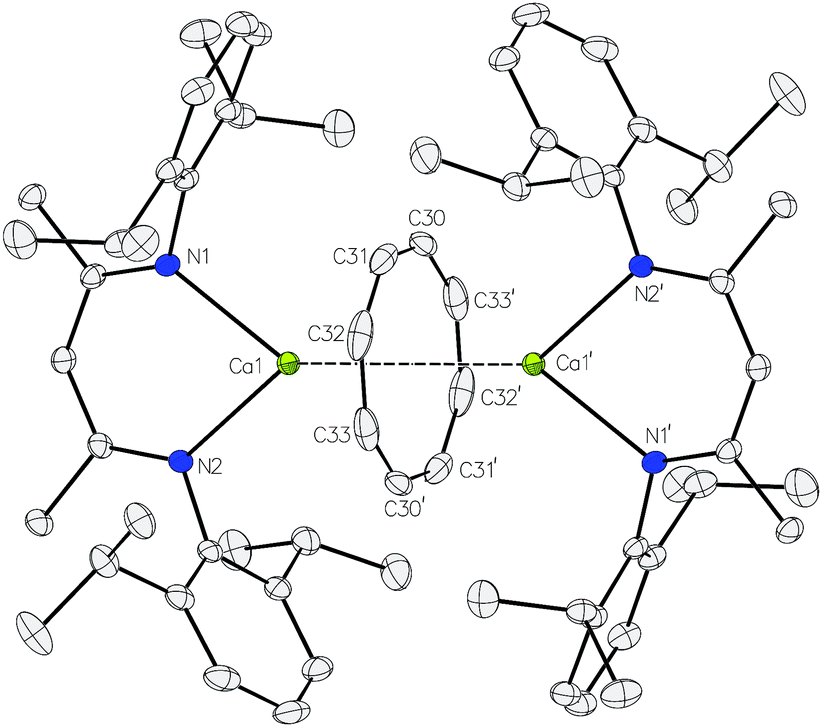 | ||
| Fig. 1 ORTEP representation of compound 3 with thermal ellipsoids at 30%. Hydrogen atoms have been removed for clarity. Selected bond lengths (Å) and bond angles (°). Ca1–N1 2.3654(11), Ca1–N2 2.3793(10), Ca1–C30 2.7200(15), Ca1–C30′ 2.7175(16), Ca1–C31 2.7121(15), Ca1–C31′ 2.7200(16), Ca1–C32 2.7215(17), Ca1–C32′ 2.7181(17), Ca1–C33 2.7121(17), Ca1–C33′ 2.7181(17), Ca1–COTcent 2.0144(4), N1–Ca1–N2 77.59(4), Ca1–COTcent–Ca1′ 180.00(2). Symmetry operation to generate primed atoms 1 − x, 1 − y, 1 − z. | ||
The asymmetric unit of compound 3 comprises half of a dimer wherein a crystallographic inversion centre coincident with the centroid of the C8 ring serves to generate the remainder of the molecule. The inverse sandwich structure of 3 is, thus, defined by an η8-interaction between the [COT]2− dianion and the calcium centres with a Ca1–COTcent distance [2.0144(4) Å] and a Ca1–COTcent–Ca1′ angle [180.00(2)°] that closely resemble the analogous measurements of [(i-Pr4C5H)Ca(μ-COT)Ca(i-Pr4C5H)] [1.99 and 1.98 Å; 173.0 and 175.8°] and the very recently reported [{(Me3Si)2N}{Ca(THF)}(μ-COT)(THF)Ca{N(SiMe3)2}] [2.02 and 2.04 Å; 179°].38,39 The coordination sphere of calcium is completed by the β-diketiminate ligand with orthodox calcium–nitrogen distances [2.3654(11) and 2.3793(10) Å].
As outlined above, the synthesis of compound 3 raises a number of significant questions with regard to the mode of delivery of the necessary reducing equivalents of electrons to produce the [COT]2− dianion. The potassium and sodium reduction of 1,3,5,7-C8H8 to the 10π aromatic dianion has been shown to occur in a stepwise fashion via the initial generation of the [COT]− radical anion, which is readily identifiable from its characteristic nine line EPR spectrum [a(1H) = 3.2 G (8.9 MHz)].27–29 The reaction between compound 2 and 1,3,5,7-C8H8 in benzene solution was, thus, repeated between 140 and 298 K within the cavity of an X-band CW EPR spectrometer. Although this procedure again provided for the facile generation of the [COT]2− dianion, no definitive evidence for any radical intermediates en route to compound 3 were observed. The formation of compound 3 was, therefore, studied by density functional theory (DFT, B3PW91) calculations.
Although different pathways were computed (see ESI†), only the lowest energy process is presented (Fig. 2). We have previously reported that the dissociation of the dimeric structure of compound 2 is endothermic by some 40.4 kcal mol−1.22 Consistent with the facile production of 3 (G) at room temperature, the current analysis (Fig. 2) indicates that the significantly exothermic reaction (ΔH = −57.5 kcal mol−1) takes place through the retention of the calcium hydride dimer. The rate determining process of the reaction (TS-BC) is provided by the nucleophilic delivery of hydride to one of the isolated CC bonds of the unsaturated [8]annulene. In line with the requisite room temperature conditions, this process occurs via a barrier of 21.2 kcal mol−1 to yield an initial dicalcium cyclooctatrienyl intermediate (C), which also retains a single μ2-Ca–H–Ca bridging interaction. Subsequent reaction of this {C8H9}−-containing species (D) requires the traversal of a further kinetically accessible transition state (TS-DE, 19.6 kcal mol−1) to provide an aromatisation step that may be viewed as an intramolecular deprotonation of the {C8H9}− methylene by the remaining hydridic hydrogen centre. Subsequent rearrangement of the two {(BDI)Ca} units is then facile to provide the bis-η8-inverse sandwich structure of compound 3 (G).
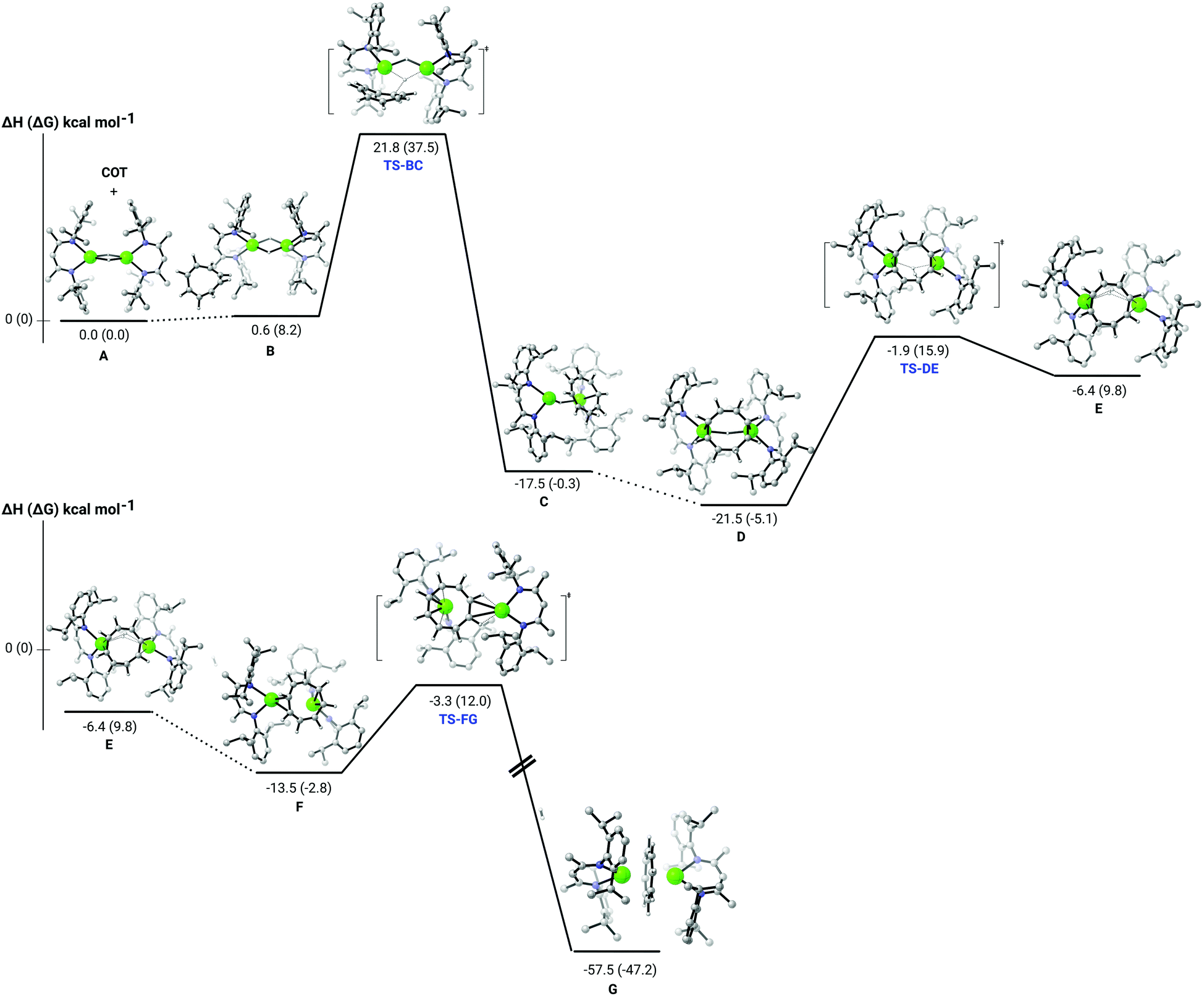 | ||
| Fig. 2 DFT (B3PW91) computed enthalpy reaction profile at room temperature for the reaction of compound 2 (A) and 1,3,5,7-C8H8. | ||
These observations underscore the ability of compound 2 to function as a potent source of polarised unsaturated insertion (2σ–2π) and metathetical (2σ–2σ) reactivity, in this case to demonstrate that the reductive aromatisation of cyclooctatetraene does not necessarily require the intermediacy of single electron or radicaloid intermediates. The broader significance of these observations may lie in their potential relevance to reductive processes mediated by other redox inactive metal hydrides. We are, therefore, continuing to consider the implications of this investigation with regard to the activation of a wider array of reducible annulene and arene substrates.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS.
- J. Spielmann and S. Harder, Chem. – Eur. J., 2007, 13, 8928–8938 CrossRef CAS.
- S. Harder, Chem. Commun., 2012, 48, 11165–11177 RSC.
- A. Causero, G. Ballmann, J. Pahl, C. Farber, J. Intemann and S. Harder, Dalton Trans., 2017, 46, 1822–1831 RSC.
- B. Maitland, M. Wiesinger, J. Langer, G. Ballmann, J. Pahl, H. Elsen, C. Farber and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 11880–11884 CrossRef CAS PubMed.
- M. Wiesinger, B. Maitland, C. Farber, G. Ballmann, C. Fischer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 16654–16659 CrossRef CAS.
- P. Jochmann, J. P. Davin, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 4452–4455 CrossRef CAS PubMed.
- V. Leich, T. P. Spaniol and J. Okuda, Inorg. Chem., 2015, 54, 4927–4933 CrossRef CAS PubMed.
- V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 4794–4797 CrossRef CAS PubMed.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS.
- D. Mukherjee, T. Hollerhage, V. Leich, T. P. Spaniol, U. Englert, L. Maron and J. Okuda, J. Am. Chem. Soc., 2018, 140, 3403–3411 CrossRef CAS PubMed.
- C. N. de Bruin-Dickason, T. Sutcliffe, C. A. Lamsfus, G. B. Deacon, L. Maron and C. Jones, Chem. Commun., 2018, 54, 786–789 RSC.
- X. H. Shi, C. P. Hou, C. L. Zhou, Y. Y. Song and J. H. Cheng, Angew. Chem., Int. Ed., 2017, 56, 16650–16653 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. A, 2010, 466, 927–963 CrossRef CAS.
- M. R. Crimmin and M. S. Hill, in Alkaline-Earth Metal Compounds: Oddities and Applications, ed. S. Harder, 2013, vol. 45, pp. 191–241 Search PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351–360 CrossRef CAS.
- A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed.
- S. Harder, B. Rösch, T. Xaver Gentner, H. Elsen, J. Langer and M. Wiesinger, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef PubMed.
- H. Bauer, K. Thum, M. Alonso, C. Fischer and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 4248–4253 CrossRef CAS PubMed.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS.
- W. J. Evans, B. M. Schmiege, S. E. Lorenz, K. A. Miller, T. M. Champagne, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 8555–8563 CrossRef CAS PubMed.
- F. J. Smentowski and G. R. Stevenson, J. Am. Chem. Soc., 1967, 89, 5120 CrossRef CAS.
- F. J. Smentowski and G. Stevenson, J. Am. Chem. Soc., 1969, 91, 7401 CrossRef CAS.
- F. J. Smentowski and G. R. Stevenson, J. Phys. Chem., 1969, 73, 340 CrossRef CAS.
- P. I. Kimmel and H. L. Strauss, J. Phys. Chem., 1968, 72, 2813 CrossRef CAS.
- G. R. Stevenson and J. G. Concepcion, J. Phys. Chem., 1972, 76, 2176 CrossRef CAS.
- G. R. Stevenson and J. G. Concepcion, J. Am. Chem. Soc., 1973, 95, 5692–5694 CrossRef CAS.
- G. R. Stevenson and I. Ocasio, Tetrahedron Lett., 1976, 427–430 CrossRef CAS.
- K. Mullen, Chem. Rev., 1984, 84, 603–646 CrossRef.
- H. Lehmkuhl, S. Kintopf and K. Mehler, J. Organomet. Chem., 1972, 46, C1–C2 CrossRef CAS.
- T. Alonso, S. Harvey, P. C. Junk, C. L. Raston, B. Skelton and A. H. White, Organometallics, 1987, 6, 2110–2116 CrossRef CAS.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973–2977 CrossRef CAS PubMed.
- M. D. Walter, G. Wolmershäuser and H. Sitzmann, J. Am. Chem. Soc., 2005, 127, 17494–17503 CrossRef CAS PubMed.
- F. M. Sroor, L. Vendier and M. Etienne, Dalton Trans., 2018, 47, 12587–12595 RSC.
- A. Causero, H. Elsen, G. Ballmann, A. Escalona and S. Harder, Chem. Commun., 2017, 53, 10386–10389 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, details of the X-ray analysis of compound 3, details of the computational analysis and atomic coordinates of the DFT computed structures. CCDC 1906088. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02418b |
| ‡ A toluene (10 ml) solution of 1,3,5,7-C8H8 (226 μl, 2.01 mmol) was added dropwise at room temperature to a stirring toluene (10 ml) of 2 (0.92 g, 1.00 mmol) and stirred overnight (ca. 16 hours). The resulting orange solution was evaporated to dryness, redissolved in hexane (20 ml), cannula filtered and concentrated. Pale-yellow crystals deposited at −35 °C overnight and were collected via cannula filtration to yield 3 (0.35 g, 34%). Colourless crystals suitable for X-ray diffraction analysis were obtained from a saturated hexane solution at −35 °C. 1H NMR (500 MHz, benzene-d6) δ 7.28–7.19 (m, 12H, Ar-H), 5.61 (s, 8H, C8H8), 4.49 (s, 2H, NC(CH3)CH), 2.61 (hept, 3JHH = 6.8 Hz, 8H, CH(CH3)2), 1.49 (s, 12H, NC(CH3)CH), 1.44 (d, 3JHH = 6.8 Hz, 24H, CH(CH3)2), 1.01 (d, 3JHH = 6.8 Hz, 24H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, benzene-d6) δ 165.1 (NC(CH3)CH), 146.9 (Cipso), 141.9 (Cortho), 124.6 (Cpara), 123.7 (Cmeta), 93.8 (NC(CH3)CH), 89.6 (C8H82−), 28.9 (CH(CH3)2), 24.6 (CH(CH3)2), 24.5 (NC(CH3)CH), 24.2 (CH(CH3)2) ppm. Despite repeated attempts, the extreme air-sensitivity of this compound precluded the acquisition of an accurate microanalysis. |
| § X-ray diffraction data for 3. C66H90Ca2N, M = 1019.57, monoclinic, P21/n, a = 14.7014(4), b = 10.3339(3), c = 19.8975(5) Å, β = 92.412(2)°, V = 3020.21(14) Å3, Z = 2, ρ = 1.121 g cm−3, temperature 150.01(10) K, R1 [I > 2σ(I)] = 0.0401, wR2 [I > 2σ(I)] = 0.1042, R1 [all data] = 0.0423, wR2 [all data] = 0.1068, measured reflections = 37686, unique reflections = 6031, Rint = 0.0617. |
| This journal is © The Royal Society of Chemistry 2019 |