
Nickel-catalysed dehydrogenative coupling of aromatic diamines with alcohols: selective synthesis of substituted benzimidazoles and quinoxalines†
Atanu
Bera
,
Motahar
Sk
,
Khushboo
Singh
and
Debasis
Banerjee
 *
*
Department of Chemistry, Laboratory of Catalysis and Organic Synthesis, Indian Institute of Technology Roorkee, Roorkee-247667, Uttarakhand, India. E-mail: dbane.fcy@iitr.ac.in
First published on 25th April 2019
Abstract
The first nickel-catalysed dehydrogenative coupling of primary alcohols and ethylene glycol with aromatic diamines for selective synthesis of mono- and di-substituted benzimidazoles and quinoxalines is reported. The earth-abundant, non-precious and simple NiCl2/L1 system enables the synthesis of N-heterocycles releasing water and hydrogen gas as byproducts. Mechanistic studies involving deuterium labeling experiments and quantitative determination of hydrogen gas evaluation were performed.
Substituted benzimidazole and quinoxaline motifs are ubiquitous in bioactive natural products and are significantly utilized for anticancer, antiviral, antitumor, antibacterial and anti-HIV related drugs.1 These N-heterocycles have found broad applications in material research and pharmaceuticals as well as in dye industries.2 Traditionally, benzimidazoles are synthesized involving the condensation of 1,2-diaminobenzene with carboxylic acid derivatives under harsh reaction conditions.3 Furthermore, the coupling of 1,2-di-aminobenzene with concentrated formic acid is another extensively used procedure for benzimidazole synthesis.3 However, these processes often require strong acidic or basic conditions as well as over stoichiometric oxidants and are limited due to the formation of stoichiometric salt waste, low atom-efficiency and functional group compatibility.4 Therefore, the development of efficient and environmentally benign technologies which could minimize the waste generation and avoid multi-step protocols is still a demanding goal.4
In this context, transition metal-catalysed de-hydrogenative coupling of renewable alcohols with 1,2-diaminobenzene would be an alternative synthetic approach for benzimidazoles, since water and hydrogen gas are only valuable byproducts.5 Since the last two decades, considerable progress has been directed for the acceptorless de-hydrogenation of alcohols with diamines for benzimidazole synthesis involving sustainable transformations, however, often limited with noble-metal catalysts and harsh reaction conditions. For instance, studies based on Ru-phosphine or Ru-NNN pincer complexes required 165–200 °C reaction temperature.6a,b Thereafter, an improved acceptorless de-hydrogenative coupling (ADC) for the benzimidazole synthesis using defined Ir-catalysts was established.6c,d Furthermore, Ru- and Ir-based heterogeneous catalysts have been developed for benzimidazole synthesis (Scheme 1a).6e,f
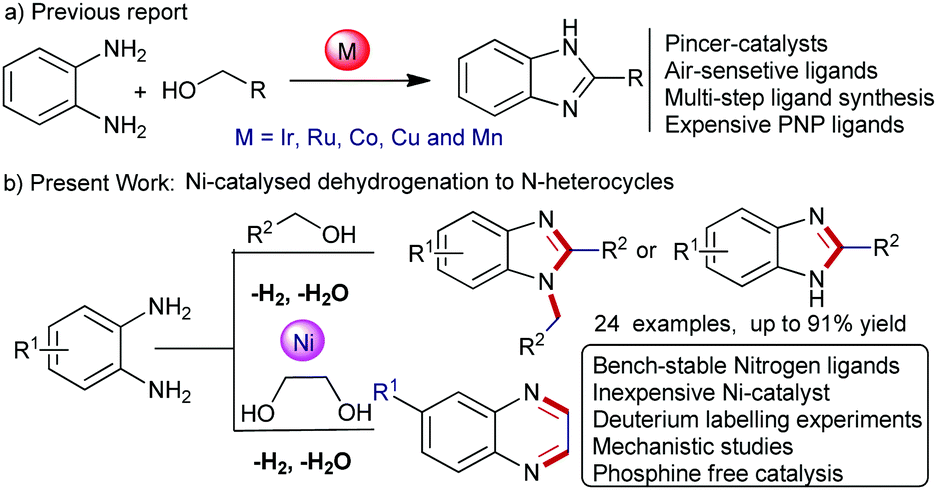 | ||
| Scheme 1 (a) The pincer complex catalysed benzimidazole synthesis. (b) The nickel-catalysed dehydrogenative couplings for the synthesis of N-heteroaromatics. | ||
Notably, the replacement of noble-metal catalysts using base-metals (Fe, Co, Mn and Ni) with equal efficiency for such key organic transformations is a challenging task. Due to their high abundance, low toxicity and inexpensive nature, the base-metal catalysts attract significant attention in homogeneous catalysis (Scheme 1a).7 Recently, de-hydrogenative condensation of alcohols with 1,2-diaminobenzene to benzimidazoles was established using Cu, Co, and Mn-based homogeneous catalysts. Nevertheless, most of these metal-complexes utilized pincer ligands, such as, tridentate NNS-ligands, PNN-ligands, and NNN-core ligands as well as triazole-core phosphine ligands or expensive phosphine ligands to achieve higher product yields.6,8 However, the multi-step synthesis and expensive nature of the pincer ligands are often a major concern in comparison to the base-metal catalysts.8 Furthermore, handling and storing of these pincer-complexes requires special attention under standard laboratory conditions.
Importantly, to date, most of these homogeneous and heterogeneous based catalytic protocols are often limited with 2-substituted benzimidazoles. However, a general catalytic protocol for 2-substituted as well as 1,2-disubstituted benzimidazoles is highly desirable. More specifically, herein we demonstrated the first nickel-catalysed general protocol for selective synthesis of substituted benzimidazoles using renewable alcohols with 1,2-diaminobenzene.9 Activation and formation of a number of bonds selectively occurred in one-pot operation using an inexpensive nickel-catalyst releasing water and hydrogen gas as byproducts. Again, this Ni-catalysed route was employed for quinoxaline synthesis. Preliminary catalytic and mechanistic studies were also performed (Scheme 1b).
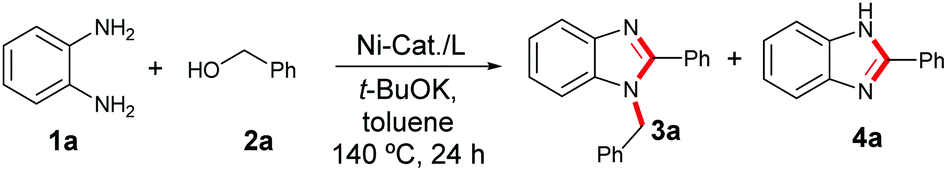
Recently, we started a program to establish Ni-catalysed (de)hydrogenative transformations using renewable alcohols or diols and a couple of novel protocols were demonstrated for the synthesis of indoles, substituted pyrroles, pyridines and quinolines.10 Herein, we explored a nickel-catalysed tandem one-pot construction of substituted benzimidazoles and quinoxalines. Remarkably, the coupling of 1,2-benzenediamines with alcohols comprises one-pot multiple step operations; de-hydrogenation of alcohol to aldehyde followed by the condensation of imine and thereafter intramolecular cyclisation and de-hydrogenation resulted in the formation of benzimidazoles (Scheme 3e).
Initially, we studied the model reaction using 1,2-benzene-diamines 1a with benzyl alcohols 2a to establish an efficient catalytic system for benzimidazole synthesis. Primarily, four different nickel pre-catalysts were employed involving 1,10-phenanthroline L1 as a ligand of our choice (Table 1, entries 1–4). Pleasingly, we observed 91% isolated yield of 1,2-disubstituted benzimidazole 3a with >49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity involving a combination of 2.5 mol% NiCl2, and 3 mol% 1,10-phenthroline L1 with 0.5 mmol of t-BuOK at 140 °C in toluene (Table 1, entry 1). Next, the influence of different nitrogen ligands L2–L5 further did not improve the product yield and the selectivity of 3a (Table 1, entries 5–8 and ESI,† Table S2). Furthermore, we examined the efficacy of different bases, such as t-BuONa, K3PO4, K2CO3 and Cs2CO3 and observed moderate product conversion to 3a (Table 1, entry 9 and ESI,† Table S3). Using different polar as well as non-polar solvents, such as n-butanol, N,N-dimethyl formamide (DMF), N,N-dimethyl acetamide (DMA), and p-xylene, proved inefficient (ESI,† Table S4). Whereas, in the case of 1,4-dioxane we observed moderate product conversion (ESI,† Table S4). As expected, product conversion is suppressed significantly when the reaction is performed at a lower reaction temperature (ESI,† Table S6). Control experiments in the absence of catalyst, ligand and base prove their significant role in achieving higher product yield and selectivity (Table 1, entries 10 and 11 and ESI,† Tables S1–S6).
:1 selectivity involving a combination of 2.5 mol% NiCl2, and 3 mol% 1,10-phenthroline L1 with 0.5 mmol of t-BuOK at 140 °C in toluene (Table 1, entry 1). Next, the influence of different nitrogen ligands L2–L5 further did not improve the product yield and the selectivity of 3a (Table 1, entries 5–8 and ESI,† Table S2). Furthermore, we examined the efficacy of different bases, such as t-BuONa, K3PO4, K2CO3 and Cs2CO3 and observed moderate product conversion to 3a (Table 1, entry 9 and ESI,† Table S3). Using different polar as well as non-polar solvents, such as n-butanol, N,N-dimethyl formamide (DMF), N,N-dimethyl acetamide (DMA), and p-xylene, proved inefficient (ESI,† Table S4). Whereas, in the case of 1,4-dioxane we observed moderate product conversion (ESI,† Table S4). As expected, product conversion is suppressed significantly when the reaction is performed at a lower reaction temperature (ESI,† Table S6). Control experiments in the absence of catalyst, ligand and base prove their significant role in achieving higher product yield and selectivity (Table 1, entries 10 and 11 and ESI,† Tables S1–S6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Ligand (mol%) | Base | Conv. (%) | |
| 3a | 4a | ||||
| Reaction conditions:a 1a (0.5 mmol), benzyl alcohol 2a (1.0 mmol), Ni-cat (0.0125 mmol), L (0.015 mmol), t-BuOK (0.5 mmol), toluene (2.0 mL) in a Schlenk tube under nitrogen atmosphere at 140 °C in an oil bath for 24 h reaction time.b Conversion was determined by GC-MS (isolated yield in parentheses, average yield of two runs).c t-BuOK (0.5 mmol) was used. L1 = 1,10-phenanthroline. L2 = 2,9-dimethyl-1,10-phenanthroline. L3 = bipyridine. L4 = 4,4′-dimethylbipyridine. L5 = 2,2′-biquinoline. | |||||
| 1 | NiCl 2 (2.5) | L1 (3) | t-BuOK | 98(91) | 2 |
| 2 | NiBr2 (2.5) | L1 (3) | t-BuOK | 65 | 5 |
| 3 | Ni(acac)2 (2.5) | L1 (3) | t-BuOK | 56 | 11 |
| 4 | NiCl2(DME) (2.5) | L1 (3) | t-BuOK | 54 | 6 |
| 5 | NiCl2 (2.5) | L2 (3) | t-BuOK | 57 | 20 |
| 6 | NiCl2 (2.5) | L3 (3) | t-BuOK | 76 | 12 |
| 7 | NiCl2 (2.5) | L4 (3) | t-BuOK | 55 | 6 |
| 8 | NiCl2 (2.5) | L5 (3) | t-BuOK | 33 | 5 |
| 9 | NiCl2 (2.5) | L1 (3) | t-BuONa | 75 | 9 |
| 10c | — | — | t-BuOK | 21 | 0 |
| 11c | NiCl2 (2.5) | — | t-BuOK | 35 | 0 |
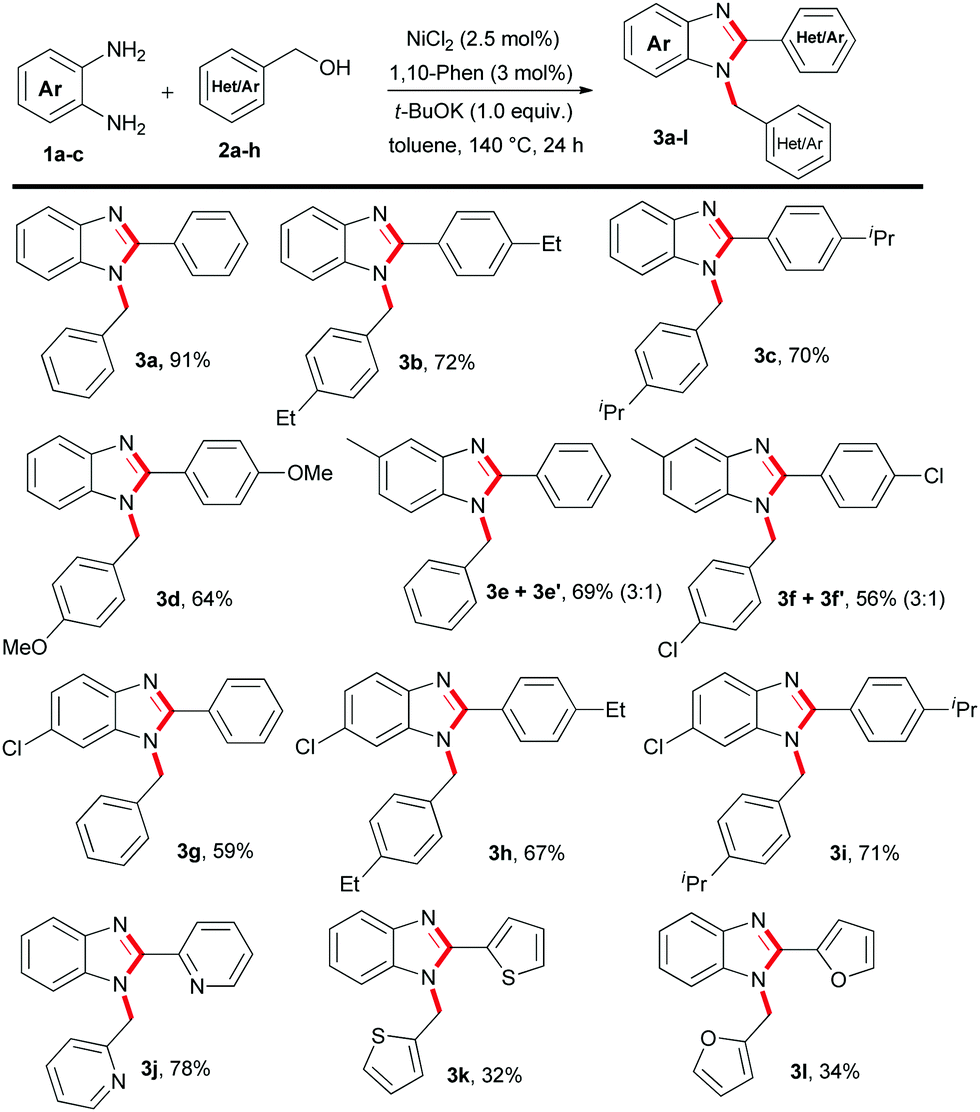
Next, a series of 1-benzyl-2-aryl-1H-benzo[d]imidazole derivatives were synthesized using 1,2-benzenediamines with substituted benzyl alcohols involving the standard catalytic conditions of Table 1 (Table 2). For instance, benzyl alcohol substituted with an alkyl or methoxy group resulted in up to 72% isolated yield of 3b–3d (Table 2). Importantly, 2-pyridinemethanol 2f efficiently transformed to 1,2-disubstituted benzimidazole 3j in 78% yield. However, the application of other heterocyclic alcohols, such as 2-thiophene-methanol 2g and 2-furfuralmethanol 2h gave a moderate yield of 3k–3l (Table 2). Moreover, 4-methyl-1,2-phenylenediamine 1b and 4-chloro-1,2-phenylenediamine 1c efficiently participated with substituted benzyl alcohols and furnished the desired benzimidazoles derivatives 3e–3i in 56–71% yield, respectively (Table 2). Notably, in the case of 4-methyl-1,2-phenylenediamine 1b an isomeric mixture of products were obtained.
| Reaction conditions:a see ESI. |
|---|
|
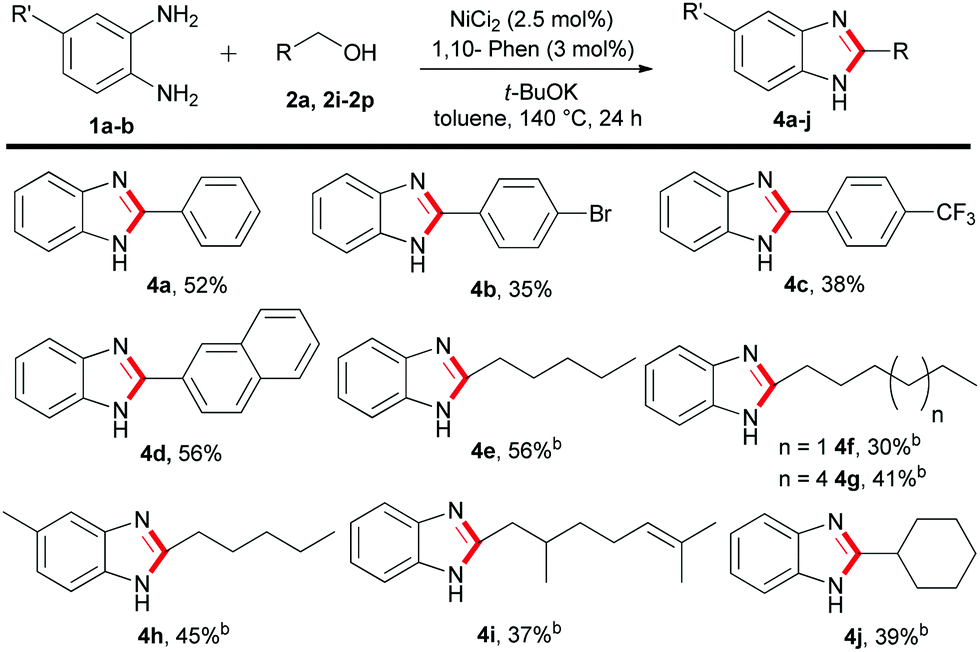
Furthermore, we explored the synthesis of 2-substituted benzimidazoles using the optimized conditions of Table 1. To our delight, a series of benzyl as well as alkyl alcohols were utilized to give the desired products in moderate to acceptable yields (Table 3). Notably, the reaction of 1a with benzyl alcohols decorated with bromide or trifluoromethyl groups gave a moderate yield of 4b–4c. However, when benzyl alcohol 2a and 2-naphthylmethanol 2k were employed, the desired 2-aryl substituted benzimidazoles 4a and 4d were obtained in 52–56% yield, respectively (Table 3). Next, we studied the reactivity of more challenging acyclic and cyclic alkyl alcohols. We were pleased to observe that long chain C6–C10 alkyl alcohols can be incorporated into the benzimidazole core with good to moderate isolated yield (Table 3, 4e–4h). Notably, the renewable terpenoid alcohol, citronellol 2o and 1-cyclohexylmethanol 2p resulted in the 2-alkylbenzimidazoles 4i–4j in acceptable yield (Table 3). This remarkable chemo-selective transformation of citronellol highlights the potential of the present protocol. Nevertheless, in all these cases, the unreacted alcohols were recovered and often a small amount of di-substituted benzimidazoles was observed as side products. At this point, it is to be noted that, variations of alcohol equivalency control the product selectivity to mono- or di-substituted benzimidazoles. More specifically, under the optimised conditions of Table 1, use of two equivalents of alcohols gave di-substituted benzimidazoles, whereas, one equivalent of alcohol is required for 2-substituted benzimidazoles.
Finally, we explored the de-hydrogenative coupling of 1,2-di-aminobenzenes with ethylene glycol for the synthesis of quinoxaline derivatives. Though long chain vicinal diols are an effective partner for quinoxaline synthesis, application of ethylene glycol is still a challenging task and rarely explored.11 Gratifyingly, using our nickel-catalyzed protocol, ethylene glycol efficiently coupled with 1a as well as 4-methoxy-1,2-diaminobenzene 1d and resulted in up to 40% yield of 5a–5b (Scheme 2). Furthermore, screening of reaction conditions, such as the variation of pre-catalysts and bases, and the effect of solvents including variable reaction temperatures did not improve the product yield further (ESI,† Tables S7–S10).
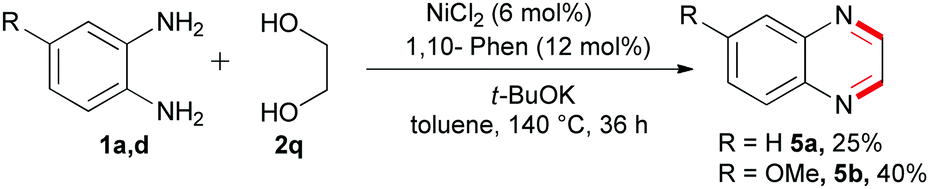 | ||
| Scheme 2 Synthesis of quinoxalines: see ESI.† | ||
The catalytic protocol is tolerant to aryl, alkyl, methoxy, halide (Cl and Br), and trifluoromethyl as well as pyridine, thiophene and furfural moieties. Importantly, the chemo-selective transformations in the presence of reducible alkene highlight the synthetic potential of the established protocol. Notably, applications of benzyl alcohols bearing functional groups, such as nitro, cyano, hydroxyl, esters, imine, amine, aldehyde, alkyne, and alkene moieties, were not successful and we observed only poor or no product conversion to the desired benzimidazoles (ESI,† Table S11).
Next, to gain more insight for such dehydrogenative couplings we explored the initial mechanistic studies for the process. To understand the involvement of the putative key Ni-intermediate species, Cat. A was independently prepared,10 and used for the model reaction of Table 1 in catalytic (2.5 mol%) and stoichiometric (100 mol%) amount. As expected, 3a was obtained in 64–90% yield along with a small amount of 4a (Scheme 3a).
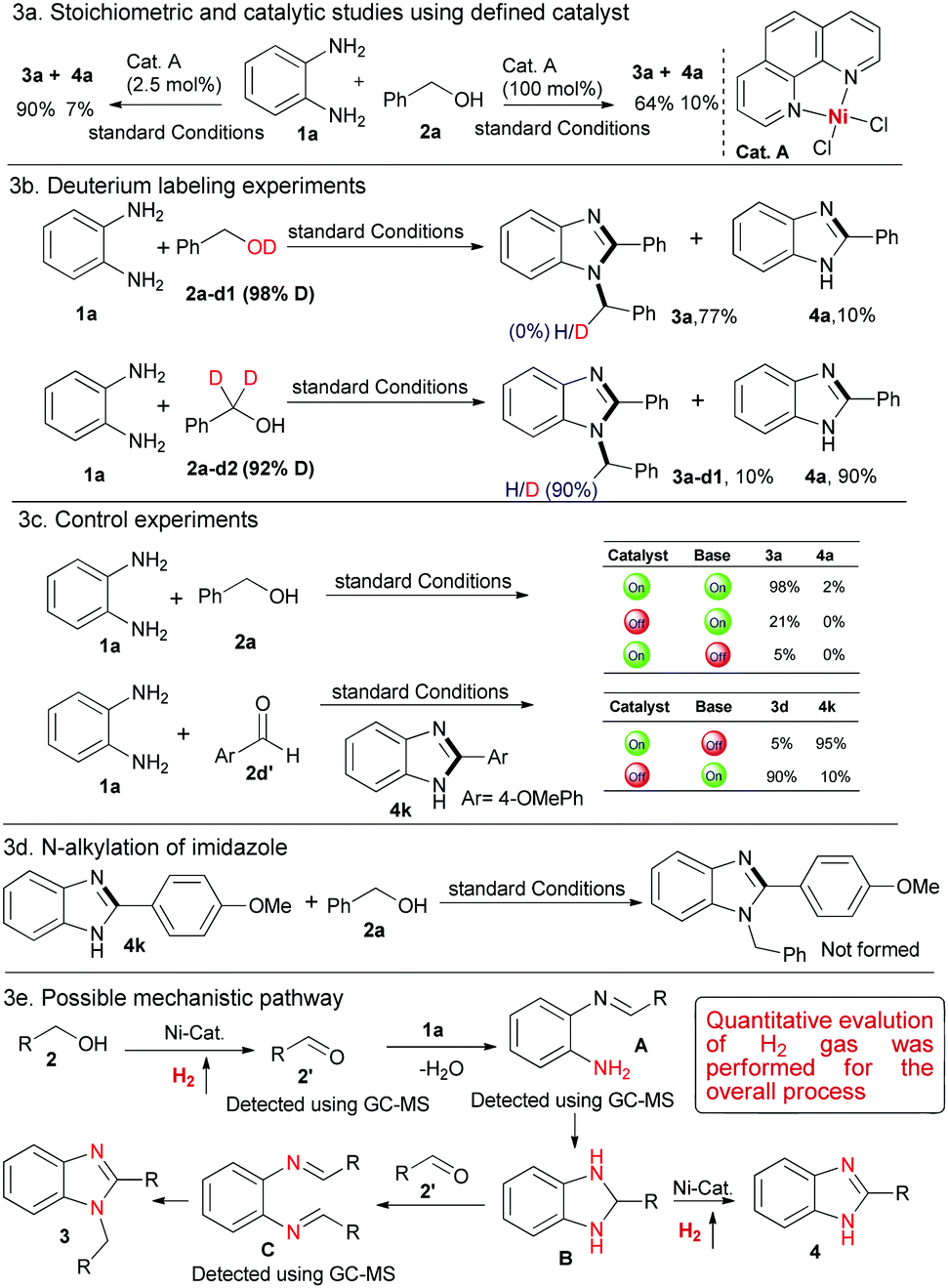 | ||
| Scheme 3 Preliminary mechanistic studies and control experiments. 1a (0.10 mmol) and 2a-d1/2a-d2 (0.20 mmol) were used (see ESI†). | ||
Furthermore, deuterium labeling experiments using 1a with 2a-d1 (98% D), resulted in 3a in 77% yield and we did not observe any deuterium incorporation at the imidazole core (Scheme 3b). However, dehydrogenative couplings of 1a with 2a-d2 (92% D) exhibited 90% deuterium incorporation in 3a-d1 along with 4a in 90% product yield (Scheme 3b and ESI,† Scheme S1). These deuterium labeling experiments evidence the participation of the benzylic C–H bond of alcohols for benzimidazole synthesis. Moreover, control experiments in the absence of base and catalyst highlight their potential role for the dehydrogenative condensation to benzimidazole synthesis (Scheme 3c). In addition, we also performed catalytic condensation of 1a with 4-methoxybenzaldehyde 2d′ under standard conditions. These experiments resulted in the formation of 3d and 4k at variable amounts, which strongly supports that the first step is the metal-catalyzed dehydrogenation of alcohol to aldehyde and thereby, base-mediated condensation/cyclisation facilitate the product formation. Notably, it also provides evidence for the potential role of the metal catalyst for bond activation, condensation followed by the construction of the new bonds for benzimidazole synthesis (Scheme 3c).
On the basis of the initial mechanistic studies and control experiments, we hereby postulated a probable catalytic pathway for the Ni-catalysed dehydrogenative condensation of aromatic diamines with alcohols (Scheme 3e).8a Initially, the Ni-catalysed dehydrogenation of primary alcohol gave aldehyde 2′ followed by condensation with 1a to imine intermediate A, which subsequently undergoes cyclisation and dehydrogenation to give product 4via intermediate B. It is noteworthy to mention that, in the GC-MS analysis of the crude reaction mixture we detected intermediate 2′ as well as intermediate A. Another possibility is that, intermediate B could couple with 2′ to intermediate C, which subsequently undergoes intra-molecular cyclisation and rearranges to 1,2-disubstituted benzimidazoles (Scheme 3e). Nevertheless, to exclude the possibility of N-alkylation of 2-arylimidazoles, 4k was allowed to react with benzyl alcohol under standard conditions and we did not observe any desired product (Scheme 3d). Moreover, during the optimization studies we detected intermediate C in the GC-MS analysis, which supports our hypotheses for 1,2-disubstituted benzimidazole synthesis. Notably, the overall process released only water and dihydrogen as side products, rendering it sustainable. Thereafter, we measured the quantitative evaluation of hydrogen gas for the process and confirmed it through gas chromatography (ESI,† Schemes S4 and S5).
In conclusion, herein we demonstrated the first Ni-catalysed selective dehydrogenative condensation of aromatic diamines with a series of primary alcohols to substituted benzimidazoles. A simple and inexpensive Ni-catalyst and 1,10-phenanthroline ligand enables a variety of functionalised benzimidazoles and quinoxalines in up to 91% yield. The catalytic system is tolerant to alkyl, alkoxy, halides, and trifluoromethyl as well as heterocyclic rings including unsaturated alcohols. Initial mechanistic investigations involving the defined Ni-catalyst and deuterium labeling experiments as well as quantitative determination of hydrogen gas were performed to establish the dehydrogenation process.
The authors thank SERB, India (ECR/2015/000600) and IIT Roorkee (SMILE-32). M. S. thanks DST/2018/IF180079 for financial support. A. B. and K. S. thank IIT-R for a fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed.
- (a) H. M. Smith, High Performance Pigments, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2002, p. 135 Search PubMed; (b) G. G. Scherer, Fuel Cells II, Springer-Verlag, Berlin, 2008, p. 65 CrossRef.
- (a) E. C. Wagner and W. H. Millett, Org. Synth., 1939, 19, 12 CrossRef CAS; (b) J. B. Wright, Chem. Rev., 1951, 48, 397 CrossRef CAS PubMed; (c) P. N. Preston, Chem. Rev., 1974, 74, 279 CrossRef CAS.
- (a) B. M. Trost, Science, 1991, 254, 147 CrossRef; (b) K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503 CrossRef CAS PubMed.
- (a) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed; (b) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed; (c) J. Moran and M. J. Krische, Pure Appl. Chem., 2012, 84, 1729 CAS.
- (a) T. Kondo, S. Yang, K. Huh, M. Kobayashi, S. Kotachi and Y. Watanabe, Chem. Lett., 1991, 1275 CrossRef CAS; (b) L. Li, Q. Luo, H. Cui, R. Li, J. Zhang and T. Peng, ChemCatChem, 2018, 10, 1607 CrossRef CAS; (c) T. Hille, T. Irrgang and R. Kempe, Chem. – Eur. J., 2014, 20, 5569 CrossRef CAS PubMed; (d) A. K. Sharma, H. Joshi, R. Bhaskar and A. K. Singh, Dalton Trans., 2017, 46, 2228 RSC; (e) K. Tateyama, K. Wada, H. Miura, S. Hosokawa, R. Abe and M. Inoue, Catal. Sci. Technol., 2016, 6, 1677 RSC; (f) K. J. Won, H. Jinling, Y. Kazuya and M. Noritaka, Chem. Lett., 2009, 920 Search PubMed; (g) Z. Xu, X. Yu, X. Sang and D. Wang, Green Chem., 2018, 20, 2571 RSC; (h) K. Tateyama, K. Wada, H. Miura, S. Hosokawa, R. Abe and M. Inoue, Catal. Sci. Technol., 2016, 6, 1677 RSC.
- R. M. Bullock, Catalysis Without Precious Metals, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) Z. Xu, D.-S. Wang, X. Yu, Y. Yang and D. Wang, Adv. Synth. Catal., 2017, 359, 3332 CrossRef CAS; (b) P. Daw, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 7456 CrossRef CAS; (c) K. Das, A. Mondal and D. Srimani, J. Org. Chem., 2018, 83, 9553 CrossRef CAS PubMed; (d) A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi and J. M. J. Williams, Org. Lett., 2009, 11, 2039 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (b) A. Valentine and P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef.
- (a) M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152 CrossRef CAS; (b) K. Singh, M. Vellakkaran and D. Banerjee, Green Chem., 2018, 20, 2250 RSC; (c) J. Das, K. Singh, M. Vellakkaran and D. Banerjee, Org. Lett., 2018, 20, 5587–5591 CrossRef CAS PubMed; (d) K. Singh, L. M. Kabadwal, S. Bera, A. Alanthadka and D. Banerjee, J. Org. Chem., 2018, 83, 15406 CrossRef CAS PubMed.
- For quinoxaline synthesis, see: (a) F. Xie, M. Zhang, H. Jiang, M. Chen, W. Lv, A. Zheng and X. Jian, Green Chem., 2015, 17, 279 RSC; (b) P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 7734 CrossRef CAS; (c) M. Mastalir, M. Glatz, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2019, 21, 1116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc02319d |
| This journal is © The Royal Society of Chemistry 2019 |