
The safety evaluation of food flavouring substances: the role of metabolic studies

Abstract
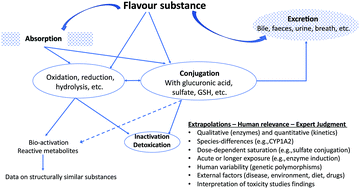
The safety assessment of a flavour substance examines several factors, including metabolic and physiological disposition data. The present article provides an overview of the metabolism and disposition of flavour substances by identifying general applicable principles of metabolism to illustrate how information on metabolic fate is taken into account in their safety evaluation. The metabolism of the majority of flavour substances involves a series both of enzymatic and non-enzymatic biotransformation that often results in products that are more hydrophilic and more readily excretable than their precursors. Flavours can undergo metabolic reactions, such as oxidation, reduction, or hydrolysis that alter a functional group relative to the parent compound. The altered functional group may serve as a reaction site for a subsequent metabolic transformation. Metabolic intermediates undergo conjugation with an endogenous agent such as glucuronic acid, sulphate, glutathione, amino acids, or acetate. Such conjugates are typically readily excreted through the kidneys and liver. This paper summarizes the types of metabolic reactions that have been documented for flavour substances that are added to the human food chain, the methodologies available for metabolic studies, and the factors that affect the metabolic fate of a flavour substance.
- This article is part of the themed collections: Recent Review Articles and Advances in Experimental and Regulatory Toxicology
 Please wait while we load your content...
Please wait while we load your content...