
A new NIR absorbing DPP-based polymer for thick organic solar cells†

Abstract
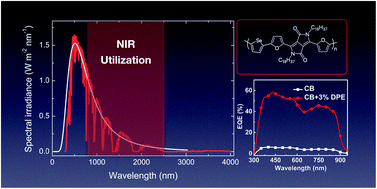
Sunlight covers a broad spectrum from ultra-violet to infrared, and low band gap materials are required to utilize the near infrared region (NIR) for better photon harvesting in organic solar cells. It has been shown that copolymers comprising diketopyrrolopyrrole-based acceptors and simple donors (thiophene or furan) achieve an absorption maximum at around 800 nm. In this study, selenophene was coupled with a diketopyrrolopyrrole based acceptor to yield a polymer (PFDPPSe) with an absorption maximum at 830 nm and an absorption onset at 930 nm. The optimized organic solar cells with PFDDPSe:PC71BM active layer blends at 210 nm showed a maximum PCE of 6.16% (average 6.02%) via solvent additive engineering with an inverted device structure. Their charge transport, recombination loss mechanism, and morphology were systematically studied. The results demonstrate that a highly efficient NIR-absorbing polymer can be achieved by the introduction of selenophene and a suitable solvent additive process for NIR organic solar cells. PFDPPSe is also one of the rare examples of a polymer with a PCE of over 6% that does not contain any thiophene-based unit in its backbone.
 Please wait while we load your content...
Please wait while we load your content...

