
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation, magnetic, and charge ordering in MnFe3O5†
K. H.
Hong
 a,
A. M.
Arevalo-Lopez
b,
M.
Coduri
c,
G. M.
McNally
a and
J. P.
Attfield
*a
a,
A. M.
Arevalo-Lopez
b,
M.
Coduri
c,
G. M.
McNally
a and
J. P.
Attfield
*a
aCentre for Science at Extreme Conditions and School of Chemistry, University of Edinburgh, Mayfield Road, Edinburgh EH9 3JZ, UK. E-mail: j.p.attfield@ed.ac.uk
bUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
cEuropean Synchrotron Radiation Facility, 71 avenue des Martyrs, Grenoble, 38000, France
First published on 16th January 2018
Abstract
The recently-discovered high pressure material MnFe3O5 displays a rich variety of magnetically ordered states on cooling. Fe spins order antiferromagnetically below a Néel transition at 350 K. A second transition at 150 K marks Mn spin order that leads to spin canting of some of the Fe spins and ferrimagnetism. A further transition at 60 K is driven by charge ordering of Fe2+ and Fe3+ over two inequivalent Fe sites, with further canting of all spins. Electrical resistivity measurements reveal semiconducting behaviour in MnFe3O5 with a change in activation energy at 285 K.
Introduction
The variable 3dn configurations of Mn and Fe cations in spinel-type and other oxides give rise to many important applications as energy materials1 and in magnetism. Many manganese oxides are used as cathode materials in rechargeable batteries, e.g. LiMn2O4-spinel, Li2MnO3, and NaMnO2. The phosphate LiFePO4 has been commercialised as a lithium battery cathode and iron oxides such as Fe2O3 and magnetite, Fe3O4, have been explored as anode materials.2 Manganese and iron based spinels are also active catalysts for oxygen reduction/evolution reactions (ORR/OER) in fuel cells, metal–air batteries, and water-splitting cells.3The spinel magnetite is also notable as the original magnetic material, and Fe3O4 has been studied intensively since Verwey's observation of a metal–insulator transition accompanied by a structural distortion.4 The low temperature structure has a complex charge and orbital ordering and weak Fe–Fe bonding interactions that form trimerons – linear orbital molecule clusters of three Fe ions.5,6 A related iron oxide, Fe4O5, was recently discovered using high temperature and high pressure synthesis,7 and has an incommensurate charge order at 150 K, below which dimeron and trimeron-like groups of Fe ions are formed.8 Subsequent work has shown that FenOn+1 homologues with n > 4 can also be made at pressure.9 M2+Fen−1On+1 analogues of these materials with M2+ = Ca were reported previously,10 and recent detailed studies of CaFe5O7 revealed a coupled structural and magnetic transition at 360 K accompanied by Fe2+/Fe3+ charge ordering.11–13
We recently reported the synthesis at high pressures of the first n > 3 M = Mn material in this family, the n = 4 member MnFe3O5.14 MnFe3O5 is isostructural with Fe4O5 and adopts the orthorhombic Sr2Tl2O5-type structure (space group Cmcm) in which divalent cations occupy triangular prismatic sites within triangular channels in a network of corner and edge-sharing octahedra. MnFe3O5 showed two magnetic transitions – an antiferromagnetic transition at 350 K and a broad ferromagnetic transition at 150 K – indicating that complex spin–spin interactions are present. We report here a high resolution powder neutron diffraction study of MnFe3O5, revealing the Mn/Fe chemical order and the temperature evolution of the magnetic behaviour between 5 and 400 K, supported by low temperature powder synchrotron X-ray diffraction and the electrical resistivity measurements.
Experimental
MnFe3O5 was synthesised using high pressure and high temperature solid state synthesis. Powders of MnO and Fe3O4 were ground together in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, and were heated at 1400 °C in a Pt capsule for 20 min under 10 GPa pressure, using a two-stage Walker-type module. Products were characterised by laboratory X-ray diffraction data collected with a Bruker D2 diffractometer using Cu-Kα radiation.
:1 ratio, and were heated at 1400 °C in a Pt capsule for 20 min under 10 GPa pressure, using a two-stage Walker-type module. Products were characterised by laboratory X-ray diffraction data collected with a Bruker D2 diffractometer using Cu-Kα radiation.
Magnetic measurements were carried out with a Quantum Design MPMS XL SQUID magnetometer. Magnetic susceptibility was recorded in zero field cooled (ZFC) and field cooled (FC) conditions between 2 and 400 K with an applied magnetic field of 5000 Oe. Hysteresis loops were measured at 2, 75, 300 and 400 K. Electrical resistivity measurements were carried out with a Quantum Design PPMS, between 260 and 380 K.
High resolution time-of-flight neutron diffraction data were collected at the WISH beamline of the ISIS facility, with 50 mg of powder from several high pressure runs packed into a vanadium can. Diffraction patterns were collected at 5, 75, 150, 300 and 400 K using a closed cycle refrigerator (CCR) with a hot stage. High resolution powder X-ray diffraction data were collected at the ID22 beamline of the ESRF with incident wavelength 0.39994 Å. A glass capillary with an outer diameter of 0.3 mm was used to contain the polycrystalline sample. Low temperature diffraction data were collected from 13 to 120 K using a liquid helium cryostat system.
Results and discussion
Crystal and magnetic structures
High resolution time-of-flight neutron diffraction experiments were carried out to determine the structure and magnetic behaviour of MnFe3O5. Crystal structure refinements show that the unit cell symmetry remains orthorhombic Cmcm at all temperatures. Occupancy refinements for the three cation sites, making use of the high neutron scattering contrast between Mn and Fe, showed that octahedral sites are occupied exclusively by Fe, and the trigonal prism site is predominantly Mn with 6.3(4)% substitution by Fe. Hence MnFe3O5 is Mn/Fe ordered and the overall Mn0.94Fe3.06O5 composition of the present sample is close to the ideal stoichiometry.Magnetisation measurements (Fig. 1) show a maximum at 350 K and a broad magnetic upturn signifying a ferro- or ferri-magnetic ordering at 150 K as reported previously.14 This is corroborated by the saturated magnetisations observed in hysteresis loops at 2 and 75 K.
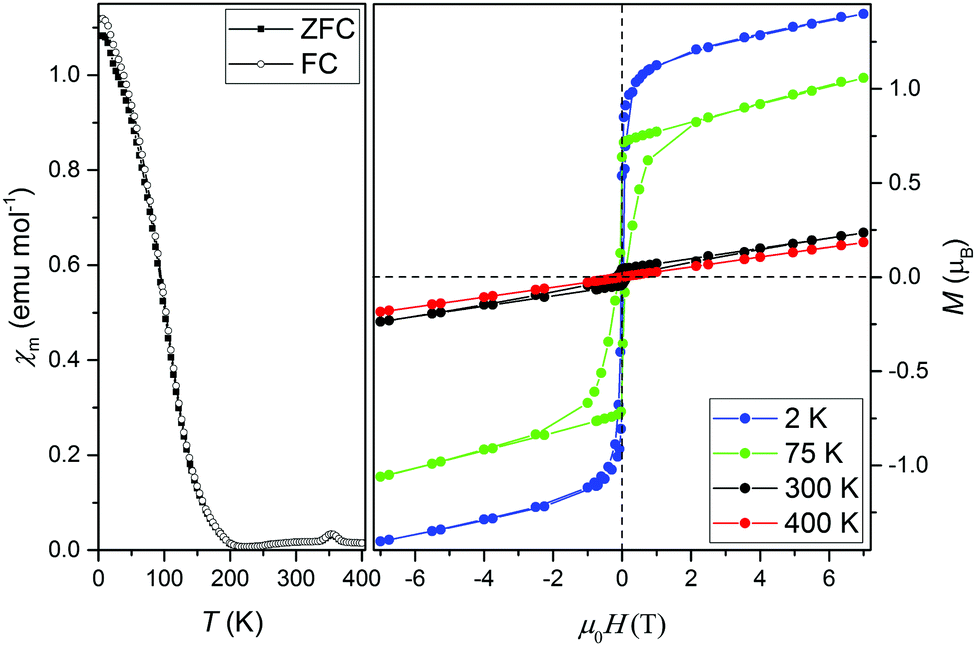 | ||
| Fig. 1 (left) Zero-field cooled and field cooled magnetisation measurements between 2 and 400 K. (right) Magnetisation-field hysteresis loops for MnFe3O5 measured at 2, 75, 300 and 400 K. | ||
The neutron diffraction patterns shown in Fig. 2 reveal the appearance of magnetic reflections when cooled below 400 K. The magnetic structures in the different regimes were solved and Rietveld fitted to the neutron diffraction patterns, as shown for the 5 K pattern in Fig. 3. The magnetic reflections from all the magnetic phases of MnFe3O5 were indexed with a propagation vector of (000), and the structures obtained at 5, 75 and 300 K are presented in Fig. 4a. At 150 and 300 K, the spins at the two independent Fe sites in MnFe3O5 are both found to be ordered antiferromagnetically parallel to the c-axis, whilst the Mn spins remain disordered. This confirms that a Néel transition is observed at TN = 350 K in the magnetisation measurements (Fig. 1).
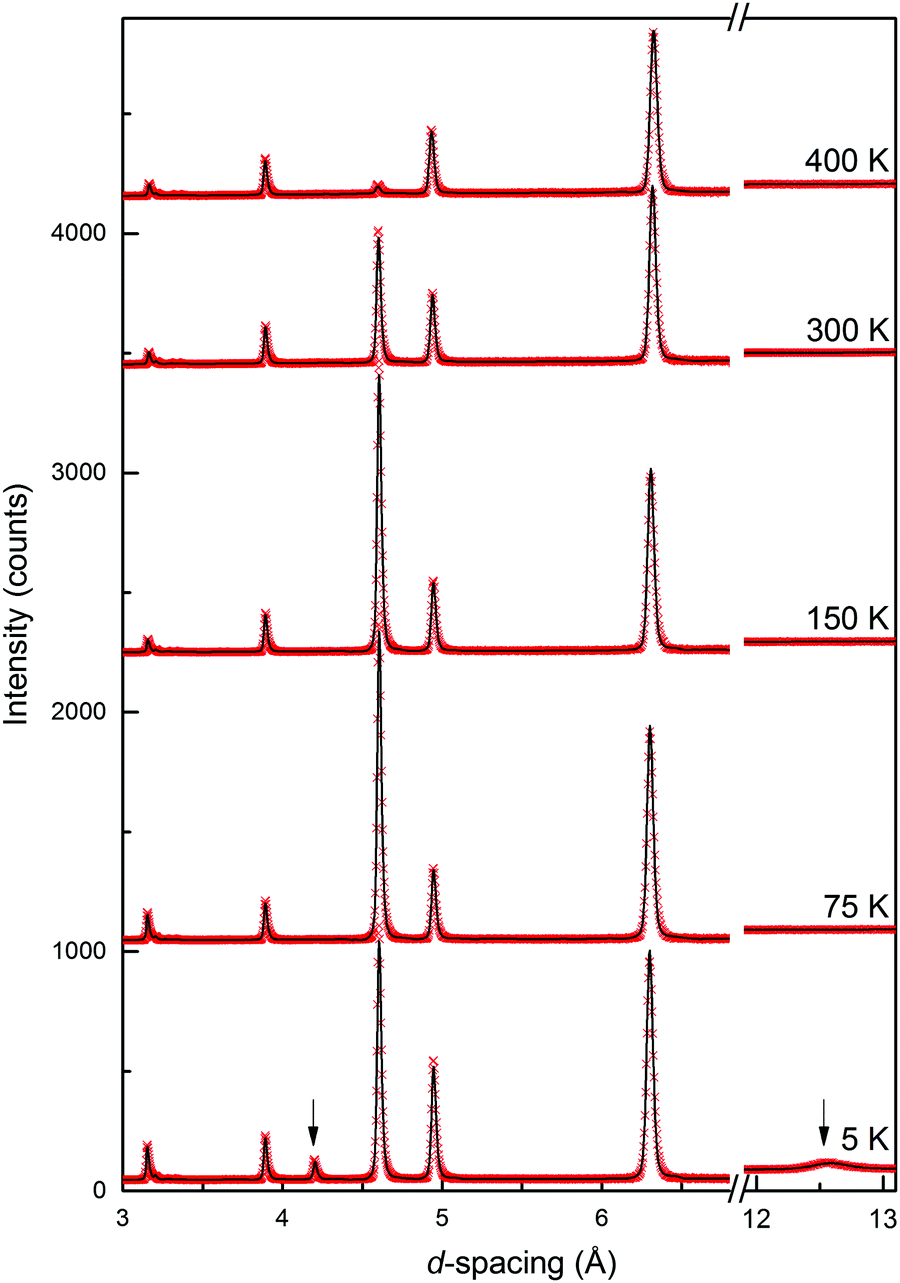 | ||
| Fig. 2 Temperature evolution of the neutron diffraction pattern of MnFe3O5. Magnetic contributions appear at 300 K, and arrows indicate additional magnetic peaks at 5 K, with hkl = (001) and (003) at d-spacing = 12.6 and 4.2 Å, respectively. | ||
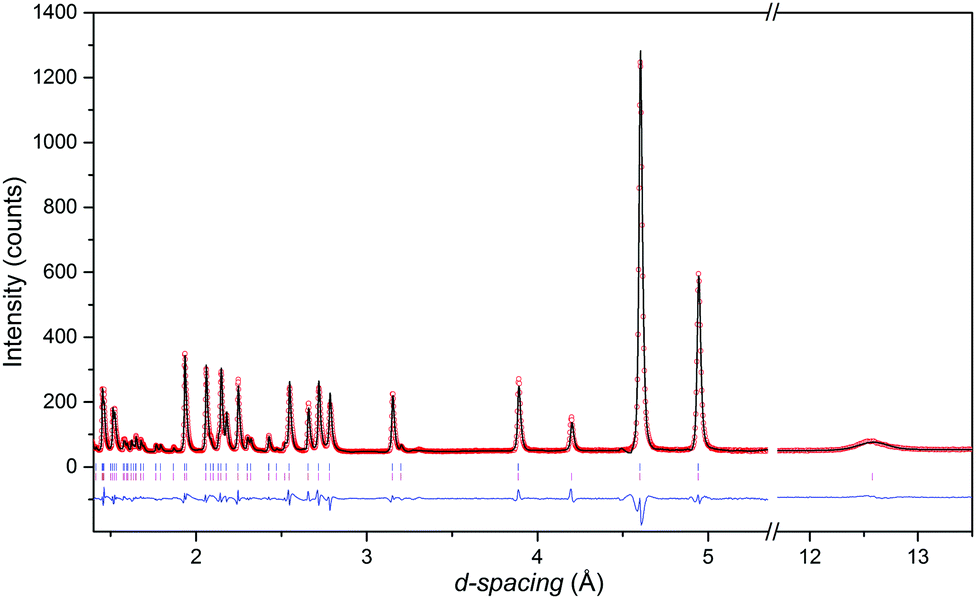 | ||
| Fig. 3 Rietveld refinement of high resolution neutron diffraction patterns of MnFe3O5 obtained at 5 K, with upper tick marks indicating nuclear peaks and lower presenting magnetic reflections. (Rwp = 12.9% and Rp = 12.6%). | ||
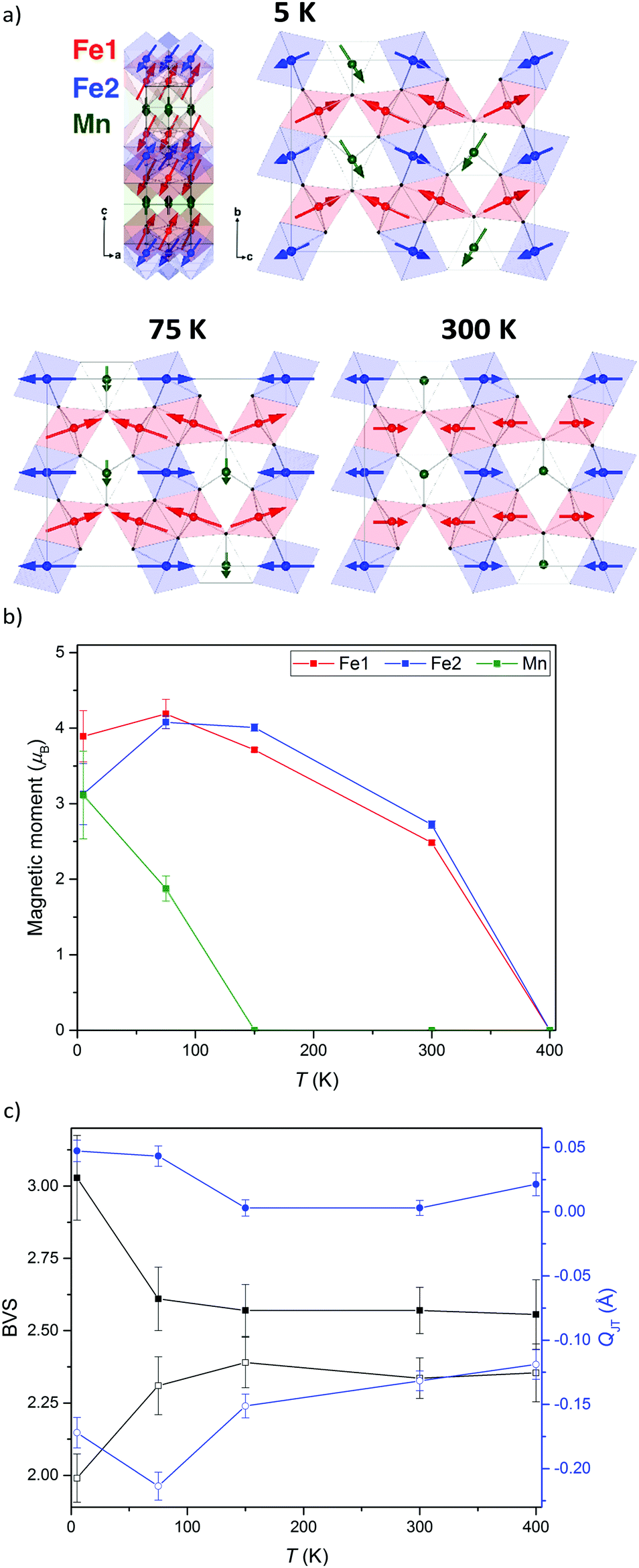 | ||
| Fig. 4 (a) Magnetic structures of MnFe3O5 at 5, 75 and 300 K. The network of FeO6 octahedra is shown, with Mn2+ in trigonal prismatic sites within channels parallel to the a-axis. (b) The temperature evolution of the ordered Mn, Fe1 and Fe2 magnetic moments. (c) Temperature evolution of BVS and QJT, with closed/open symbols representing Fe1/Fe2 sites. | ||
Fits to the 75 K diffraction data showed additional spin ordering of the Mn site, with moments aligned ferromagnetically along the b axis below TMn = 150 K. The order of the Mn site moment leads the spins of the nearest Fe site – Fe1 to cant towards the b axis. The b-components of Fe1 spins are antiparallel to those of Mn, resulting in a net magnetisation of ∼0.5 μB per MnFe3O5 formula unit, which is consistent with the increase in magnetisation on cooling from 300 to 75 K shown in Fig. 1. A similar spin canting was reported in Fe4O5.8
The additional magnetic reflections observed in the 5 K neutron diffraction patterns reveal another change in the magnetic structure in MnFe3O5. The onset for this third magnetic transition appears to be the divergence between zero-field cooled and field cooled susceptibilities at ∼60 K in the susceptibility data (Fig. 1). An increase in the ordered Mn moment leads to further canting of all the spins, with both of the Fe sites canted antiferromagnetically towards the a axis and ferromagnetically along b. In addition, the Mn spins become canted antiferromagnetically to the c axis. The magnetic components of the Mn and the Fe2 sites on the b axis are antiparallel to Fe1, as shown in Fig. 4a. This enhances the magnetisation along the b axis to ∼0.6 μB per MnFe3O5 formula unit. The thermal evolution of the magnitude of the ordered moments in MnFe3O5 are shown in Fig. 4b, and the values of the ordered components and other refinement results are given in ESI.†
The 300 K magnetic structure reveals dominant antiferromagnetic Fe1–Fe2 interactions; from direct exchange via overlap of half-filled t2g orbitals through edge-sharing of FeO6 octahedra, and through superexchange mediated by Fe–O–Fe connections at shared corners. Each Mn spin is coupled to 4 Fe1 and 4 Fe2 spins via Mn–O–Fe bridges, and frustration of both of these interactions leads to an almost perpendicular alignment of the Mn moments at 75 K, although Fe1 spin canting occurs such that their components in the b-direction are antiferromagnetically coupled to the Mn spins. Further canting occurs at 5 K as the Mn spins become more fully ordered and Fe1 and Fe2 spins cant out of the bc-plane. This magnetic order breaks the mirror-plane symmetries of the lattice perpendicular to the a and c axes, so an exchange-strictive distortion might be expected but is not observed within the resolution of the present data.
The possibility of internal electronic distortions was investigated by using the Bond Valence Sum (BVS) method to estimate oxidation states for the Fe sites via a standard interpolation method15,16 with bond distances derived from the neutron refinements. The BVS results in Fig. 4c show that both Fe1 and Fe2 sites have mixed Fe2+/Fe3+ charge states at 75–400 K, but charge ordering is evident at 5 K with Fe1 and Fe2 respectively tending to Fe3+ and Fe2+ states. The effect of Jahn Teller distortion (QJT) is also calculated for both Fe sites. Charge localisation as Fe2+ is expected to lead to Jahn Teller compression of the Fe2O6 octahedron (negative values of the QJT parameter reported in ref. 5), and although no large changes are observed on cooling below 75 K, the negative QJT for Fe2 (Fe2+) and near zero value for Fe1 (Fe3+) corroborate the BVS charge ordering results. Hence a Fe2+/Fe3+ charge ordering transition occurs at TCO = 60 K in MnFe3O5, with concomitant spin canting. As the Fe2+ and Fe3+ states respectively localise at inequivalent Fe2 and Fe1 sites there is no symmetry-breaking distortion associated with the charge order, or with the Fe2+ orbital order, although the associated 5 K spin order does break the Cmcm lattice symmetry as noted above.
Our previous study of the crystal structure using powder synchrotron X-ray diffraction data showed that anisotropic thermal expansion of the lattice parameters is observed over the temperature range 90–400 K. Further data collected between 15 and 120 K here confirm that the orthorhombic Cmcm structure persists to low temperatures. A discontinuity in the refined lattice parameters and cell volume is observed at 60 K, (Fig. 5) corresponding to the divergence in the ZFC and FC magnetisation measurements. This likely marks the onset of the charge ordering observed in the 5 K but not the 75 K neutron diffraction data.
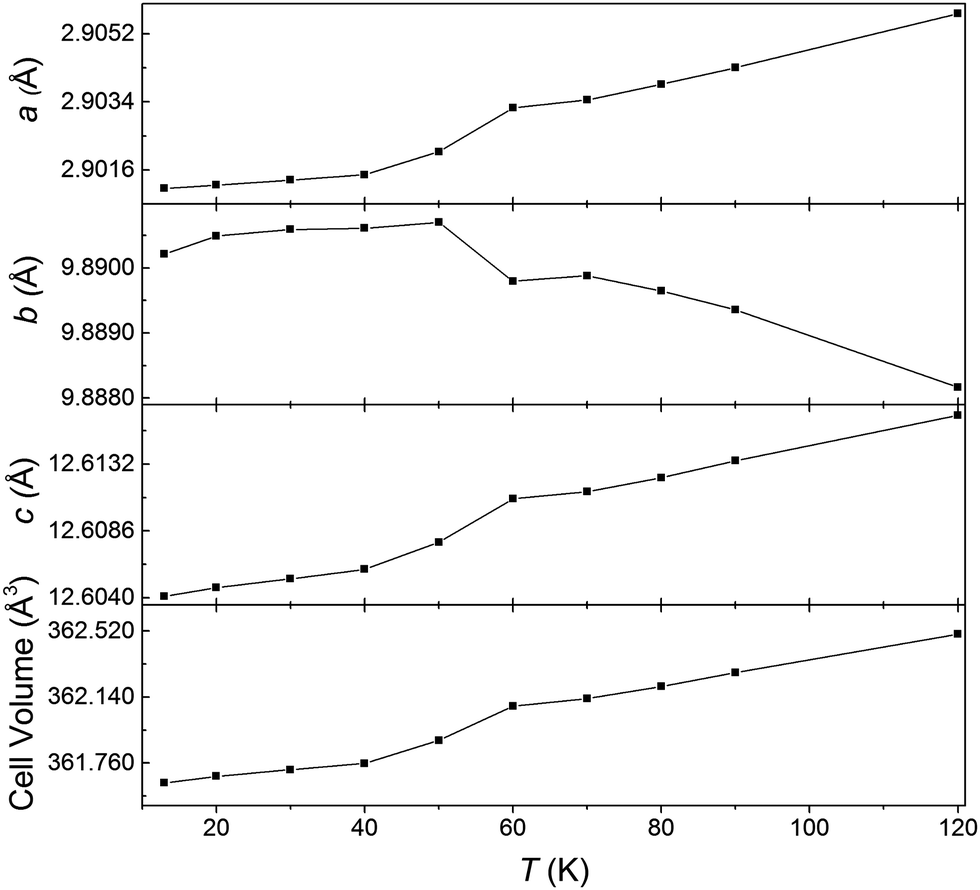 | ||
| Fig. 5 Changes in the lattice parameters and cell volume obtained from powder synchrotron X-ray diffraction experiments. | ||
Electrical properties
Electrical resistivity measurement of a polycrystalline pellet of MnFe3O5 shows semiconducting behaviour, with the resistivity increasing when cooled (Fig. 6). The sample resistance was too great to be measured below 260 K. Two linear regions are observed in the inset plot of log(resistivity) vs. inverse temperature, with a change of slope near 285 K. Fitting the Arrhenius equation ρ = Aexp(Ea/kBT) to the two linear regions gives the activation energy Ea as 210 meV above and 280 meV below the 285 K crossover. This temperature does not match any of the observed magnetic transition temperatures, and may correspond to a change from defect-dominated to intrinsic bandgap conduction.
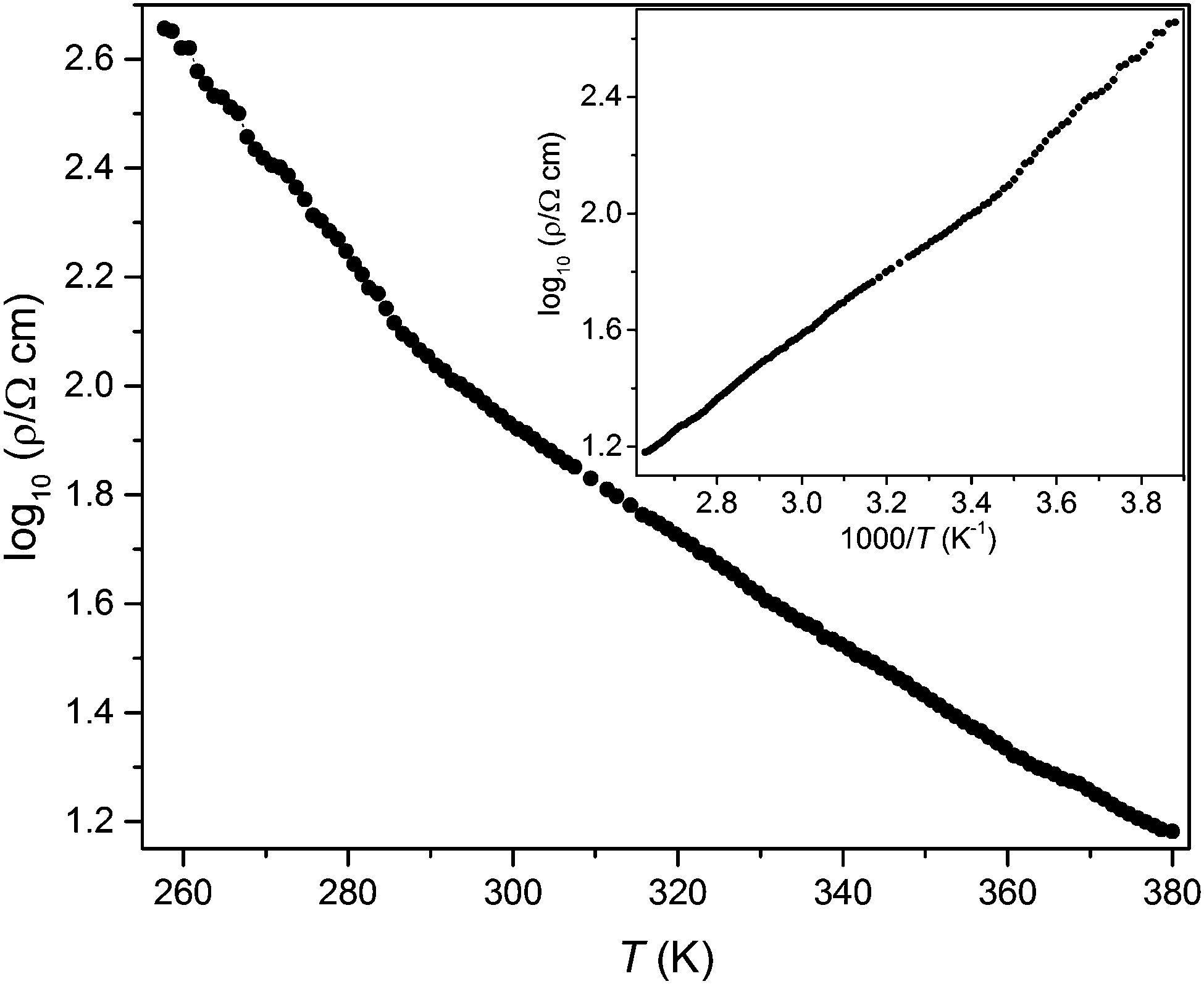 | ||
| Fig. 6 Log of electrical resistivity of MnFe3O5 measured between 260 and 380 K, with insert showing the plot against reciprocal temperature. | ||
The presence of structural channels within a framework of redox-active FeO6 octahedra, and moderate electrical conductivity, suggests that MnFe3O5 would be worth investigating as a battery electrode material. Full reduction of Fe3+ to Fe2+ through lithium insertion would give Li2MnFe3O5, and as cycling between this and the parent phase does not involve oxidation of Mn2+ then structural degradation from formation of Jahn–Teller active Mn3+ is avoided. However, alternative methods to high pressure synthesis are likely to be needed to make sufficient quantities of MnFe3O5 for practical battery research.
Conclusions
MnFe3O5 is found to have a rich magnetic ordered behaviour between 5 and 400 K. On cooling from high temperatures, the moments at the iron sites order with an antiferromagnetic arrangement at TN = 350 K, but additional order of Mn moments below TMn = 150 K leads to canting of Fe1 spins and a significant ferrimagnetic moment. A similar canting of Fe (and Re) spins due to low temperature order of Mn2+ moments was reported in the double perovskite Mn2FeReO6.17 Fe2+/Fe3+ charge ordering drives a further transition at TCO = 60 K with additional canting of all spins. Similar charge and orbital orders are observed in Fe3O4 and Fe4O5. Electrical resistivity measurements reveal semiconducting behaviour in MnFe3O5 and a small change in the activation energy at 285 K may correspond to a crossover from defect to intrinsic conduction regimes. MnFe3O5 is worth further investigation as a potential lithium battery electrode material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from European Research Council (ERC), Engineering and Physical Sciences Research Council (EPSRC) and Science, Technology Facilities Council (STFC) and the ESRF for provision of beamtime. We would also like to thank Andy Fitch (ESRF), Pascal Manuel (ISIS) and James Cumby, Alexander J. Browne, Giuditta Perversi and Paul M. Sarte (Edinburgh) for assistance provided.References
- N. Yabuuch and S. Komaba, Sci. Technol. Adv. Mater., 2014, 15, 043501 CrossRef PubMed.
- L. Zhang, H. B. Wu and X. W. Lou, Adv. Energy Mater., 2013, 4, 1300958 CrossRef.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- E. J. W. Verwey, Nature, 1939, 144, 327–328 CrossRef CAS.
- M. S. Senn, J. P. Wright and J. P. Attfield, Nature, 2012, 481, 173–176 CrossRef CAS PubMed.
- G. Perversi, J. Cumby, E. Pachoud, J. P. Wright and J. P. Attfield, Chem. Commun., 2016, 52, 4864 RSC.
- B. Lavina, P. Dera, E. Kim, Y. Meng, R. T. Downs, P. F. Weck, S. R. Sutton and Y. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17281–17285 CrossRef CAS PubMed.
- S. V. Ovsyannikov, M. Bykov, E. Bykova, D. P. Kozlenko, A. A. Tsirlin, A. E. Karkin, V. V. Shchennikov, S. E. Kichanov, H. Gou, A. M. Abakumov, R. Egoavil, J. Verbeeck, C. McCammon, V. Dyadkin, D. Chernyshov, S. van Smaalen and L. S. Dubrovinsky, Nat. Chem., 2016, 1–8 Search PubMed.
- B. Lavina and Y. Meng, Sci. Adv., 2015, 1, e1400260 Search PubMed.
- O. Evrard, B. Malaman and F. Jeannot, J. Solid State Chem., 1980, 35, 112–119 CrossRef CAS.
- C. Delacotte, F. Hüe, Y. Bréard, S. Hébert, O. Pérez, V. Caignaert, J. M. Greneche and D. Pelloquin, Inorg. Chem., 2014, 53, 10171–10177 CrossRef CAS PubMed.
- C. Delacotte, Y. Bréard, V. Caignaert, V. Hardy, J. M. Greneche, S. Hébert, E. Suard and D. Pelloquin, Key Eng. Mater., 2014, 617, 237–240 CrossRef CAS.
- C. Delacotte, Y. Bréard, V. Caignaert, V. Hardy, J. M. Greneche, S. Hébert, E. Suard and D. Pelloquin, J. Solid State Chem., 2017, 247, 13–19 CrossRef CAS.
- K. H. Hong, G. M. McNally, M. Coduri and J. P. Attfield, Z. Anorg. Allg. Chem., 2016, 642, 1355–1358 CrossRef CAS.
- J. P. Attfield, Solid State Sci., 2006, 8, 861–867 CrossRef CAS.
- I. D. Brown, J. Appl. Crystallogr., 1996, 29, 479–480 CrossRef CAS.
- A. M. Arévalo-López, G. M. McNally and J. P. Attfield, Angew. Chem., Int. Ed., 2015, 54, 12074–12077 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of crystallographic results and powder synchrotron profile fit. See DOI: 10.1039/c8tc00053k |
| This journal is © The Royal Society of Chemistry 2018 |