
The failure of rodent carcinogenesis as a model for Man
Abstract
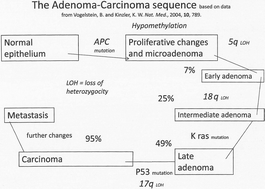
Recent advances in our understanding of the process of carcinogenesis in Man have required revision of our thinking about the classical initiation/promotion sequence; understanding must now encompass the roles of both genetic and epigenetic change, realisation of the importance of the variable genetic backgrounds of the tumour bearers in any group and an understanding of the importance of random genetic events over time. The behavior of tumours, once established, is more complex than has been thought. Current views of the processes involved are not modelled in toxicity testing programmes.
- This article is part of the themed collection: Advances in Experimental and Regulatory Toxicology
 Please wait while we load your content...
Please wait while we load your content...