
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the variability in oxidation states of magnetite nanoparticles by single-particle spectroscopy†
A.
Fraile Rodríguez
 *ab,
C.
Moya
ab,
M.
Escoda-Torroella
ab,
A.
Romero
ab,
A.
Labarta
ab and
X.
Batlle
ab
*ab,
C.
Moya
ab,
M.
Escoda-Torroella
ab,
A.
Romero
ab,
A.
Labarta
ab and
X.
Batlle
ab
aDepartament de Física de la Matèria Condensada, Universitat de Barcelona, Martí i Franqués 1, 08028 Barcelona, Spain. E-mail: arantxa.fraile@ub.edu
bInstitut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, Spain
First published on 10th January 2018
Abstract
We have studied the electronic and chemical properties of a variety of ensembles of size- and shape-selected Fe3O4 nanoparticles with single-particle sensitivity by means of synchrotron-based X-ray photoemission electron microscopy. The local X-ray absorption spectra reveal that the oxidation states and the amount and type of cations within the individual nanoparticles can show a striking local variability even when the average structural and magnetic parameters of the monodisperse ensembles appear to be compatible with those of conventional homogeneous magnetite nanoparticles. Our results show the key role played by oleic acid concentration in the reaction mixture on the formation and compositional homogeneity within individual nanoparticles. When the concentration of oleic acid is high enough, the nanoparticles are composed of a Fe3O4 core surrounded by a thin γ-Fe2O3 shell. However, at a low concentration of the fatty acid, the Fe3O4 nanoparticles are likely inhomogeneous with small inclusions of FeO and Fe phases, as a result of an uncontrolled reduction of Fe3+ cations. All the foregoing underlines the importance of combining both advanced synthesis techniques and complementary single-particle investigations performed on a statistically significant number of particles so as to improve the understanding and control over electronic and magnetic phenomena at the nanoscale.
Introduction
Magnetic nanoparticles (NP) are extremely versatile building blocks with applications ranging from biomedicine1–4 to energy, magnetic data storage, catalysis, and spin-filter devices.5–10 In addition, they are excellent model systems to study unique magnetic phenomena taking place at the nanoscale, such as single domain states, superparamagnetism (SPM), as well as the occurrence of enhanced values of magnetic moments and anisotropy energies.11 In particular, magnetite (Fe3O4) and maghemite (γ-Fe2O3) are among the most commonly studied systems due to their good magnetic performance, ease of production and functionalization by chemical routes,12 and low toxicity.13 However, for optimal performance, good control over the magnetic and structural properties as well as the surface chemistry is critical, and great efforts have been made for developing methods to obtain magnetic NP with high crystal quality, low size dispersion and enhanced magnetic response.11,13 Moreover, interpreting and controlling the magnetic and electronic properties of iron oxide NP, especially at these very small sizes, still remain a challenge because of their crucial dependence on composition, structure, surface chemistry, and interparticle interactions. All of these contributions are difficult to disentangle, thus limiting the ability to quantitatively assess their actual effects on the functional response of the NP. For example, a detailed knowledge of the specific surface modifications of iron-oxide NP is relevant in their functionalization using biomolecular interactions,14 or in the manipulation of interfacial cross-coupling between magnetic and electrical properties in multiferroic nanostructures.15 Furthermore, good control over the magnetic anisotropy in such systems could be of potential interest for ultra-high density storage media and for magnetic tags in biological assays.16,17 At a more fundamental level, several questions still remain open about Fe3O4, including the half-metallic character18 or understanding what hinders the experimental realization of the theoretically predicted large spin polarization.19Meeting the above challenges requires both advanced synthesis methods enabling obtaining size-selected, chemically homogeneous Fe3O4 NP with good magnetic properties and a precise and systematic characterization of the NP system. Regarding the first issue, chemical methods based on the high-temperature decomposition of iron organometallic precursors in organic media with accurate control of the reaction temperature and the surfactant concentration have proven successful to synthesize Fe3O4 NP with high crystal quality, well-controlled shapes and sizes in the range from a few nm to about 50 nm, and reduced magnetic disorder.13 However, even for highly monodisperse particles, a strong correlation has been found between their macroscopic magnetic behavior and the structural and compositional features of the individual NP.20–22 This calls for the use of single-particle characterization techniques with elemental selectivity and high sensitivity to magnetic and electronic properties.23–26 For example, a 3D visualization of the iron oxidation state in FeO/Fe3O4 core–shell nanocubes has recently been achieved using electron energy loss tomography.26 Such advanced transmission electron microscopy (TEM)-based methods have the main advantage of combining an excellent spatial resolution (Å) with a fair energy resolution (about 0.5 eV). However, both the measurements and analyses are laborious, and thus the studies are typically restricted to a very limited number of particles.
In the present work, we investigate the electronic structure and distribution of Fe oxide phases within individual Fe3O4 NP by means of X-ray photoemission electron microscopy (X-PEEM) in a large number of particles. This is a non-invasive, element-specific technique providing quantitative information about the chemical composition, electronic structure, and magnetism of individual NP, both static23,27,28 and time-resolved.24,29 Using this unique approach, we find that NP that appear homogeneous in crystal quality and macroscopic magnetic response in a monodisperse ensemble can exhibit a striking size-independent coexistence of NP with distinct Fe oxide phases as a result of the uncontrolled reduction of Fe3+ during the reaction process.
Experimental section
Synthesis of Fe3O4 NP
Two samples of nominal Fe3O4 NP with mean diameters of 15 and 24 nm, – in the following referred as S1 and S2, respectively – were synthesized by slightly modifying a previously reported procedure20 by using iron(III) acetylacetonate (Sigma-Aldrich, 99%) as an organometallic precursor and oleic acid (Sigma-Aldrich, 90%) and benzyl ether (Sigma-Aldrich, 98%) as a surfactant and organic solvent with a high boiling point, respectively. All the reactants were used in the synthesis without further purification.Sample S1
15 nm NP were obtained by mixing 0.71 g of Fe(acac)3 and 2.5 g of oleic acid (8 mmol) in 20 ml of benzyl ether. The reaction mixture was degassed at 60 °C for 30 minutes, and it was then heated up to 200 °C and kept at this temperature for 2 hours under a nitrogen atmosphere and vigorous stirring. Thereafter, the solution was heated up to reflux at 6.7 °C min−1 for 1 hour. Finally, the solution was cooled down to room temperature and transferred to a 50 ml centrifuge tube together with a 20 ml mixture of hexane and acetone with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. After three times centrifugation at 9500 rpm for 15 minutes a black powder was obtained, which was then re-dispersed in hexane.
:3. After three times centrifugation at 9500 rpm for 15 minutes a black powder was obtained, which was then re-dispersed in hexane.
Sample S2
Bigger NP were obtained using the same experimental procedure than the above but reducing the amount of oleic acid in the reaction mixture to 2.1 g (6.7 mmol).Structural and magnetic characterization of monodisperse ensembles of NP
The size and shape of the NP were determined using transmission electron microscopy (MT80-Hitachi microscope and Philips CM30) by placing a drop of a diluted suspension of NP in hexane onto standard C-coated Cu grids. The size distributions were determined through analysis of ensembles of around 2000 particles in randomly selected areas of the samples (see Table 1 and Fig. S1, ESI†). The particles were found to be pseudo-spherical (S1) and truncated octahedra (S2) (see Fig. 1(c) and (d)). High resolution TEM images (JEOL 2100 microscope) of S1 showed highly crystalline NP up to the particle surface (see Fig. 1(a)). In contrast, a large fraction of S2 NP exhibited crystallographic domains and or boundaries (see Fig. S2, ESI†). The latter is also confirmed by dark field TEM images showing inhomogeneous contrast within the NP, which is indicative of different crystallographic domains and/or different Fe phases inside the NP (Fig. S3, ESI†).| Samples | D TEM (nm) | σ TEM (nm) | D XRD (nm) | M s at 5 K (emu g−1) | H c at 5 K (kOe) | M s at 300 K (emu g−1) | H c at 300 K (kOe) |
|---|---|---|---|---|---|---|---|
| S1 | 16.2 | 9.8 | 14(1) | 91(1) | 0.32(0.02) | 82(1) | 0.0090 |
| S2 | 24.8 | 15.0 | 25(0.5) | 95(2) | 0.40(0.01) | 87(2) | 0.0094 |
 | ||
| Fig. 1 Structural and magnetic characterization. (a and b) High-resolution TEM images of samples S1 and S2, respectively. (c and d) TEM images at medium-resolution of the corresponding samples. Scale bars are as follows: (a) 4 nm, (b) 7 nm, (c) and (d) 75 nm. | ||
The crystal phase of the iron oxide NP was identified by powder X-ray diffraction. The patterns were collected in a PANalytical X’Pert PRO MPD diffractometer by using Cu Kα radiation (λ = 1.5418 Å), within 5° and 120° in 2θ with a step size of 0.033°. The crystal size was determined by using a Rietveld analysis on the XRD patterns, indexing the Bragg's planes to face center cubic inverse spinel magnetite (Fd3m), with a lattice constant a = 8.359 Å (Fig. 2(a)). The organic fraction of the samples was evaluated by thermogravimetric analysis (TGA) in a TGA-SDTA 851e/SF/1100 (Mettler Toledo) at a heating rate of 10 °C min−1 from room temperature up to 800 °C under a nitrogen atmosphere (see Fig. S4, ESI†). Magnetization measurements in powder samples were performed with a Quantum Design SQUID magnetometer (Fig. 2(b)). Hysteresis loops, M(H), were recorded at several temperatures within 5 and 300 K under a maximum magnetic field of ±50 kOe to study the saturation magnetization Ms and the coercive field Hc. Ms was obtained by extrapolation of the high-field region of M(H) to zero field, assuming the high-field behavior M(H) = Ms + χ·H where χ is a residual susceptibility (Table 1).30–32Ms values were normalized to the magnetic content by subtracting the organic fraction determined by TGA measurements. The Ms of both samples at 5 K (Table 1) were close to that of bulk Fe3O4,33,34 indicating a nearly perfect ferrimagnetic order throughout the whole volume of the NP. It is also worth noting that the actual shape of the hysteresis loops of both samples, showing saturation at low fields and absence of high closure fields, is the expected signature for bulk-like magnetic behavior, as well established for magnetite NP.30 In these highly crystalline samples, above the Verwey temperature, well defined magneto-crystalline axes are expected to favor spins pointing along the [111] directions,21,35 as shown in Fig. 1(a) and (b) and previously reported in highly crystalline samples synthesized by thermal decomposition methods.12,20,30,33 The coercive field (Hc) was obtained as Hc = (|Hc+| + |Hc−|)/2, where Hc+ and Hc− were the intercepts of the hysteresis loop with the positive and the negative sides of the H-axis, respectively. The Hc values at 5 K (Table 1) were in agreement with those published elsewhere for NP of similar sizes.12,20 Hysteresis loops were also recorded at 5 K after cooling the sample under 10 kOe (FC) from room temperature (Fig. S5, ESI†) in order to elucidate the existence of any changes in the internal magnetic configuration of the particles due to the presence of distinct coexisting magnetic phases.11,25,36–38 The hysteresis loops before and after FC for S1 look similar enough so as to discard the presence of inhomogeneous particles containing ferrimagnetic, antiferromagnetic (FeO in wustite phase) and/or ferromagnetic phases in close contact with each other. However, the FC hysteresis loop for S2 shows significant narrowing at low fields which may be attributed to the weak coupling among magnetic phases within a nanoparticle with a different magnetic anisotropy. The NP in both samples are magnetically blocked at room temperature (see e.g. the ZFC–FC magnetization curves for sample S2 in Fig. S6, ESI†).
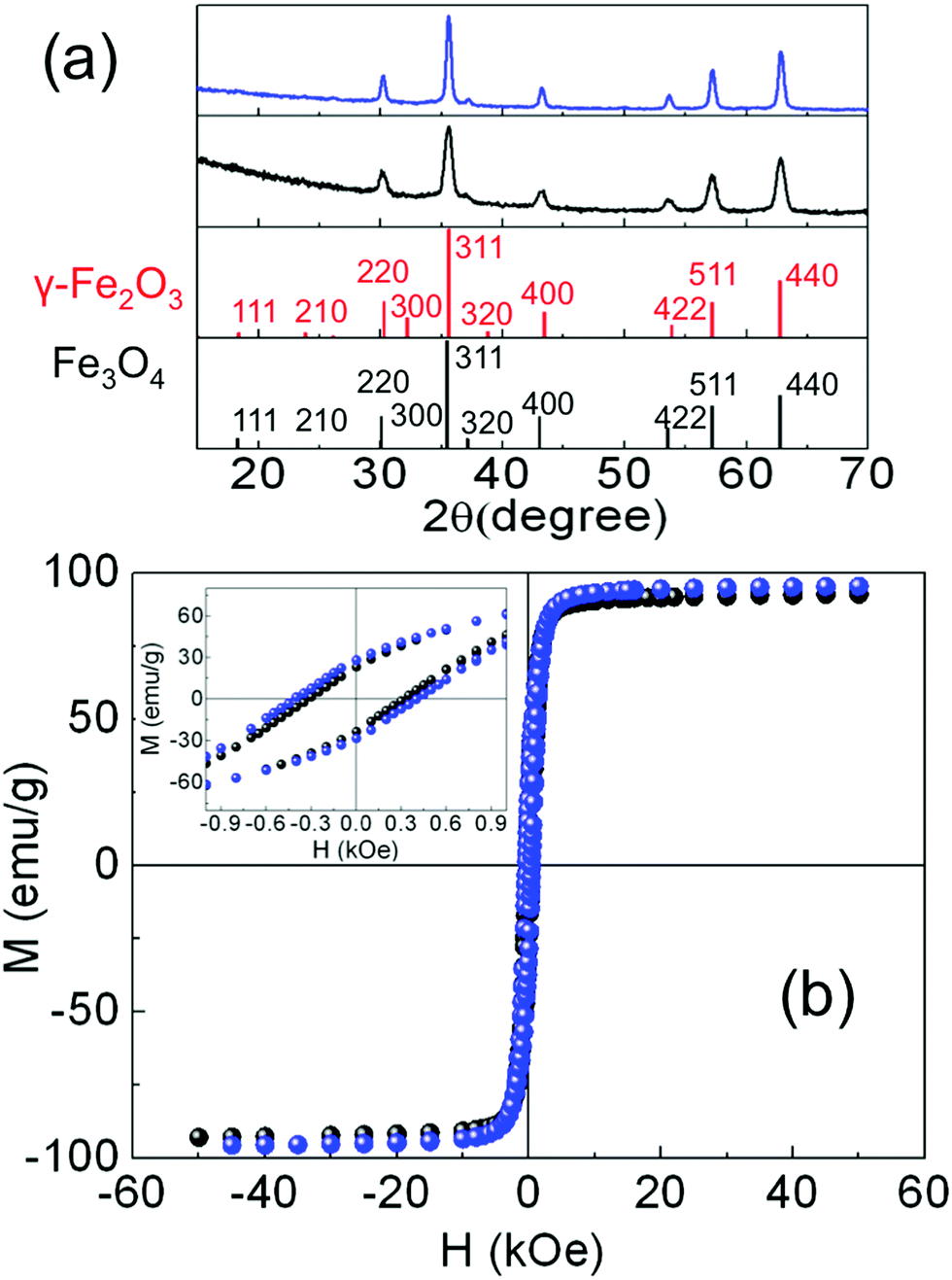 | ||
| Fig. 2 (a) X-ray diffraction patterns with the indexation of the Bragg peaks to an inverse-spinel structure for S1 (black line) and S2 (blue line). Vertical lines at the bottom correspond to X’Pert High Score Plus patterns for γ-Fe2O3 (red) and Fe3O4 (black). (b) Hysteresis loops at 5 K. Black spheres: sample S1, blue spheres: sample S2. Inset: Detail of the low field region. | ||
The above spatially averaged characterizations on ensembles with a large number of particles would lead us to conclude that the structural and magnetic features for the different samples are rather uniform, regardless of the tuning of the synthesis parameters. However, as will be shown in the following, distributions of the actual chemical composition of the NP are present, even for such highly monodisperse (both in size and shape) ensembles of NP with consistently good crystallinity. Such heterogeneous physicochemical properties can only be disentangled through the local information obtained from single-particle experiments.
X-ray imaging of single NP
For the PEEM experiments, NP monolayers of the samples were prepared by either drop casting or spin coating under a N2 atmosphere of highly diluted NP suspensions onto bare SiOx (for sample S1) and carbon-coated SiOx (for sample S2) substrates. The typical particle density on the substrates for all cases was limited to a few particles per μm2. The reason for this was twofold. First, single-particle detection by X-ray PEEM with a lateral resolution of about 50 nm is warranted; and second, it was aimed to avoid interparticle interactions in order to distinguish the intrinsic properties of the individual NP from the complex magnetic behavior of dense particle ensembles.27,39 The stochastic nature of the deposition process leads to random lateral distributions and random orientations of the crystallographic axes of the NP with respect to the substrate. After deposition, the samples were transferred to the PEEM chamber (LEEM III, Elmitec GmbH) of the Surface/Interface: Microscopy (SIM) beamline of the Swiss Light Source, Paul Scherrer Institut, Switzerland.28 The base pressure of the microscope was 5 × 10−10 mbar for the present experiments. All experiments were carried out at room temperature. The PEEM technique provides a full-field magnified image of the emitted secondary photoelectrons28 with a probing depth of a few nm at the L2,3 edges of Fe,40 thus allowing a spatial map of the absorption of the particles.To univocally determine the presence and position of the iron oxide NP, X-ray PEEM elemental contrast maps of the iron oxide nanoparticles were first obtained by a pixel-wise division of two PEEM images successively recorded at the resonant Fe L3 absorption edge (∼709 eV) and at the pre-edge region (∼705 eV). Representative data are shown in Fig. 3(b). Such a procedure ensures that the observed contrast truly originates from the iron oxide NP and reduces topographic contrast. In addition, it reduces illumination inhomogeneity, thus significantly decreasing the systematic errors in X-ray absorption, which is of crucial importance to obtain reliable data. Considering that the particles are randomly distributed with a typical density of 1–2 particles per μm2, each bright spot should ideally correspond to an individual iron oxide NP, such as particle A in Fig. 3(b). However, dimers or close-lying particles (particles B and C, respectively, in Fig. 3(b)) are also present in our samples. Consequently, only truly individual and well separated particles were considered in the analysis to avoid misinterpretations. The random disposition of the particles on the substrates reflects the stochastic nature of the deposition process and the limited mobility of the particles on the surface.
 | ||
| Fig. 3 (a) AFM image of iron oxide NP (sample S1) with circles marking the positions of a few particles. The insets show high resolution AFM images of three particles tagged as A (with a height of 15 ± 1 nm), B, and C in (a). (b) Elemental contrast PEEM image of the same sample area as in (a), obtained by dividing images successively recorded at the Fe L3 absorption edge (709 eV) and at the pre-edge region (705 eV). The scale bars are 2 μm. | ||
AFM characterization of single NP
While X-ray PEEM yields information about the electronic properties of well-separated individual NP, its limited lateral resolution prevents a detailed morphological characterization. Thus, after the PEEM experiments were carried out, the samples were further studied by ex situ atomic force microscopy (AFM) (Fig. 3(a)) to accurately determine the size (height) of the very same individual NP investigated with PEEM (Fig. 3(b)). The identification of the same areas of the samples in the two microscopes was achieved using suitable marks on the substrates (Cr/Au lithographic markers in Fig. 3(a)).X-ray absorption spectroscopy of single NP
Isotropic local X-ray absorption spectroscopy (XAS) images were obtained by recording sequences of X-ray PEEM images around the Fe L2,3 edges using linear, s-polarized light and analyzing the isotropic (non-magnetic) intensity as a function of the photon energy.41 The field of view was 20 μm. The images in each energy scan were first aligned to correct for sample drifts during acquisition. Subsequently, the spectra of single NP were obtained by extracting the image intensity from square pixel areas centered on the bright spots in the PEEM images and normalized to the pixel area. The spectra were then normalized by dividing the signal from the single NP by a background signal from neighboring areas without particles. All of the particles within the field of view were probed simultaneously, thus, systematic errors arising from either the measurements (e.g. due to an inhomogeneous illumination across the PEEM images) or the analysis procedure (e.g. caused by image drift correction) were comparable for all the analyzed particles. Although the particles are smaller than the lateral resolution, individual spectra can be obtained when the particles are separated from their nearest neighbors by a sufficiently large distance.27,41 In our case this is on average between 0.5 and 1 μm (see Fig. 3(a)). Note that for smaller interparticle distances (see e.g. particle C in Fig. 3(a) and (c)), the absorption intensity profiles of the close-lying particles overlap and thus the resulting spectral features do not represent the actual single-particle characteristics, but instead, some weighted average of the spectral features from different close-lying particles. Data was collected from different areas of the samples to ensure sufficient data statistics on single particles. The spectra of the individual particles were rescaled between zero at the pre-edge (705 eV, no absorption) and one at the post-edge (730 eV, finite non-resonant absorption). This normalization procedure makes the XAS from different particles and samples directly comparable. Nevertheless, it should be noted that a quantitative interpretation of X-ray PEEM spectra is not straightforward. In particular, the analysis procedures employed to obtain the local spectra can significantly alter the final result.41 Therefore, careful data processing was performed to ensure that consistent, reproducible spectra were computed, as discussed elsewhere.41 XMCD spectra of individual iron oxide NP can also be obtained by collecting successive stacks of images for right (σ+) and left (σ−) circularly polarized X-rays around the Fe L2,3 edges.41 However, the low signal-to-noise ratio (SNR) of our XMCD spectra (not shown) hinders any attempts to get rough estimates of the spin and orbital moments from the individual NP. Therefore, in this work, we focus only on the study of the electronic and chemical structure from the isotropic XAS spectra.Analysis of the local XAS spectra
In Fig. 4, illustrative examples of the Fe L-edge XAS spectra of single NP for samples S1(a) and S2(b–e) are shown. For each sample, the analysis was performed on a number of NP of similar size: 24 NP of 15 ± 1 nm for sample S1 and 33 NP of 24 ± 1 nm for sample S2. Iron forms several oxide compounds, such as FeO, Fe3O4, γ-Fe2O3, and α-Fe2O3 where FeO and α-Fe2O3 are antiferromagnetic while Fe3O4 and γ-Fe2O3 are ferrimagnetic. The Fe L-edge XAS fine structure is a signature of the structural differences between the oxide phases and can thus be used to identify them. There are two main peaks corresponding to the L3 (∼709 eV) and L2 edges (∼722 eV), respectively, and several shoulder peaks (indicated by thin vertical lines in Fig. 4) with different energy positions and relative peak intensities depending on the iron oxide phase. To quantify the amount of these oxides present in selected individual particles within the samples, the measured local isotropic XAS spectra were fitted to a weighted linear combination of the reference bulk spectra of different iron oxide species taken from ref. 40: Fe, FeO, Fe3O4, and γ-Fe2O3. The assignment of the oxide phases shown in Fig. 4 has been chosen to achieve the best compromise between the most relevant criteria, as discussed in the literature.42 These are namely the shapes of the spectral features (thin vertical lines in Fig. 4), the branching L3:L2 ratio, the shift between the first and main peak at the L3 edge, the occurrence of a L3 single peak, and the disappearance of the L2 edge splitting. Note that no single criterion is able to discriminate one oxide species over another due to the large variability of experimental data and peak-fitting methods in the literature.43 These difficulties together with the low SNR of the measured single-particle XAS spectra limit the average accuracy in the assignment of the oxide phases to about 8% in our case.
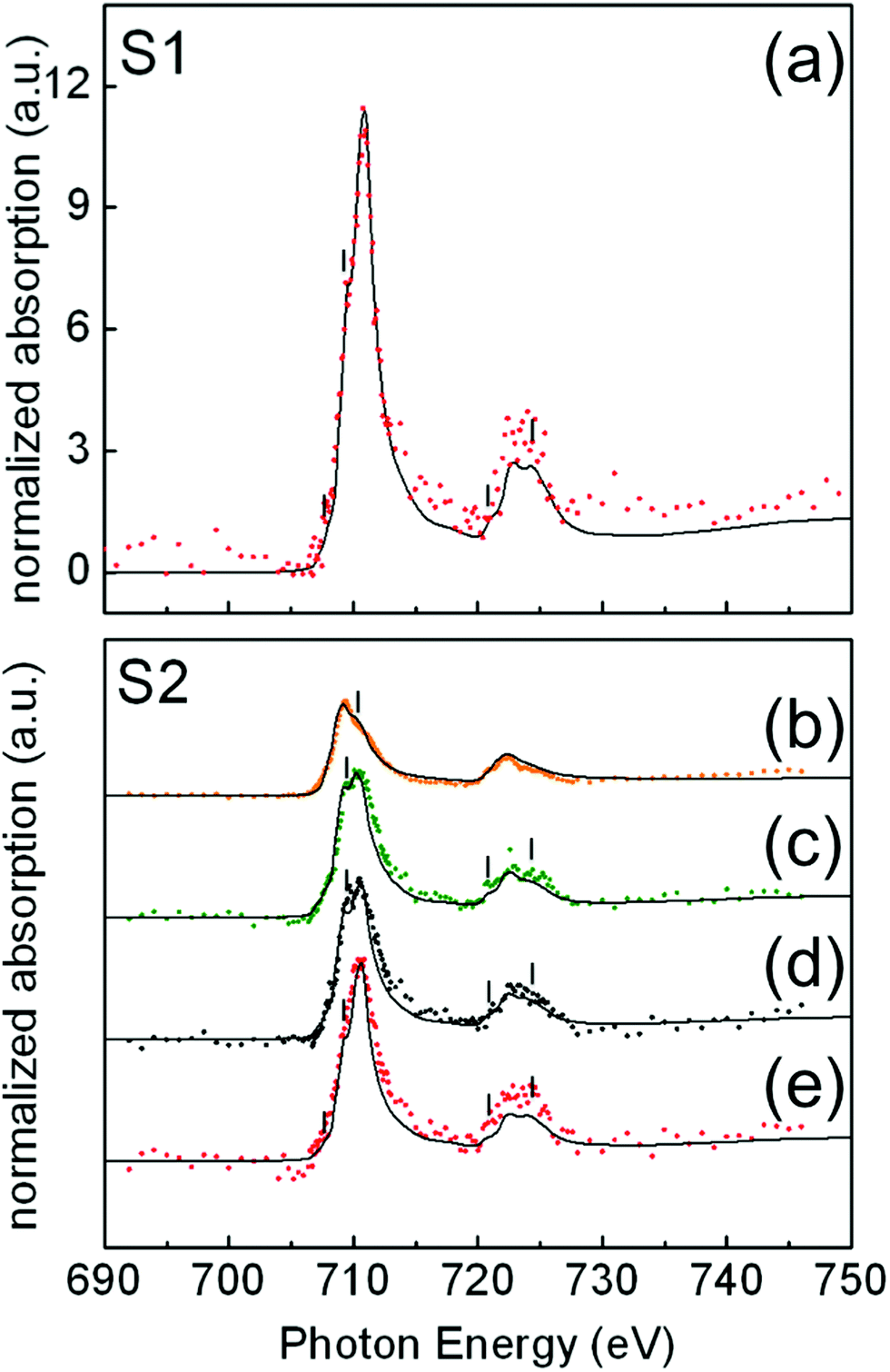 | ||
| Fig. 4 Normalized XAS spectra of representative single NP for samples S1(a) and S2(b–e), obtained from a series of X-PEEM images recorded around the Fe L2,3-edges, compared to the best spectral fits (continuous lines) obtained as the weighted sum of reference bulk spectra for different iron species. (a) 80% Fe3O4 + 20% γ-Fe2O3; (b) 15% Fe3O4 + 85% Fe; (c) 50% Fe3O4 + 50% FeO; (d) 50% Fe3O4 + 30% FeO + 20% γ-Fe2O3; (e) 80% Fe3O4 + 20% γ-Fe2O3. | ||
In the case of the 15 nm S1 NP, data show that all particles have a similar composition. For all the analyzed particles, the best spectral fits were compatible with a XAS signal arising from the weighted sum of 80% Fe3O4 and 20% γ-Fe2O3 (Fig. 4). In Fig. 4(a), we show a representative example of a fit to the measured XAS for a single NP, showing good agreement.
In contrast, in the case of 24 nm S2 NP, we found that particles of the same size and high crystal quality (see Fig. 1(b) and (d)) were composed of different Fe oxide species (Fig. 4(b)–(e)). The XAS spectra for about 40% of the NP were compatible with a composition of 80% Fe3O4 + 20% γ-Fe2O3 (Fig. 4(e)); about 40% with 50% Fe3O4 + 50% FeO (Fig. 4(c)); about 10% of the NP with 15% Fe3O4 + 85% Fe (Fig. 4(b)); and about 10% with 50% Fe3O4 + 30% FeO + 20% γ-Fe2O3 (Fig. 4(d)). Note that the occurrence of a single L3 peak and the symmetric shape of the L2 edge are features that are characteristic of over-reduced phases such as FeO and Fe,40,44 and are consistent with our spectra in Fig. 4(b) and (c). In contrast, the presence of multiplet features and the splitting of both the L3 and L2 peaks indicate a predominance of the Fe3O4 and γ-Fe2O3 phases39,44 in agreement with our spectra in Fig. 4(d) and (e).
An accurate volume determination of each oxide phase is difficult. First, due to the intrinsic limitations of the experimental probe arising from the fact that the local intensity of the incident X-rays on the particles does not simply decay exponentially with the penetration depth, but is further modulated by the varying absorption cross sections of the different layers at a given photon energy, so the emitted electrons also experience a material dependent attenuation when leaving the sample.40 In addition, for NP of the sizes that we are investigating here (>12 nm) deposited on substrates, a large fraction of the volume of the NP is hardly probed.45 Second, if the volume of the parasitic phases is sufficiently small, it is difficult to discern whether they stabilize in layered-like structures or in the form of small inclusions of the Fe phases embedded within the Fe3O4 NP, as will be further discussed later (Fig. 5). We have estimated the diameters of the Fe and FeO inclusions to be very small (<1 nm), based on the bulk densities of the different Fe phases (5.2 g cm−3, Fe3O4; 4.9 cm3, γ-Fe2O3; 5.74 g cm−3, FeO; 7.84 g cm−3, Fe)46 and the Ms values at 5 K (Fig. 2(b)). Therefore, neither XRD (Fig. 2(a)) nor high-resolution TEM data (Fig. 1(b)) enable discriminating between Fe3O4 and γ-Fe2O3, although signatures from these phases as well as from over-reduced Fe phases have been confirmed through high resolution STEM and EELS characterization (Fig. S7, ESI†).
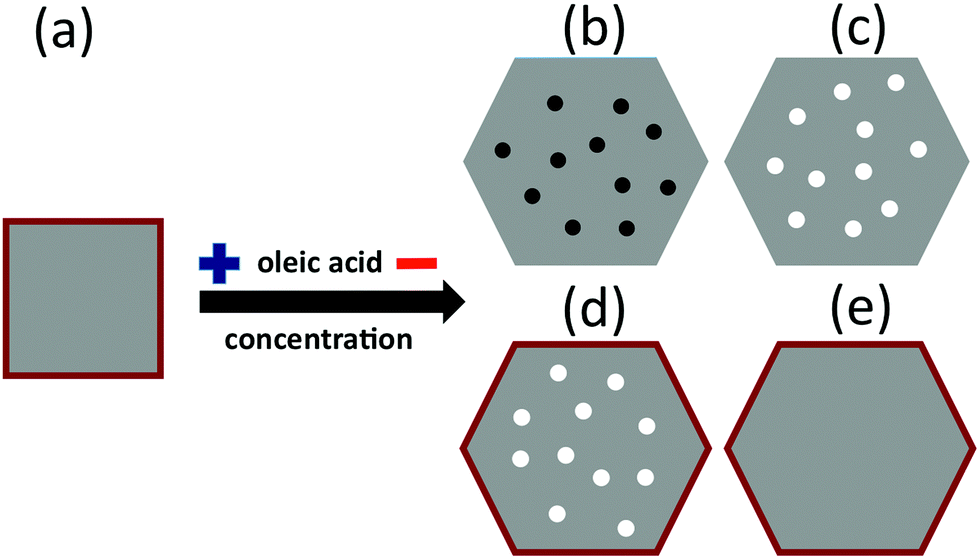 | ||
| Fig. 5 Schematic representation of the role of oleic acid concentration on the formation of the different Fe phases in the synthesis of Fe3O4 NP. For sample S1: (a) 100% of NP are composed of a homogeneous Fe3O4 core (gray) surrounded by a thin γ-Fe2O3 layer (red). For sample S2: (b) 10% of Fe3O4 NP (gray) containing small inclusions of Fe (black), (c) 40% of NP containing small inclusions of FeO (white), (d) 10% of NP containing small inclusions of FeO and a thin γ-Fe2O3 surface layer (red), (e) 40% of NP composed of a homogeneous Fe3O4 core surrounded by a thin γ-Fe2O3 surface layer. Note that the high oleic acid concentration of sample S1 generally yields cubic NP; only below a certain size threshold do they become pseudospherical as is our case. | ||
In the case of the 15 nm S1 NP, since in γ-Fe2O3, iron is in a higher oxidation state than in Fe3O4, it is reasonable to assume that the particles stabilize in the form of a Fe3O4 (core)/γ-Fe2O3 shell structure, as often reported in the literature for magnetite NP prepared by similar methods (see e.g.ref. 25 and 47). The same argument applies for the 24 nm S2(e) particles. In these two cases, we can estimate the relative oxide layer thickness corresponding to the topmost part of the particle that is locally probed by XAS using the electron escape depths from the literature – 35 Å for γ-Fe2O3, and 50 Å for Fe3O440 – and a well-established procedure introduced by Regan et al.40 The thickness of the γ-Fe2O3 shell for S1 and S2(e) NP was estimated to be about 0.7 nm. It should be pointed out that the thin γ-Fe2O3 shell at the surface of the S1 and S2(e) particles may coexist with the oxygen contribution from the carboxylic groups of the oleic acid bonded to the surface Fe ions.25
In contrast to the homogeneous Fe3O4 (core)/γ-Fe2O3 shell structure of the S1 and S2(e) particles, for S2(b), S2(c), and S2(d) particles, iron can also be found in low oxidation states such as FeO or metallic Fe. It is thus reasonable to assume that Fe3O4 stabilizes around the more reduced phases in the three cases, e.g., in the form of inhomogeneous Fe3O4 NP throughout which small core inclusions of FeO or Fe are present (Fig. 5). This scenario would still be compatible with the XAS signal percent distributions in Fig. 4 and with the STEM and EELS data (Fig. S7, ESI†). The over-reduced Fe phases hinder the formation of the over-oxidized γ-Fe2O3 phase at the surface of the S2(b) and (c) particles.
The relatively low SNR of the experimental single-particle spectra in Fig. 4 limits a quantitative, accurate determination of the cation site occupancies by fitting them to a linear combination of ligand field multiplet simulated spectra.44 This in turn prevents a reliable comparison with the data available in the literature for bulk or nanostructured Fe3O4.39,40,48 Furthermore, it should be noted that neither finite-size nor surface effects are responsible for the large variation in phase homogeneity between 15 nm S1 particles (homogeneous) and 24 nm S2 particles (highly heterogeneous), since both sizes are well above those at which these effects become prevalent (around 5–6 nm).34 Besides, the NP in both cases show high crystallinity up to the surface, and comparable values and shapes of the magnetic parameters (Table 1) and hysteresis loops (Fig. 2(b)), respectively. These facts rule out the occurrence of any effects associated with either the finite size or the surface of the NP.
Role of the oleic acid concentration on the type of Fe oxide phases
Our single-particle spectra suggest that the concentration of the oleic acid in the reaction mixture has a remarkable effect on the nature and distribution of the various Fe oxide phases within individual NP. Typically, the role of the oleic acid on the synthesis of Fe3O4 NP is threefold. At the beginning of the reaction, oleic acid reacts with iron acetylacetonate(III) to form an intermediate [Fe(oleate)x]3+ complex.49 Then, the temperature of the reaction mixture is increased to reflux temperature where the [Fe(oleate)x]3+ decomposes49 yielding Fe3O4 nuclei that are partially coated by unreacted oleic acid. This coating has a key role in self-regulating the subsequently growth of the nuclei to form the final particles.35 Finally, the oleic acid layer binds to the particle surface by adhesion or covalent bonding both enhancing surface magnetization and preventing further oxidation.25In addition, one can obtain good control of the particle growth by only varying the concentration of the oleic acid in the reaction mixture, thus obtaining a large variation of the structural properties of the particles. In particular, at low concentrations of the oleic acid with respect to the iron acetylacetonate(III) ([Fe(acac)3] > 4[oleic acid]),12 NP are obtained by the formation and further decomposition of various intermediate iron complexes. Thus, not only the aforementioned formation of [Fe(oleate)x]3+ – which further decomposes giving rise to homogeneous Fe3O4 structures – takes place, but also secondary reactions between the Fe(acac)3 and the benzyl-ether – that is not actually a non-inert solvent – yield other intermediate compounds49 that produce inhomogeneous oxide NP with poor control of the iron oxidation state.50 As a result, inhomogeneous particles composed of different Fe species are obtained as a function of the concentration of oleic acid in the reaction mixture. When the concentration of oleic acid is high enough, the NP are composed of a nucleus of Fe3O4 with an oxidized shell of γ-Fe2O3, as commonly found.25 However, at low concentrations of the fatty acid, the Fe3O4 NP are likely inhomogeneous with small inclusions of FeO31 and Fe50 (Fig. 5), as a result of the uncontrolled reduction of Fe3+ ions.
Conclusions
We have studied the electronic and chemical properties of ensembles of size- and shape-selected Fe3O4 NP by means of synchrotron-based X-ray spectro-microscopy using PEEM with single-particle sensitivity. The local XAS spectra reveal that the oxidation states and cation distribution of the individual nanoparticles may show a striking local variability even when the average structural and magnetic parameters of monodisperse ensembles appear homogeneous and size-and sample-independent. Our results show the key role of oleic acid concentration during the synthesis reaction on the formation and distribution of the Fe phases within the individual NP. When the concentration of oleic acid is high enough, the NP are composed of a Fe3O4 core surrounded by a thin oxidized shell of γ-Fe2O3. In contrast, at low concentration of the fatty acid, the Fe3O4 NP are likely inhomogeneous with small inclusions of FeO31 and Fe49 phases, as a result of the uncontrolled reduction of Fe3+. Our work demonstrates the relevance of single-particle spectroscopies performed on a statistically significant number of particles to bring about a paradigm shift in the understanding of NP with heterogeneous physicochemical properties. Finally, our results have broad implications for applications focusing on the modification and functionalization of magnetic nanoparticles. They also highlight that a deeper knowledge of the formation pathways, phase coexistence and surface chemistry of magnetic nanoparticles is still required.51Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Spanish MINECO (MAT2015-68772-P), Catalan DURSI (2014SGR220) and European Union FEDER funds. A. F. R. acknowledges the support from the Spanish MICIIN “Ramón y Cajal” Program. Part of this work was performed at the Surface/Interface: Microscopy (SIM) beamline of the Swiss Light Source (SLS), Paul Scherrer Institut, Switzerland. The STEM and EELS studies have been conducted in the Laboratorio de Microscopias Avanzadas (LMA) at Instituto de Nanociencia de Aragon (INA) – Universidad de Zaragoza, Spain.Notes and references
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- J. M. Pérez, L. Josephson and R. Weissleder, ChemBioChem, 2004, 5, 261 CrossRef PubMed.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252 CrossRef CAS PubMed.
- R. Mejías, S. Pérez-Yagüe, A. G. Roca, N. Pérez, A. Villanueva, M. Cañete, S. Mañes, J. Ruiz-Cabello, M. Benito, A. Labarta, X. Batlle, S. Veintemillas-Verdaguer, M. P. Morales, D. F. Barber and C. J. Serna, Nanomedicine, 2010, 5, 397 CrossRef PubMed.
- J. Bansmann, S. H. Baker, C. Binns, J. A. Blackman, J.-P. Bucher, J. Dorantes-Dávila, V. Dupuis, L. Favre, D. Kechrakos and A. Kleibert, et al. , Surf. Sci. Rep., 2005, 56, 189 CrossRef CAS.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed.
- N. Jones, Nature, 2011, 472, 22 CrossRef CAS PubMed.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371 RSC.
- Spintronics for Next Generation Innovative Devices, ed. K. Sato and E. Saitoh, John Wiley & Sons, 2015 Search PubMed.
- B. Q. Geng, Z. L. Ding and Y. Q. Ma, Nano Res., 2016, 9, 2772 CrossRef CAS.
- X. Batlle and X. Labarta, J. Phys. D: Appl. Phys., 2002, 35, R15 CrossRef CAS.
- P. Guardia, N. Pérez, A. Labarta and X. Batlle, Langmuir, 2010, 26, 5843 CrossRef CAS PubMed.
- P. Tartaj, M. P. Morales, S. Veintemillas-Verdaguer, T. González-Carreño and C. J. Serna, J. Phys. D: Appl. Phys., 2003, 36, R182 CrossRef CAS.
- P. Nicolás, M. Saleta, H. Troiani, R. Zyssleer, V. Lassalle and M. L. Ferreira, Acta Biomater., 2010, 9, 4754 CrossRef PubMed.
- Y. Wang, J. Hu, Y. Li and C.-W. Nan, NPG Asia Mater., 2010, 2, 61 CrossRef.
- B. D. Terris and T. Thomson, J. Phys. D: Appl. Phys., 2005, 38, R199 CrossRef CAS.
- D. L. Huber, Small, 2005, 1, 482 CrossRef CAS PubMed.
- M. Fonin, Y. S. Dedkov, R. Pentcheva, U. Rüdiger and G. Güntherodt, J. Phys.: Condens. Matter, 2007, 19, 315217 CrossRef CAS PubMed.
- H. C. Wu, C. Ó. Coileáin, M. Abid, O. Mauit, A. Syrlybekov, A. Khalid, H. Xu, R. Gatensby, J. J. Wang, L. Yang, G. S. Duerberg, H.-Z. Zhang, M. Abid and I. Shvets, Sci. Rep., 2015, 5, 15984 CrossRef CAS PubMed.
- C. Moya, X. Batlle and A. Labarta, Phys. Chem. Chem. Phys., 2015, 17, 27373 RSC.
- C. Moya, Ó. Iglesias-Freire, N. Pérez, X. Batlle, A. Labarta and A. Asenjo, Nanoscale, 2015, 7, 8110 RSC.
- C. Moya, G. Salas, M. P. Morales, X. Batlle and A. Labarta, J. Mater. Chem. C, 2015, 3, 4522 RSC.
- A. Fraile Rodríguez, A. Kleibert, J. Bansmann, A. Voitkans, L. J. Heyderman and F. Nolting, Phys. Rev. Lett., 2010, 104, 127201 CrossRef PubMed.
- A. Balan, P. M. Derlet, A. Fraile Rodríguez, J. Bansmann, R. Yanes, U. Nowak, A. Kleibert and F. Nolting, Phys. Rev. Lett., 2014, 112, 107201 CrossRef PubMed.
- J. Salafranca, J. Gazquez, N. Pérez, A. Labarta, S. T. Pantelides, S. J. Pennycook, X. Batlle and M. Varela, Nano Lett., 2012, 12, 2499 CrossRef CAS PubMed.
- P. Torruella, R. Arenal, F. De La Peña, Z. Saghi, L. Yedra, A. Eljarrat, L. López-Conesa, M. Estrader, A. López-Ortega, G. Salazar-Alvarez, J. Nogués, C. Ducati, P. A. Midgley, F. Peiró and S. Estradé, Nano Lett., 2016, 16, 5068 CrossRef CAS PubMed.
- F. Kronast, N. Friedenberger, K. Ollefs, S. Gliga, L. Tati-Bismath, R. Thies, A. Ney, R. Weber, C. Hassel, F. M. Römer, A. V. Trunova, C. Wirtz, R. Hertel, H. A. Dürr and M. Farle, Nano Lett., 2011, 11, 1710 CrossRef CAS PubMed.
- L. Le Guyader, A. Kleibert, A. Fraile Rodríguez, S. El Moussaoui, A. Balan, M. Buzzi, J. Raabe and F. Nolting, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 371 CrossRef CAS.
- M. Rohmer, M. Bauer, T. Leissner, Ch. Schneider, A. Fischer, G. Niedner-Schatteburg, B. v. Issendorff and M. Aeschlimann, Phys. Status Solidi B, 2010, 247, 1132 CAS.
- P. Guardia, B. Batlle-Brugal, A. G. Roca, O. Iglesias, M. P. Morales, C. J. Serna, A. Labarta and X. Batlle, J. Magn. Magn. Mater., 2007, 316, e756 CrossRef CAS.
- P. Guardia, A. Labarta and X. Batlle, J. Phys. Chem. C, 2011, 115, 390 CAS.
- X. Batlle, M. García del Muro, J. Tejada, H. Pfeiffer, P. Görnert and E. Sinn, J. Appl. Phys., 1993, 74, 3333 CrossRef CAS.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, Wiley-IEEE Press, 2011 Search PubMed.
- X. Batlle, N. Pérez, P. Guardia, O. Iglesias, A. Labarta, F. Bartolomé, L. M. García, J. Bartolomé, A. G. Roca, M. P. Morales and C. J. Serna, J. Appl. Phys., 2011, 109, 07B524 CrossRef.
- F. Heider, A. Zitzelsberger and K. Fabian, Phys. Earth Planet. Inter., 1996, 93, 239 CrossRef CAS.
- J. Nogués, J. Sort, V. Langlais, V. Skumryev, S. Suriñach, J. S. Muñoz and M. D. Baró, Phys. Rep., 2005, 422, 65 CrossRef.
- M. Estrader, A. López-Ortega, I. V. Golosovsky, S. Estradé, A. G. Roca, G. Salazar-Alvarez, L. López-Conesa, D. Tobia, E. Winkler and J. D. Ardisson, et al. , Nanoscale, 2015, 7, 3002 RSC.
- O. Iglesias, X. Batlle and A. Labarta, J. Phys.: Condens. Matter, 2007, 19, 406232 CrossRef PubMed.
- F. Jimenez-Villacorta, C. Prieto, Y. Huttel, N. D. Telling and G. van der Laan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 172404 CrossRef.
- T. J. Regan, H. Ohldag, C. Stamm, F. Nolting, J. Lüning, J. Stöhr and R. L. White, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 214422 CrossRef.
- A. Fraile Rodríguez, A. Kleibert, J. Bansmann and F. Nolting, J. Phys. D: Appl. Phys., 2010, 43, 474006 CrossRef.
- J. G. Chen, Surf. Sci. Rep., 1997, 30, 1 CrossRef CAS.
- C. Wang, D. R. Baer, J. E. Amonette, M. H. Engelhard, J. Antony and Y. Qiang, J. Am. Chem. Soc., 2009, 131, 8824 CrossRef CAS PubMed.
- A. Kleibert, J. Bansmann and K.-H. Meiwes-Broer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125423 CrossRef.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, Wiley-IEEE Press, Hoboken, New Jersey, 2011 Search PubMed.
- J. P. Crocombette, M. Pollak, F. Jollet, N. Thromat and M. Gautier-Soyer, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 3143 CrossRef CAS.
- X. Zhu, S. S. Kalirai, A. P. Hitchcock and D. A. Bazylinski, J. Electron Spectrosc. Relat. Phenom., 2015, 199, 19 CrossRef CAS.
- N. Pérez, F. López-Calahorra, A. Labarta and X. Batlle, Phys. Chem. Chem. Phys., 2011, 13, 19485 RSC.
- J. Park, K. An, Y. Hwang, J. E. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed.
- P. Guardia, J. Pérez-Juste, A. Labarta, X. Batlle and L. M. Liz-Marzán, Chem. Commun., 2010, 46, 6108 RSC.
- S. P. Schwaminger, D. Bauer, P. Fraga-García, F. E. Wagner and S. Berensmeier, CrystEngComm, 2017, 19, 246 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Particle size distributions. Thermogravimetric curves. Field-cooled hysteresis loops. Low resolution dark field TEM images. High resolution STEM images and EELS spectra. See DOI: 10.1039/c7tc03010j |
| This journal is © The Royal Society of Chemistry 2018 |