
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature dependence of the photo- and electroluminescence of poly(p-phenylene vinylene) based polymers†
Irina
Rörich
ab,
Ann-Kathrin
Schönbein
a,
Deepthi Kamath
Mangalore
a,
Anielen
Halda Ribeiro
a,
Christian
Kasparek
a,
Christian
Bauer
a,
N. Irina
Crăciun
a,
Paul W. M.
Blom
a and
Charusheela
Ramanan
 *a
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: ramanan@mpip-mainz.mpg.de
bDutch Polymer Institute, P.O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 4th September 2018
Abstract
We show that the exciton transport and decay processes in two poly(p-phenylene vinylene) (PPV) based semiconducting polymers exhibit distinct temperature dependence based on the energetic disorder of the polymer. Time-resolved photoluminescence (TRPL) spectroscopy and Monte Carlo exciton diffusion simulations are used to disentangle the contributions from radiative and non-radiative decay processes, with the latter including non-radiative decay due to exciton diffusion toward trap sites. In a highly disordered polymer, the exciton lifetime and quantum yield are nearly temperature independent. In the case of a less disordered polymer, the exciton lifetime and quantum yield increase with decreasing temperature, due to both freezing out vibrations and less exciton quenching by slowing down the diffusion toward trap sites. We further demonstrate that the temperature dependence of the electroluminescence of polymer light-emitting diodes comprising these polymers is directly correlated with the photoluminescence behavior.
Introduction
Conjugated polymer materials for thin film optoelectronics are subject to energetic disorder arising from both molecular conformational irregularities as well as the sensitivity of these amorphous systems to film processing conditions. As a result, these organic semiconductors are commonly associated with strongly dispersive charge carrier mobilities and exciton diffusion characteristics as compared to their inorganic counterparts.1–3 In order to take advantage of the attractive properties of these systems, such as low-cost manufacturing and mechanical flexibility, it is essential to understand the interplay between energetic disorder and processes affecting optoelectronic performance. Herein, we characterized the effects of energetic disorder on photo and electroluminescence behavior in two poly(p-phenylene vinylene) (PPV) based semiconducting polymers used in polymer light-emitting diodes (PLEDs).The device operation of a PLED is determined by three processes: charge carrier (electron and hole) injection from the electrodes, charge transport through the polymer active layer, and charge recombination generating an exciton, which can then decay radiatively.4–6 Charge carrier and exciton transport in organic semiconductors can be regarded as a hopping process between localized states on conjugated chain segments that are inhomogeneously distributed in size and energy within a Gaussian density of states (DOS).1,7–9 The width of the Gaussian DOS (σ) represents the degree of energetic disorder in the system. The three processes that drive PLED operation depend on the DOS: charge injection is mediated by the relative energy level matching to the electrodes, and charge transport and successive exciton diffusion, prior to radiative decay, depends on the energy level distribution. An exciton introduced into an arbitrary energy level of the DOS will subsequently migrate toward the lower-energy tail via dispersive relaxation steps. At quasi-equilibrium, the exciton reaches the energy level of the highest population of states, located at −σ2/kT, where the hopping is determined by thermally-activated transfer.1,9 Thus, the energetic disorder resulting from molecular conformation, structural defects, and the variety of inter- and intrachain interactions due to the irregular morphologies, will govern the exciton diffusion and decay characteristics in PPV films for PLEDs.10,11 Time-resolved photoluminescence (PL) experiments enable characterization of the exciton diffusion behavior in conjugated polymers. An exciton, formed by either photo-excitation or Langevin recombination in a PLED, will decay back to the ground state within a lifetime τf. The PL quantum yield (ϕf) represents the ratio between radiative and non-radiative processes.12 In a thin film with significant chromophore–chromophore interactions, energy-transfer will occur via hopping along inter- and intrachain segments until the exciton decays either radiatively or non-radiatively. Thus, the PL lifetime τf and quantum yield ϕf are governed by the intrinsic radiative (kr) and non-radiative (knr) decay rates, as well as the exciton diffusion with a rate kdiff, which represents the non-radiative decay related to exciton quenching at defect sites (Fig. 1, eqn (1) and (2)).13
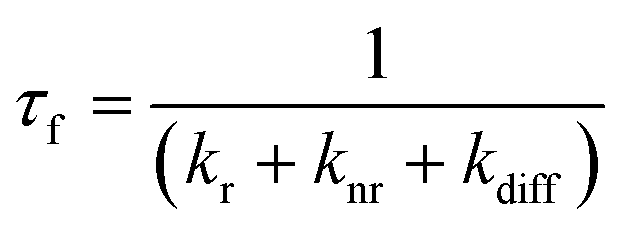 | (1) |
 | (2) |
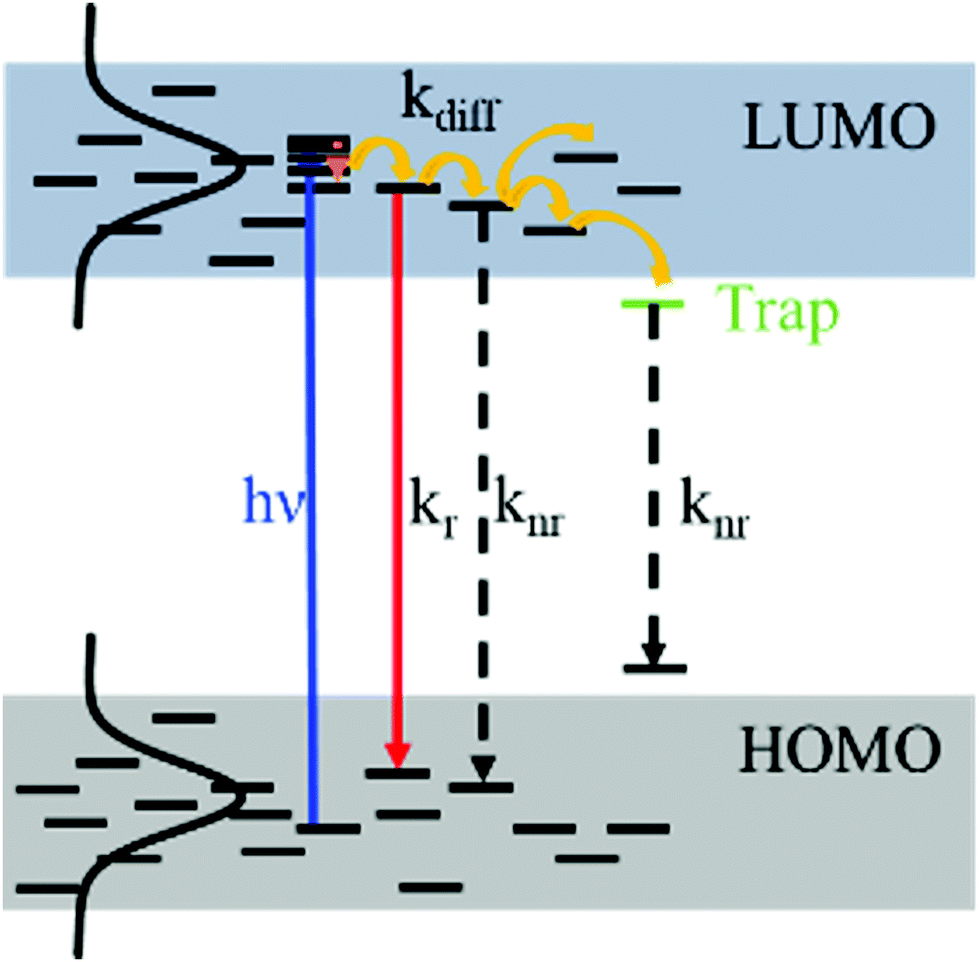 | ||
| Fig. 1 The photophysical processes of excitons in a conjugated polymer thin film, which determine the exciton lifetime τf and the photoluminescence quantum yield, ϕf. | ||
Previous work demonstrates that τf and ϕf directly correlate with the energetic disorder of conjugated polymers, where increased disorder slows down the diffusion of the exciton toward non-radiative quenching sites and strongly enhances both τf and ϕf.14 This is consistent with diffusion-limited exciton quenching at defects in organic semiconductors.15 It should be noted that while the electronic DOS relevant for charge transport and exciton DOS are not the same, both represent the energy level distribution in the polymer. Consistent with this and our previous results, Markov et al. observed a three orders of magnitude enhanced charge carrier mobility in PPV derivatives with decreasing energetic disorder.16
Although charge transfer and exciton diffusion processes are well studied in disordered conjugated polymers, a significant open question remains as to how the PL characteristics of a polymer influence the PLED conversion efficiency. For example, the PLED efficiency is charge carrier mobility independent, and is therefore expected to be temperature independent with regards to the active layer material.17,18 However, the PL behavior of a polymer thin film may vary with temperature as the chromophore–chromophore interactions will affect the balance of kr, knr, and kdiff. Since the PLED efficiency is governed by the formation of excitons, it follows that the respective temperature dependencies of the PLED and PL characteristics should have a direct correlation.
Here, we investigate this relationship by measuring the temperature-dependence of the PL and PLED efficiencies in the PPV derivatives BEH-PPV (poly[2,5-bis(2′-ethylhexyloxy)-1,4-phenylene vinylene]) and SY-PPV (SuperYellow copolymer, Merck) (see Fig. 2 for chemical structures). These materials exhibit different degrees of energetic disorder, as the width of the charge carrier Gaussian DOS (σelectrical) is determined to be 0.092 and 0.140 eV for BEH-PPV and SY-PPV, respectively.16 Furthermore, the exciton lifetime is strongly enhanced with increasing disorder, ranging from 180 ps for BEH-PPV to 1.9 ns for SY-PPV.14 In the new work presented here, time-resolved photoluminescence spectroscopy (TRPL) at varying temperature shows a correlation between the conformational energetic disorder and the balance between radiative (kr) and non-radiative decay processes (knr and kdiff). The thin film of SY-PPV, a highly disordered polymer, exhibits a nearly temperature independent photoluminescence quantum yield, while the more ordered BEH-PPV demonstrates a significant decrease in non-radiative quenching with decreasing temperature (decrease of kdiff and knr). We then fabricated PLEDs with these materials in order to correlate device behavior with the results from our optical experiments. We found that PLEDs based on the disordered SY-PPV show temperature independent current efficiencies, whereas the better ordered BEH-PPV demonstrates enhanced PLED efficiency with lowering temperature, consistent with the temperature dependence of the photoluminescence quantum yield. In addition, we carried out Monte Carlo simulations to model how the exciton diffusion in the semiconductors depends on both the energetic disorder and the concentration of non-radiative quenching-sites. The results are discussed within the context of the diffusion-limited exciton quenching model.
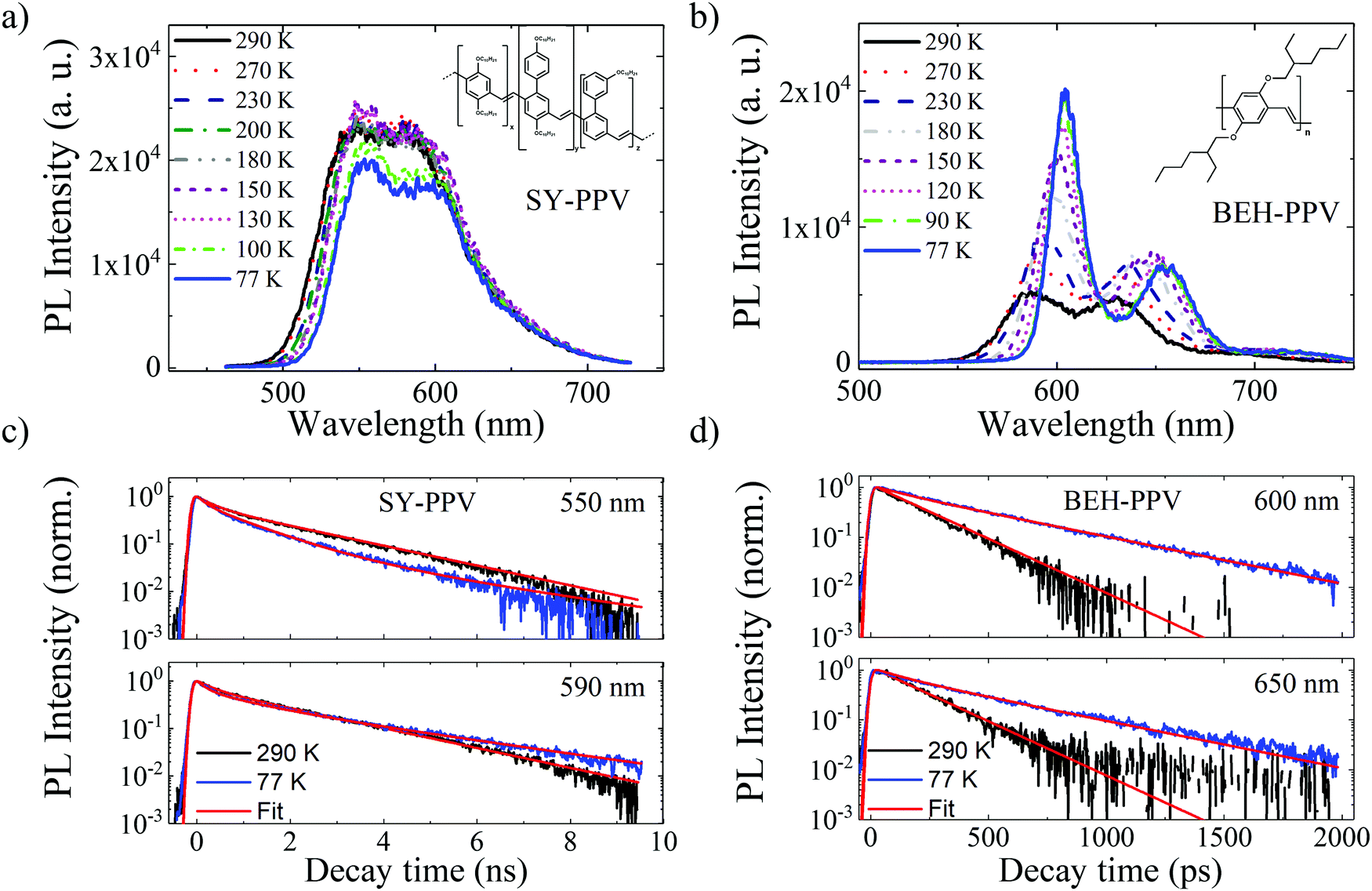 | ||
| Fig. 2 (a and b) Temperature dependent photoluminescence spectra and (c and d) associated normalized PL decay traces at specified wavelengths for SY-PPV and BEH-PPV. Spectral traces are taken at the maximum from the time-resolved measurements. Fits to the decay traces are from global analysis as described in the text, and are further detailed in Tables 1 and 2. | ||
Experimental
Materials and thin film preparation
Super Yellow (SY-PPV) was purchased from Merck KGaA (PDY-132, Mw > 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000 g mol−1, Mn > 400000 g mol−1) and phenyl-C61-butyric acid methyl ester (PCBM) was purchased from Solene BV. Both were used without further purification. Poly[2,5-bis(2′-ethylhexyloxy)-1,4-phenylene vinylene] (BEH-PPV) was synthesized in house as follows: 8.76 g (16.8 mmol) 1,4-bis(bromomethyl)-2,5-bis(2-ethyl-hexyloxy)benzene (premonomer) was dissolved in 500 mL dry, degassed tetrahydrofuran (THF) in a 1 L three necked flask and cooled down to −95 °C under Ar. In a second flask a base solution of 9.28 g (82.7 mmol) potassium tert-butoxide was dissolved in 55 mL dry, degassed THF and also cooled down to −95 °C under Ar. Under vigorous stirring, the base solution was added to the premonomer solution via syringe in order to maintain the Ar atmosphere. The reaction mixture was left to warm up to room temperature over 5 hours. After 24 hours of total stirring time, the flask was equipped with a reflux condenser and the reaction mixture heated to reflux for 24 hours. Subsequently the polymer was precipitated in 3 L methanol, filtered off and dried. The resultant solid was dissolved in 700 mL degassed chloroform to give a concentration of 7 mg mL−1. The polymer solution was again heated to reflux for 48 hours, precipitated in methanol, filtered off, and dried. 4.66 g of a red compact solid was obtained with 77% yield. GPC-MALLS (THF, 30 °C): Mw = 640370 g mol−1, Mn = 293120 g mol−1, PDI = 2.18.
300000 g mol−1, Mn > 400000 g mol−1) and phenyl-C61-butyric acid methyl ester (PCBM) was purchased from Solene BV. Both were used without further purification. Poly[2,5-bis(2′-ethylhexyloxy)-1,4-phenylene vinylene] (BEH-PPV) was synthesized in house as follows: 8.76 g (16.8 mmol) 1,4-bis(bromomethyl)-2,5-bis(2-ethyl-hexyloxy)benzene (premonomer) was dissolved in 500 mL dry, degassed tetrahydrofuran (THF) in a 1 L three necked flask and cooled down to −95 °C under Ar. In a second flask a base solution of 9.28 g (82.7 mmol) potassium tert-butoxide was dissolved in 55 mL dry, degassed THF and also cooled down to −95 °C under Ar. Under vigorous stirring, the base solution was added to the premonomer solution via syringe in order to maintain the Ar atmosphere. The reaction mixture was left to warm up to room temperature over 5 hours. After 24 hours of total stirring time, the flask was equipped with a reflux condenser and the reaction mixture heated to reflux for 24 hours. Subsequently the polymer was precipitated in 3 L methanol, filtered off and dried. The resultant solid was dissolved in 700 mL degassed chloroform to give a concentration of 7 mg mL−1. The polymer solution was again heated to reflux for 48 hours, precipitated in methanol, filtered off, and dried. 4.66 g of a red compact solid was obtained with 77% yield. GPC-MALLS (THF, 30 °C): Mw = 640370 g mol−1, Mn = 293120 g mol−1, PDI = 2.18.
Thin films for optical characterization were prepared from 6 mg mL−1 (SY-PPV) and 5.2 mg mL−1 (BEH-PPV) polymer or polymer:PCBM blend solution in chlorobenzene (Sigma-Aldrich, anhydrous 99.8%) on 1 × 2 cm quartz substrates. The substrates were cleaned with acetone and then propanol in an ultrasonic bath and dried for 20 minutes at 140 °C. The films were spin-coated (60 s 1000 rpm; 30 s 4000 rpm) in a nitrogen atmosphere. The polymer layer thicknesses were determined by using a surface profilometer (Bruker, DektakXT), yielding 80 nm for SY-PPV and 160 nm for BEH-PPV. The films were measured the same day they were prepared, in order to limit polymer degradation.
Steady-state absorption and photoluminescence
Steady-state absorption was measured on a Perkin-Elmer Lambda 900 UV/VIS/NIR spectrometer. Steady-state photoluminescence was measured on a J&M Tidas FL3095SL spectrofluorometer.Time-resolved photoluminescence measurements
TRPL was measured with a C5680 Hamamatsu streak camera system. Optical samples were photoexcited at λex = 400 nm with the frequency-doubled output from a Ti:sapphire laser (Coherent, Libra HE, 3.5 mJ, 100 fs) with a repetition rate of either 1 kHz (10 ns time window, 0.12 ns instrument response) or 80 MHz (2 ns time window, 14 ps instrument response). The laser power at the sample was 65 μW at 1 kHz for SY-PPV, and 225 μW at 80 MHz for BEH-PPV, as these powers avoided annihilation effects (Fig. S1, ESI†). The laser fluence at the sample was <1 μJ cm−2. In order to limit polymer degradation due to oxygen exposure, the samples were loaded into a continuous flow nitrogen cooled cryostat (Optistat CF-V2, Oxford) equipped with temperature controller (Mercury iTC, Oxford) and measured under a dynamic vacuum of 10−4–10−5 mbar for the entire temperature range. Each sample was first measured at 298 K and then cooled to 77 K. The sample was then heated to each measurement temperature at a rate of ∼1 K min−1, and the temperature stabilized for 5 minutes before running the experiment. At the end of the heating procedure, the measurement at 298 K was repeated. The consistency of the 298 K measurements provided good evidence that the sample did not intrinsically change over the course of the experiments.Data analysis and simulations
The photoluminescence decay dynamics are derived from global analysis19 of the TRPL data was done using the R-package TIMP software20 with the graphical interface Glotaran 1.5.1.21 Regarding the Monte Carlo simulation, the utilized scheme models the PL decay in semiconductor–quencher mixtures in order to extract the exciton diffusion parameters, where the excitons and the quencher molecules, depending on their concentration, are randomly placed in a cubic simulation box of 50 × 50 × 50 nm length as balls of 1 nm diameter. The simulation supposes normal diffusion, which is modeled as a random walk with a constant exciton hopping distance (“hop size”). For each time iteration δt every exciton is moved in a random 3D direction for a fixed distance δs, which correlates with the exciton diffusion coefficient as follows: D = δs2/6δt. Radiative recombination is assumed if an exciton has not been quenched after time τi, which is fixed at the beginning of the simulation. Thus, the simulation is based on the exciton-diffusion limited quenching model at non-radiative quenchers. A comprehensive description of the Monte Carlo simulation is described elsewhere,22,23 and the software package can be downloaded from the internet.24 In the case of a monoexponential PL decay, the Stern–Volmer analysis is used to extract the exciton diffusion parameters, as described previously in literature.12,14,15PLED device fabrication and characterization
The PLED devices were fabricated on glass-ITO (tin-doped indium oxide) substrates according to the device structure ITO/PEDOT:PSS (55 nm)/PPV/Ba (5 nm)/Al (100 nm). The substrates were cleaned with acetone and propanol in an ultrasonic bath, dried and treated with ultraviolet (UV)-ozone. PEDOT:PSS (Heraeus Clevios™ Al 4083) was spin-coated (60 s 1200 rpm; 20 s 4000 rpm) after UV-ozone treatment. The PPV layers were spin-coated in a nitrogen atmosphere. The layer thickness of BEH-PPV was 165 nm, obtained from 5.2 mg mL−1 chlorobenzene solution and spin-speeds of 1000 rpm for 60 s followed by 4000 rpm for 30 s. The layer thickness of SY-PPV was 80 nm, obtained from a 6 mg mL−1 chlorobenzene solution (60 s 1000 rpm; 30 s 4000 rpm). The SY-PPV device fabricated from a 5.5 mg mL−1 toluene solution (20 s 1000 rpm; 60 s 250 rpm), gave a film thickness of 100 nm. The Ba/Al cathode was thermally evaporated onto the PPV layer (chamber pressure 10−7 mbar). The current efficiencies were measured with a Keithley 2400 source meter in nitrogen atmosphere. The electroluminescence spectra were measured at 7 V with an Ocean optics (USB4000-UV-Vis-ES) spectrometer.Results
Temperature-dependent time-resolved photoluminescence
The PL spectrum of SY-PPV collected at the maximum of the TRPL intensity (Fig. 2a) exhibits a broad line shape from 500–700 nm, which fits to a sum of three Gaussian peaks (Table S1, ESI†), ascribed to the electronic S0,0 and vibronic S0,1 and S0,2 transitions. The peak positions are in good agreement with previous reports,25 as well as the steady-state PL measurements (Fig. S2, ESI†). The PL spectrum red-shifts with lowering temperature. The peak intensity stays relatively constant from 290–130 K, and then slightly decreases at 100 and 77 K. The PL spectrum of BEH-PPV spans 550–750 nm (Fig. 2b) and is similarly characterized by one primarily electronic transition (S0,0) and two vibronic peaks (S0,1, S0,2). The peak positions from a fit to a sum of three Gaussians are shown in Table S2 (ESI†). With lowering temperature, the PL spectrum of BEH-PPV red-shifts and the peak intensity increases. The relative intensity between the electronic (S0,0) and vibronic peaks (S0,x) also changes with decreasing temperature. At 298 K, the S0,0 and S0,1 transitions are of similar intensity, while at 77 K, the S0,0 transition is more than twice as intense as the S0,1 transition, in agreement with previous work. This indicates a decrease in electron-vibrational coupling with decreasing temperature.26 The narrowing of the fluorescence peaks with lowering temperature is more evident in BEH-PPV as opposed to SY-PPV.The comparison of the normalized PL decay kinetics measured at the S0,0 (550 nm) and S0,1 (590 nm) transitions (Fig. 2c, black and blue traces) demonstrate that the SY-PPV temperature-dependence depends on the emission wavelength. At 550 nm, the 77 K trace decays faster than the 290 K trace, while at 590 nm, the traces are mostly similar, with the 290 K trace decaying faster by a small amount. Fig. 2c shows only the lowest and highest measured temperatures for the S0,0 and S0,1 transition, for clarity, but the observed trend is consistent throughout the entire measured temperature range (Fig. S3, ESI†). The normalized BEH-PPV PL decay traces (Fig. 2d), monitored at 600 and 650 nm, demonstrate that the PL lifetime at 77 K is longer than at 290 K at all wavelengths. This trend is consistent through the measured temperature range (Fig. S4, ESI†).
The TRPL decay dynamics were modeled with a global analysis scheme, which takes into account the decay dynamics across the entire spectral range.19,21 The fits are represented as red lines in Fig. 2c and d and the lifetimes and relative amplitudes are summarized in Tables 1 and 2. For both materials, the data were fit to a sum of a fast and slow exponential contribution. The measured exciton lifetime (τf) is then determined from a weighted average of the two kinetic components, (eqn (3))
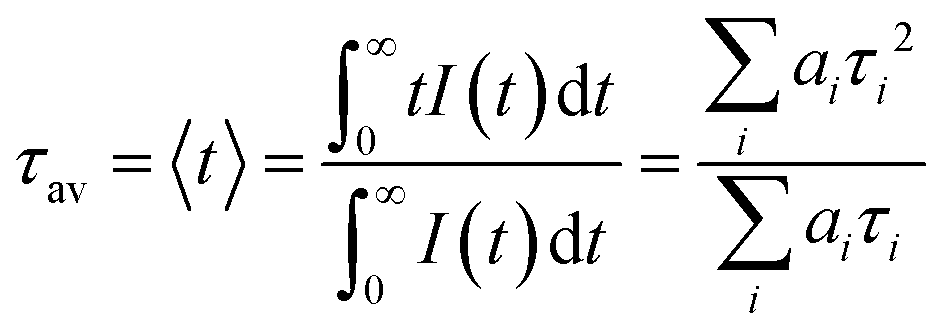 | (3) |
| T (K) | A 1 | τ 1 (ps) | A 2 | τ 2 (ps) | τ f (ps) | ϕ T/ϕ290K |
|---|---|---|---|---|---|---|
| 290 | 0.39 | 360 | 0.61 | 2080 | 1900 | 1.00 |
| 270 | 0.42 | 370 | 0.58 | 2190 | 1990 | 1.02 |
| 230 | 0.46 | 410 | 0.54 | 2390 | 2140 | 0.99 |
| 200 | 0.47 | 420 | 0.53 | 2480 | 2200 | 0.98 |
| 180 | 0.49 | 440 | 0.51 | 2540 | 2240 | 0.96 |
| 150 | 0.51 | 450 | 0.49 | 2570 | 2250 | 1.01 |
| 130 | 0.51 | 460 | 0.49 | 2600 | 2260 | 0.99 |
| 100 | 0.53 | 480 | 0.47 | 2680 | 2320 | 0.84 |
| 77 | 0.51 | 490 | 0.49 | 2710 | 2360 | 0.78 |
| T (K) | A 1 | τ 1 (ps) | A 2 | τ 2 (ps) | τ f (ps) | ϕ T/ϕ290K |
|---|---|---|---|---|---|---|
| 290 | 0.89 | 160 | 0.11 | 370 | 210 | 1.00 |
| 270 | 0.91 | 180 | 0.09 | 450 | 240 | 1.32 |
| 230 | 0.90 | 220 | 0.10 | 550 | 290 | 1.49 |
| 180 | 0.87 | 270 | 0.13 | 670 | 370 | 1.70 |
| 150 | 0.83 | 290 | 0.17 | 630 | 390 | 1.84 |
| 120 | 0.79 | 300 | 0.21 | 600 | 410 | 1.76 |
| 90 | 0.62 | 190 | 0.38 | 480 | 370 | 1.71 |
| 77 | 0.56 | 130 | 0.44 | 480 | 390 | 1.76 |
Exciton diffusion coefficient and diffusion length
As a next step, we investigated the spatial and temporal evolution of the singlet exciton population, characterized by the exciton diffusion coefficient D, as a function of temperature. We measured fluorescence quenching dynamics of thin polymer films with randomly distributed PCBM molecules, known to be efficient exciton quenchers.9,14,28–30 Thin films of SY-PPV and BEH-PPV, each blended with known concentrations of PCBM, were prepared from chlorobenzene solution. The average fluorescence lifetime was determined from TRPL experiments, using the same procedure described in the previous section. The results were analyzed by Monte Carlo simulation, which is appropriate for multi-exponential decays, to return the exciton diffusion coefficient D.22,23 The exciton diffusion length LD, which corresponds to the average distance an exciton can diffuse during its lifetime τf, is calculated as follows,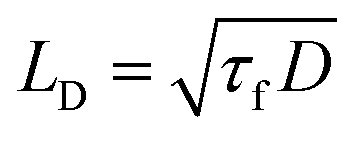 | (4) |
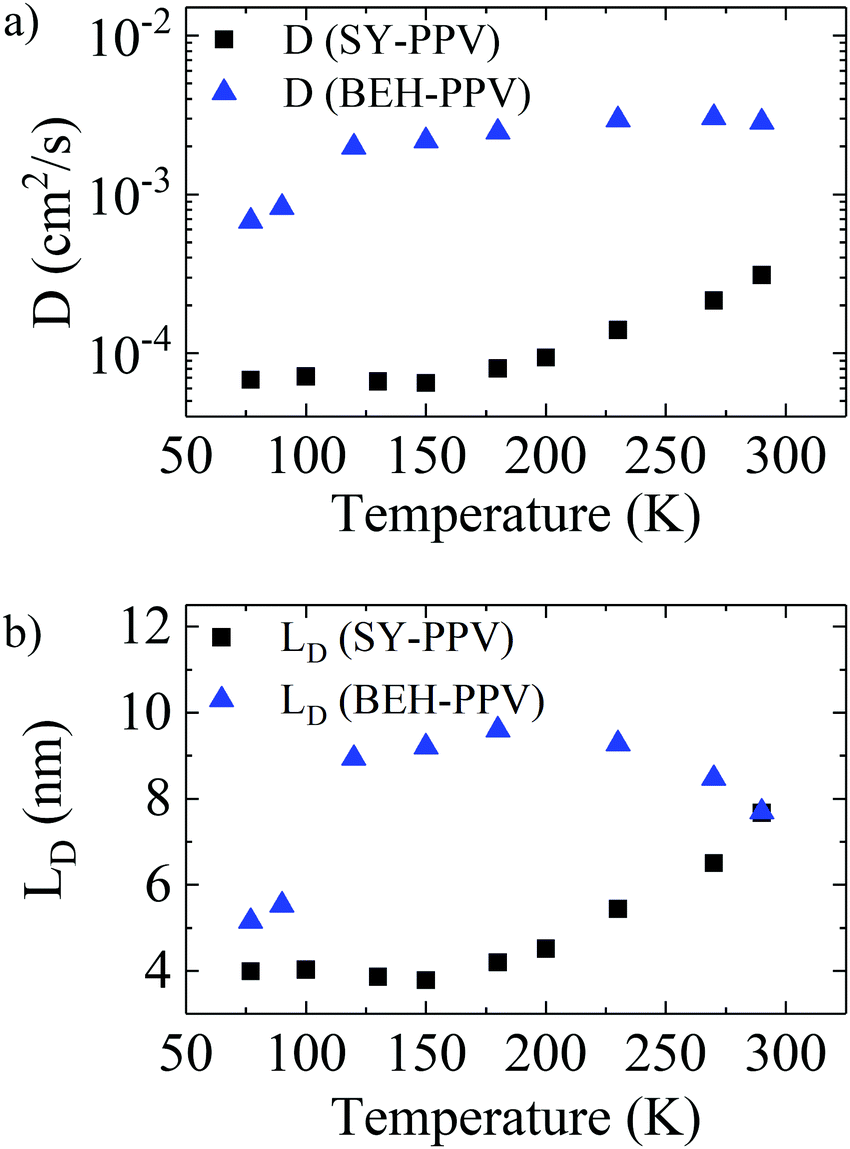 | ||
| Fig. 3 Temperature-dependence of (a) exciton diffusion coefficient D and (b) exciton diffusion length LD for SY-PPV and BEH-PPV, determined from time-resolved fluorescence quenching experiments of thin films. | ||
The better ordered BEH-PPV exhibits overall higher exciton diffusion coefficients and exciton diffusion lengths than the less well-ordered SY-PPV. With lowering temperature, SY-PPV shows a significant reduction of the exciton diffusion parameters between 298–180 K, with D decreasing from 3.1 × 10−4 cm2 s−1 to 8.0 × 10−5 cm2 s−1 and LD decreasing from 7.7 nm to 4.2 nm. Further cooling from 180 K to 77 K reveals nearly constant exciton diffusion parameters for SY-PPV. In contrast, the exciton diffusion parameters for the better ordered BEH-PPV display less temperature dependence in the higher temperature range (298–120 K), while in the lower temperature range 100–77 K, there is greater dependence on temperature, with D decreasing from 2.5 × 10−3 cm2 s−1 to 6.8 × 10−4 cm2 s−1 and LD decreasing from 9.6 nm to 5.2 nm. Both polymers exhibit an approximately 75% reduction in D at 77 K vs. 290 K.
For BEH-PPV, we also compared these results with values derived from a monoexponential fit to the fluorescence decay, which we have reported before for this polymer at room temperature.14 The temperature-dependent exciton diffusion parameters, in this case determined by the Stern–Volmer equation, and exciton lifetimes determined from monoexponential fits of the PL decays are shown in Fig. S5 (ESI†). The results show a similar trend of D and LD in BEH-PPV compared to the results determined from global analysis and Monte Carlo simulations (Fig. 3), with slightly higher values for LD. As D is quite similar for the two fits, the difference in LD is most likely due to the longer fluorescence lifetime from the monoexponential fits, which do not account for the increased energy transfer between polymer chains at lower temperatures.
Temperature-dependent polymer light-emitting diode efficiencies
PLEDs of each SY-PPV and BEH-PPV were fabricated in order to test the temperature dependence of the device efficiencies. The device structure ITO/PEDOT:PSS/PPV/Ba/Al was used for both active layer materials. The SY-PPV active layer is 80 nm thick and the BEH-PPV layer is 165 nm thick. Fig. 4 shows the temperature dependent device current efficiencies vs. voltage. The current efficiency is defined as photon/charge carrier. It is calculated by dividing the photocurrent density by the electrical current density (Fig. S6, ESI†). This unitless value is a measure for the relation between the number of emitted photons and the number of charges that pass through the device for a given time interval. The SY-PPV PLED exhibits no significant change in current efficiency with decreasing temperature in the range of 295–233 K.17 We also confirmed this in a device made from toluene, which is the more commonly used solvent for processing SY-PPV31 (Fig. S7, ESI†). The current efficiency of the BEH-PPV PLED, however, increases by more than a factor of two when the device temperature is decreased between 295–215 K. Additionally, the rise of current efficiency with increasing voltage becomes steeper as the BEH-PPV device temperature is lowered.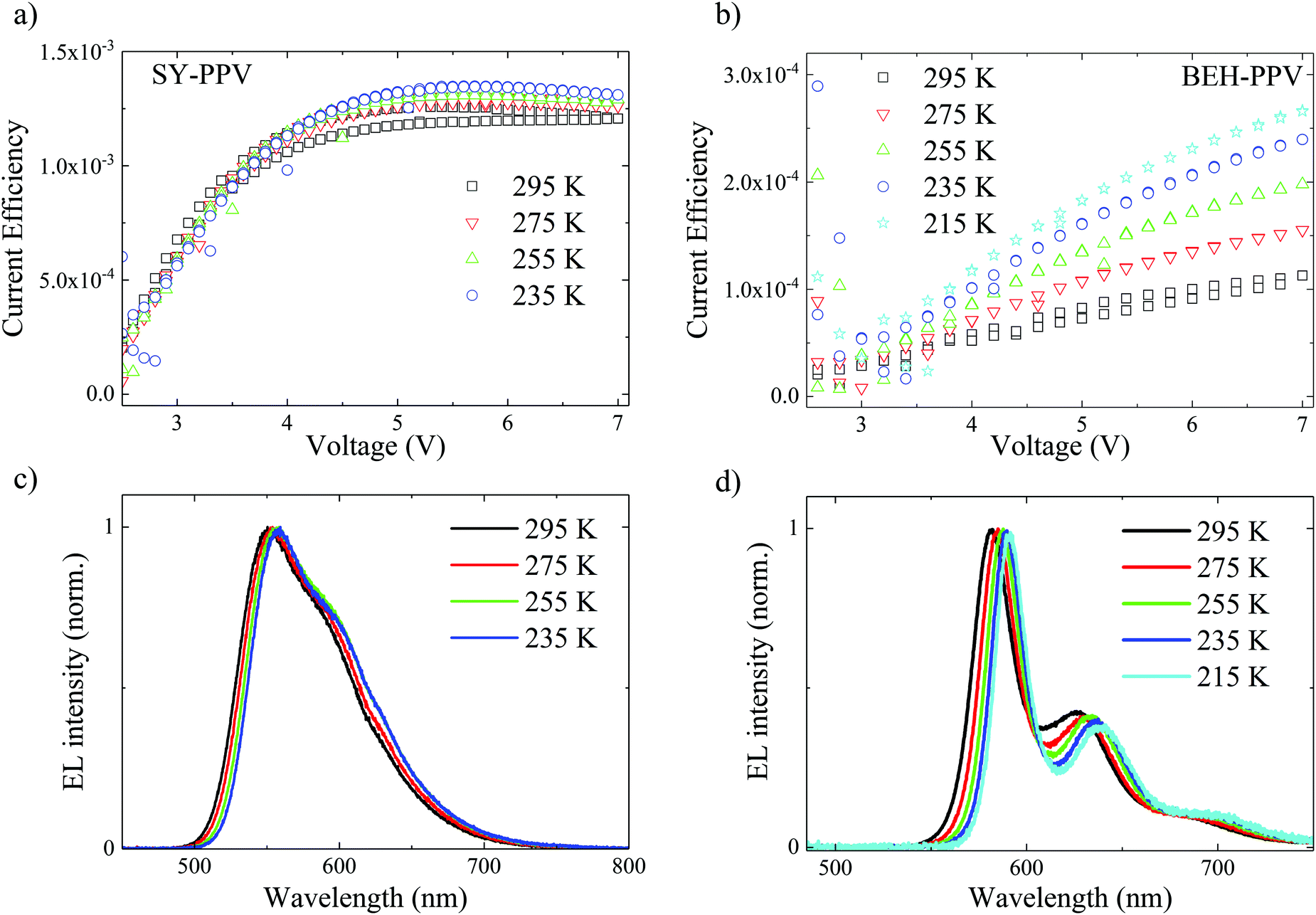 | ||
| Fig. 4 Temperature dependent current efficiencies vs. voltage of PLEDs comprised of (a) SY-PPV and (b) BEH-PPV along with electroluminescence spectra of the (c) SY-PPV and (d) BEH-PPV PLED measured at 7 V, normalized to the spectral maximum. | ||
Temperature dependent EL spectra of SY-PPV and BEH-PPV are shown in Fig. 4c and d. The SY-PPV EL appears qualitatively similar to the PL, while the BEH-PPV EL demonstrates a different spectral line shape than PL spectra in the same temperature range, with the S0,0 peak exhibiting always higher intensity than S0,1. To understand this further, we compared the steady-state PL of the pristine polymer films to that within the PLED stack (Fig. S2, ESI†). Under the same measurement conditions, the BEH-PPV PL spectrum within the PLED stack shows a change in peak ratio relative to the pristine BEH-PPV film. In SY-PPV, there is also a small shift and narrowing of the PL spectrum within the PLED stack. This EL spectral line shape is consistent with an optical microcavity effect in the PLED, wherein the near-field coupling results in a narrowing of the spectral line shape and enhances the spontaneous emission rate.2,32 Our results in Fig. 4 and Fig. S2 (ESI†) show that this is a much stronger effect in the 165 nm BEH-PPV device than in the 80 nm SY-PPV device.
Discussion
The BEH-PPV and SY-PPV polymers studied here have different DOS characteristics due in part to their molecular conformations. The chemical structures in Fig. 2 show that BEH-PPV has two symmetrical alkoxy side chains and the configurational freedom of the individual chains is limited, leading to less energy spread of the Gaussian density of states,33 and thus a narrow width of the energy distribution. In contrast, the random copolymer SY-PPV exhibits high configurational freedom of individual conformational subunits, resulting in more structural disorder and thus a broader width of the Gaussian DOS. The width of the excitonic DOS (σoptical) is calculated by assuming that the energy position of the S0,0 transition from our PL measurements corresponds to the level of the most populated states, which lies at the equilibrium level −σ2/kT below the center of the DOS.1,9 The slope of the relationship between the S0,0 peak position vs. 1/T allows us to extract σoptical. From this we find that energetic disorder of BEH-PPV (σoptical = 0.033 eV) is indeed less than that of SY-PPV (σoptical = 0.048 eV). The half-width of the excitonic (optical) Gaussian DOS is about 3 times smaller than the half-width of the charge carrier (electrical) DOS extracted from charge transport measurements, for BEH-PPV σelectrical = 0.092 eV and SY-PPV σelectrical = 0.140 eV, respectively.16 Previous work showed that σelectrical is larger than σoptical due to the different nature of interactions between charge carriers and excitons within the DOS.1 Nonetheless, BEH-PPV reproducibly exhibits a narrower DOS than SY-PPV.Singlet excited states in disordered polymers are generated at an arbitrary energy-site within the high-energy tail of the Gaussian DOS and simultaneously undergo downhill migration toward lower energy-sites.7,34–36 The down-hill migration, also known as spectral diffusion, ends when the excitons approach the energy level of the most populated excited states, provided that the time needed to approach quasi-equilibrium is shorter than the intrinsic exciton lifetime. At room temperature, spectral diffusion is followed by temperature activated hopping, where balanced downward and thermally activated upward hopping occurs. With decreasing temperature, the equilibrium level shifts deeper into the tail of the Gaussian DOS, causing a red-shift of the PL spectrum.9,25,36 There, the probability to transfer to nearby energy-sites decreases, resulting in a slower exciton transfer. Additionally, thermally activated hopping becomes less pronounced. Thus, the exciton diffusion characteristics (and quantum yield) of a conjugated polymer thin film depend strongly on the disorder parameter σ and the temperature T,37 as well as on the starting singlet excitation energy within the DOS and the coupling strength between the energy sites.38
Temperature-dependent exciton decay characteristics
The differences in energetic disorder between BEH-PPV and SY-PPV manifest in the temperature dependent PL and exciton diffusion characteristics as measured in thin films. The exciton decay processes in these systems are governed by radiative decay, non-radiative decay, and exciton diffusion to non-radiative traps (Fig. 1). The TRPL experiments show that, with decreasing temperature, both the SY-PPV and BEH-PPV films demonstrate longer fluorescence lifetimes. However, while the relative quantum yield for SY-PPV is fairly constant until very low temperatures, the one of BEH-PPV increases along with the lifetime.The excited state transfer rates for the two polymers were calculated in order to quantitatively describe the exciton decay processes (Tables 3, 4 and Fig. S8, ESI†). The lifetime τi correlates with the transfer rate ki with the following relation: τi = 1/ki. The values were determined as follows: the radiative decay rate kr is first calculated by eqn (5),
| kr = ϕf/τf = (ϕT/ϕ290K·ϕabs)/τf | (5) |
| T (K) | ϕ f | k f (s−1) | k r (s−1) | k diff (s−1) | k nr (s−1) |
|---|---|---|---|---|---|
| 290 | 0.60 | 5.3 × 108 | 3.2 × 108 | 5.2 × 107 | 1.6 × 108 |
| 270 | 0.61 | 5.0 × 108 | 3.1 × 108 | 3.6 × 107 | 1.6 × 108 |
| 230 | 0.59 | 4.7 × 108 | 2.8 × 108 | 2.3 × 107 | 1.7 × 108 |
| 200 | 0.59 | 4.5 × 108 | 2.7 × 108 | 1.5 × 107 | 1.7 × 108 |
| 180 | 0.57 | 4.5 × 108 | 2.6 × 108 | 1.3 × 107 | 1.8 × 108 |
| 150 | 0.60 | 4.5 × 108 | 2.7 × 108 | 1.1 × 107 | 1.7 × 108 |
| 130 | 0.60 | 4.4 × 108 | 2.6 × 108 | 1.1 × 107 | 1.7 × 108 |
| 100 | 0.51 | 4.3 × 108 | 2.2 × 108 | 1.1 × 107 | 2.0 × 108 |
| 77 | 0.47 | 4.2 × 108 | 2.0 × 108 | 1.1 × 107 | 2.1 × 108 |
| T (K) | ϕ f | k f (s−1) | k r (s−1) | k diff (s−1) | k nr (s−1) |
|---|---|---|---|---|---|
| 290 | 0.06 | 5.0 × 109 | 3.0 × 108 | 4.3 × 109 | 3.6 × 108 |
| 270 | 0.08 | 4.7 × 109 | 3.7 × 108 | 4.1 × 109 | 2.1 × 108 |
| 230 | 0.09 | 3.9 × 109 | 3.5 × 108 | 3.4 × 109 | 1.4 × 108 |
| 180 | 0.10 | 2.9 × 109 | 2.9 × 108 | 2.5 × 109 | 1.3 × 108 |
| 150 | 0.11 | 2.7 × 109 | 2.9 × 108 | 2.2 × 109 | 1.4 × 108 |
| 120 | 0.11 | 2.9 × 109 | 3.0 × 108 | 2.4 × 109 | 2.0 × 108 |
| 90 | 0.10 | 2.7 × 109 | 2.8 × 108 | 1.8 × 109 | 6.5 × 108 |
| 77 | 0.11 | 2.7 × 109 | 2.9 × 108 | 1.7 × 109 | 7.5 × 108 |
In order to disentangle knr from kdiff, we used the exciton diffusion model to simulate quenching efficiencies Q (Q = 1 − NR/N0) for a range of diffusion coefficients D. The solid lines in Fig. 5 show the dependence of Q as determined by the ratio of the number of radiatively decaying excitons (NR) and the number of excitons that would decay radiatively in the absence of a quencher (N0). The curves represent Q for a constant intrinsic exciton lifetime τr = 3 ns. The model also assumes a constant number of exciton defects (background quenchers). In SY-PPV an electron trap density of 1 × 1017 cm−3 was determined by charge transport measurements,40 and in BEH-PPV a trap concentration of 6.5 × 1017 cm−3 was determined by using Stern–Volmer analysis, as described previously.14Fig. 5 shows that Q increases with enhanced D, since a high exciton diffusion coefficient represents a fast exciton diffusion toward traps, leading to more efficient exciton quenching, and vice versa. The quenching efficiency is then retrieved from the calculated value of D from TRPL, and kdiff is determined by kf × Q.
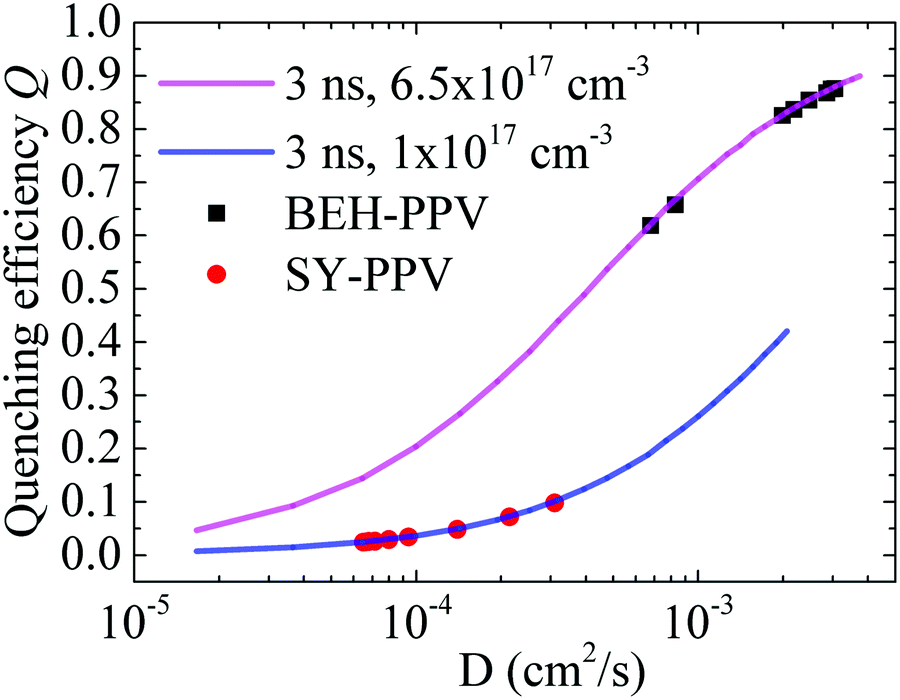 | ||
| Fig. 5 Exciton diffusion coefficient D dependent quenching efficiency Q for SY-PPV and BEH-PPV. The amount of background quenchers equals 1 × 1017 cm−3 and 6.5 × 1017 cm−3 for SY-PPV and BEH-PPV, respectively. The black and red points correspond to the values of D determined from the experimental results (Fig. 3). | ||
Fig. 5 shows that, at room temperature, almost 90% of the excitons in BEH-PPV are quenched at a defect, whereas for SY-PPV, this value is less than 10%. As seen in Tables 3 and 4, kdiff of BEH-PPV is two orders of magnitude higher than that of SY-PPV. As a result, the non-radiative losses in BEH-PPV are almost fully dominated by diffusion-limited quenching at the defects present in the polymer. Furthermore, both PPV derivatives demonstrate a decrease in kdiff with lowering temperature, in agreement with the diffusion limited exciton quenching model. With the values of kr and kdiff known, the non-radiative transfer rates knr were determined using eqn (1) and (2).
The calculated values show that kr in SY-PPV slowly decreases with decreasing temperature, while in BEH-PPV it barely changes. This is consistent with the predominantly radiative PL observed in BEH-PPV. Considering the non-radiative processes, exciton diffusion towards defects (kdiff) is largely determining the PL quenching in BEH-PPV (Fig. 5). In contrast, in SY-PPV knr is the dominant non-radiative loss process.
The increase of knr with a concomitant slight decrease of kr in SY-PPV is ascribed to increased population of conformational subunits that have higher contribution from non-radiative decay processes, such as may be expected with increased interchain interactions. Also, kdiff remains temperature-independent below 180 K. The decrease in the exciton diffusion rate kdiff with lower temperature leads to a slight increase in the overall (average) exciton lifetime from 1.9 ns to 2.4 ns going from 290 K to 77 K. The wavelength dependent exciton lifetimes are attributed to different sub-populations of emitting species with varying degrees of radiative and non-radiative emission, where at higher energies the radiative species dominates the fluorescence. At lower energies, there are greater contributions due to spectral diffusion caused by energy transfer from shorter to longer conformational subunits as well as from energy transfer to interchain excimer like species, which will have a lower oscillator strength and subsequently longer PL lifetime.25,27 The exciton decay processes in SY-PPV balance out to give a relative quantum yield ϕf, which is fairly temperature independent.
In BEH-PPV, the decrease of knr (300–150 K) results from the decrease in internal vibrational energy redistribution pathways due to the changes in inter- and intrachain packing. This is consistent with the decreased electron-vibrational coupling at lower temperatures, evidenced by the changing peak ratio between S0,0 and S0,1 (Fig. 2d). At lower temperatures (<150 K), knr increases. Previous work, including vibrational spectroscopy, has shown that the electronic-vibrational coupling in BEH-PPV is temperature dependent, with even additional vibronic peaks being resolved at temperatures lower than what we have studied here.26 Such an effect was also observed in MEH-PPV.41 The increase in knr at lower temperatures is therefore attributed to increased energy transfer processes from shorter to longer conformational subunits as well as a possible change in the electron-vibrational coupling. The increased excited state lifetime at lower temperatures in BEH-PPV mainly originates from the decrease in exciton quenching due to slower diffusion toward traps (decrease in kdiff), since kdiff is an order of magnitude larger as compared to knr. The reduction of these non-radiative processes at lower temperatures, dominated by a reduced kdiff, then give rise an increase in the relative quantum yield ϕf as observed in BEH-PPV.
The electronic and optical properties of conductive polymer films may also be influenced by thermal treatment at or around the glass transition temperature, Tg.42,43 For PPV based polymers, Tg values reported in literature are above room temperature.44,45 We therefore do not expect this to influence our results, where the highest temperature measured is 298 K.
Downhill migration depends on energetic disorder
We found further, that at higher temperatures, the less disordered polymer BEH-PPV exhibits less temperature dependence on D and kdiff. This is consistent with the low energetic disorder in this system. Since the quasi-equilibrium state is due to the narrow σ close to the center of the DOS, fast energy transfer between the conformational subunits proceeds, represented by a high exciton diffusion coefficient at room temperature (2.9 × 10−3 cm2 s−1). While cooling down to the characteristic temperature (<120 K), the exciton diffusion coefficient decreases slightly to 2.0 × 10−3 cm2 s−1, which is consistent with the decrease in kdiff. Below 120 K, where thermally activated hopping is absent, the exciton diffusion coefficient drops down to 8.3 × 10−4 cm2 s−1 and kdiff to 1.79 × 109 s−1, respectively. Due to the narrow energy spread in BEH-PPV, with decreasing temperature there are still enough possible energy-sites for the excitons to jump while the equilibrium-state shifts down into the Gaussian tail. Thus, the exciton diffusion coefficient of BEH-PPV at 120 K is still one order of magnitude higher than the one of SY-PPV at room temperature (3.1 × 10−4 cm2 s−1), while kdiff is two orders of magnitude higher (see also Tables 3 and 4).In contrast to the exciton diffusion coefficient, the exciton diffusion length LD of BEH-PPV first increases while cooling to 180 K. This increase from 7.7 nm to 9.6 nm is a consequence of the increased exciton lifetime resulting from less competitive non-radiative decay rates with lowering temperature (see eqn (5)). Then, in between 180 K and 120 K the LD of BEH-PPV slightly decreases. The previously noted change in electronic-vibrational coupling could affect the disorder parameter σ and therefore also the temperature dependence of LD. Finally, at a characteristic temperature of 100 K, the exciton diffusion is fully determined by downhill migration and the exciton diffusion length drops off from 8.9 nm to 5.2 nm.
Since the thermal equilibrium state −σ2/kT shifts with increasing disorder strength σ deeper into the tail states of the Gaussian DOS, the probability to find a nearby energy-site to which a jump is probable decreases noticeably in the case of a more disordered polymer.37 Therefore, in SY-PPV the exciton migration toward quenching defects is much slower at room temperature, leading to an order of magnitude longer exciton lifetime (1.9 ns) compared to BEH-PPV (200 ps).14 At room temperature, the slow exciton diffusion in the disordered SY-PPV is represented by a low exciton diffusion coefficient of 3.1 × 10−4 cm2 s−1 and the two orders of magnitude lower transfer rate kdiff (Tables 3 and 4) compared to BEH-PPV. The decrease in D and LD, while cooling to 180 K (Fig. 3), is a direct consequence of the down-shift of the equilibrium level into the tail of the Gaussian DOS, where the energy difference between occupied and neighboring sites increases, leading to an even lower D (8.0 × 10−5 cm2 s−1) and kdiff. This lower D along with a nearly constant exciton lifetime results in a decrease of LD from 7.7 nm to 4.2 nm in the temperature range of 290–180 K. Below the characteristic temperature (180 K), where thermally activated hopping is switched off, the exciton diffusion becomes fully determined by downhill migration, represented by temperature-independent D (∼6.8 × 10−5 cm2 s−1) and LD (∼3.9 nm).
The better ordered BEH-PPV exhibits PL characteristics that are temperature sensitive and are dominated by the exciton diffusion-limited quenching model. The increase in LD of BEH-PPV with lowering temperature is governed by the increase in τf, while in the case of SY-PPV temperature-dependence of LD is dominated by the temperature-dependence of D. Our findings demonstrate that the temperature at which thermally activated hopping diminishes scales with the energetic disorder. Better ordered PPV derivatives correlate with lower transition temperatures between thermally activated hopping and downhill migration.
Origin of PLED efficiency enhancement in BEH-PPV at low temperature
PLEDs based on these materials demonstrate that BEH-PPV exhibits temperature dependent efficiencies while SY-PPV does not. The current efficiency (CE) in PLEDs is defined as light output divided by current (exciton/charge carrier), and describes the relation between the number of emitted photons divided by the number of charges that passed the device in the same time interval. Since the charge current and the recombination prefactors of the radiative Langevin and nonradiative Shockley–Read–Hall recombination scale linearly with mobility, upon dividing these rates for current and light output, the mobility drops out and the efficiency is expected to be mobility independent.6,40,46 In a space-charge limited device such as PLEDs, the number of charge carriers is mainly determined by the applied voltage and not by the temperature. Therefore, the current efficiency in PLEDs is reported to be temperature-independent.17 Previous work on the temperature-dependent characteristics of PLEDs have focused on the role of the charge-injection barrier with temperature.18,47However, the PLED efficiency also depends on the decay dynamics of a formed exciton,17,48 which is described by the PL efficiency. The temperature-independence of the CE in the high disordered polymer SY-PPV PLED (Fig. 4a) is explained as follows: after formation of the exciton, it subsequently diffuses along the conjugated chain segments until it reaches a radiative or non-radiative recombination center and decays either radiatively or non-radiatively. The increased population of non-luminescent conformational subunits present in the SY-PPV copolymer compensates for the decrease in exciton diffusion toward non-radiative quenching-sites with lowering temperature. Therefore, the current efficiency, which directly correlates with the PL efficiency ϕf, shows no temperature-dependence in the SY-PPV based PLED.
In contrast, the increased current efficiency in the less disordered BEH-PPV PLED in the temperature-range of 295–215 K (Fig. 4b) is a direct consequence of enhanced ϕf, resulting from less competitive non-radiative diffusion-dominated decay processes with lowering temperature. We note that the temperature-dependence of the current efficiency in BEH-PPV is stronger than that of the relative quantum yield ϕf. This can be attributed to the difference in the relative intensity of peaks in the EL spectra compared to the PL spectra due to interference in the optical microcavity of the EL device. This will influence the device efficiency and the rate of spontaneous emission.32 In the optical microcavity, the BEH-PPV EL spectra still demonstrate decreased electron-vibrational coupling with decreasing temperature. This is evidenced by the increasing ratio between the S0,0 and S0,1 peaks (Fig. 4d), and is consistent with the decrease in knr noted in this temperature range in the PL measurements.
In addition to the different responses to temperature, the SY-PPV PLED exhibits an order of magnitude higher CE than the BEH-PPV PLED. The lower CE in BEH-PPV is a direct result of the faster exciton diffusion (kdiff) towards non-radiative traps, and is consistent with the photoluminescence measurements. As mentioned above, Fig. 5 demonstrates that, at room temperature, the exciton quenching at defects is 90% in BEH-PPV and 10% in SY-PPV. As a result, while the same amount of excitons are formed in the two materials for a given current, the amount of photons generated differs by an order of magnitude due to the difference in the kinetically competitive quenching at non-radiative traps. This results in a higher CE in the more energetically disordered SY-PPV.
Conclusions
We have shown that the temperature-dependent excited state lifetime, relative quantum yield, and exciton diffusion characteristics, i.e. exciton diffusion coefficient and exciton diffusion length, depend on the energetic disorder of the conjugated polymer system. The less disordered polymer BEH-PPV shows a decrease in non-radiative decay due to both freezing out vibrations (influences knr) with decreasing temperature and less exciton quenching due to slowing down the diffusion toward traps (decrease in kdiff), leading to increase in exciton lifetime and relative quantum yield. Since the CE of a PLED is directly correlated to the PL efficiency, we found in that the PLED based on the better ordered polymer showed enhanced CE at low temperatures due to reduced quenching at defect centers. Polymers with higher energetic disorder, as in SY-PPV, show different temperature-dependence of the exciton diffusion properties. With lowering temperature the exciton lifetime slightly increases, while the relative quantum yield decreases, due to different conformational subunits and increased competitions from radiative and non-radiative sites. As a consequence, PLEDs based on strongly disordered polymers show nearly temperature independent conversion efficiencies and PLQYs, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work of I. R. is part of the research program of the Dutch Polymer Institute (project #763). The authors are grateful to Gert-Jan A. H. Wetzelaer and Jasper Michels for helpful discussions and to Dominik Gehrig, Simon A. Bretschneider, Frank Keller, and Hans-Jürgen Guttmann for their technical support. Open Access funding provided by the Max Planck Society.Notes and references
- H. Bässler, Phys. Status Solidi, 1993, 175, 15–56 CrossRef.
- A. Köhler and H. Bässler, Electronic Processes in Organic Semiconductors, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed.
- T. Tiedje and A. Rose, Solid State Commun., 1981, 37, 49–52 CrossRef.
- D. D. C. Bradley, Synth. Met., 1993, 54, 401–415 CrossRef.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef.
- M. Kuik, G.-J. A. H. Wetzelaer, H. T. Nicolai, N. I. Craciun, D. M. De Leeuw and P. W. M. Blom, Adv. Mater., 2014, 26, 512–531 CrossRef PubMed.
- B. Movaghar, M. Grünewald, B. Ries, H. Bassler and D. Würtz, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 5545–5554 CrossRef.
- M. Grünewald, B. Pohlmann, B. Movaghar and D. Würtz, Philos. Mag. B, 1984, 49, 341–356 Search PubMed.
- O. V. Mikhnenko, F. Cordella, A. B. Sieval, J. C. Hummelen, P. W. M. Blom and M. A. Loi, J. Phys. Chem. B, 2008, 112, 11601–11604 CrossRef PubMed.
- S. Athanasopoulos, E. Hennebicq, D. Beljonne and A. B. Walker, J. Phys. Chem. C, 2008, 112, 11532–11538 CrossRef.
- T. Q. Nguyen, V. Doan and B. J. Schwartz, J. Chem. Phys., 1999, 110, 4068–4078 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer, Dordrecht, 1999 Search PubMed.
- O. V. Mikhnenko, P. W. M. Blom and T.-Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- I. Rörich, O. V. Mikhnenko, D. Gehrig, P. W. M. Blom and N. I. Crăciun, J. Phys. Chem. B, 2017, 121, 1405–1412 CrossRef PubMed.
- O. V. Mikhnenko, M. Kuik, J. Lin, N. van der Kaap, T.-Q. Nguyen and P. W. M. Blom, Adv. Mater., 2014, 26, 1912–1917 CrossRef PubMed.
- D. E. Markov, C. Tanase, P. W. M. Blom and J. Wildeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045217 CrossRef.
- P. W. M. Blom, M. J. M. De Jong and S. Breedijk, Appl. Phys. Lett., 1997, 71, 930–932 CrossRef.
- S. J. Martin, J. M. Lupton, I. D. W. Samuel and A. B. Walker, J. Phys.: Condens. Matter, 2002, 14, 9925–9933 CrossRef.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef PubMed.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2007, 18, 1–46 CrossRef.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- O. V. Mikhnenko, H. Azimi, M. Scharber, M. Morana, P. W. M. Blom and M. A. Loi, Energy Environ. Sci., 2012, 5, 6960 RSC.
- O. V. Mikhnenko, J. Lin, Y. Shu, J. E. Anthony, P. W. M. Blom, T.-Q. Nguyen and M. A. Loi, Phys. Chem. Chem. Phys., 2012, 14, 14196–14201 RSC.
- O. V. Mikhnenko, Software for Monte Carlo simulations, http://mikhnenko.com/eDiffusion/ Search PubMed.
- E. W. Snedden, L. A. Cury, K. N. Bourdakos and A. P. Monkman, Chem. Phys. Lett., 2010, 490, 76–79 CrossRef.
- F. A. C. Oliveira, L. A. Cury, A. Righi, R. L. Moreira, P. S. S. Guimarães, F. M. Matinaga, M. A. Pimenta and R. A. Nogueira, J. Chem. Phys., 2003, 119, 9777–9782 CrossRef.
- S. Gambino, A. K. Bansal and I. D. W. Samuel, Org. Electron., 2013, 14, 1980–1987 CrossRef.
- A. Bruno, L. X. Reynolds, C. Dyer-Smith, J. Nelson and S. A. Haque, J. Phys. Chem. C, 2013, 117, 19832–19838 CrossRef.
- A. Haugeneder, M. Neges, C. Kallinger, W. Spirkl, U. Lemmer, J. Feldmann, U. Scherf, E. Harth, A. Gügel and K. Müllen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15346–15351 CrossRef.
- S. R. Scully and M. D. McGehee, J. Appl. Phys., 2006, 100, 034907 CrossRef.
- D. Abbaszadeh, A. Kunz, G.-J. A. H. Wetzelaer, J. J. Michels, N. I. Crăciun, K. Koynov, I. Lieberwirth and P. W. M. Blom, Nat. Mater., 2016, 15, 628–633 CrossRef PubMed.
- M. Furno, R. Meerheim, S. Hofmann, B. Lüssem and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 115205 CrossRef.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef.
- B. Mollay, U. Lemmer, R. Kersting, R. F. Mahrt, H. Kurz, H. F. Kauffmann and H. Bässler, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10769–10779 CrossRef.
- R. Kersting, U. Lemmer, R. F. Mahrt, K. Leo, H. Kurz, H. Bässler and E. O. Göbel, Phys. Rev. Lett., 1993, 70, 3820–3823 CrossRef PubMed.
- M. Anni, M. E. Caruso, S. Lattante and R. Cingolani, J. Chem. Phys., 2006, 124, 134707 CrossRef PubMed.
- S. Athanasopoulos, E. V. Emelianova, A. B. Walker and D. Beljonne, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 195209 CrossRef.
- S. T. Hoffmann, H. Bässler, J. M. Koenen, M. Forster, U. Scherf, E. Scheler, P. Strohriegl and A. Köhler, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 115103 CrossRef.
- M. R. Andersson, G. Yu and A. J. Heeger, Synth. Met., 1997, 85, 1275–1276 CrossRef.
- Q. Niu, G.-J. A. H. Wetzelaer, P. W. M. Blom and N. I. Crăciun, Adv. Electron. Mater., 2016, 2, 1600103 CrossRef.
- M. A. T. Da Silva, I. F. L. Dias, J. L. Duarte, E. Laureto, I. Silvestre, L. A. Cury and P. S. S. Guimarães, J. Chem. Phys., 2008, 128, 094902 CrossRef PubMed.
- J. Liu, T.-F. Guo and Y. Yang, J. Appl. Phys., 2002, 91, 1595–1600 CrossRef.
- D. Kajiya, T. Koganezawa and K. Saitow, AIP Adv., 2015, 5, 127130 CrossRef.
- O. Schäfer, A. Greiner, J. Pommerehne, W. Guss, H. Vestweber, H. Y. Tak, H. Bässler, C. Schmidt, G. Lüssem, B. Schartel, V. Stümpflen, J. H. Wendorff, S. Spiegel, C. Möller and H. W. Spiess, Synth. Met., 1996, 82, 1–9 CrossRef.
- R. I. Venkatanarayanan, S. Krishnan, A. Sreeram, P. A. Yuya, N. G. Patel, A. Tandia and J. B. McLaughlin, Macromol. Theory Simul., 2016, 25, 238–253 CrossRef.
- M. Kuik, L. J. A. Koster, G.-J. A. H. Wetzelaer and P. W. M. Blom, Phys. Rev. Lett., 2011, 107, 256805 CrossRef PubMed.
- J. M. Lupton and I. D. W. Samuel, Synth. Met., 2000, 111–112, 381–384 CrossRef.
- P. W. M. Blom and M. J. M. De Jong, IEEE J. Sel. Top. Quantum Electron., 1998, 4, 105–112 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc01998c |
| This journal is © The Royal Society of Chemistry 2018 |