
Sky-blue thermally activated delayed fluorescence material employing a diphenylethyne acceptor for organic light-emitting diodes†
Zuozheng
He‡
,
Xinyi
Cai‡
,
Zhiheng
Wang
,
Yunchuan
Li
,
Zhida
Xu
,
Kunkun
Liu
,
Dongcheng
Chen
and
Shi-Jian
Su
 *
*
State Key Laboratory of Luminescent Materials and Devices, Institute of Polymer Optoelectronic Materials and Devices, South China University of Technology, Guangzhou, 510640, P. R. China. E-mail: mssjsu@scut.edu.cn
First published on 20th November 2017
Abstract
The strong electronegativity of sp-hybridized carbons, contributed by their large s character (50%), inspired the development of diphenylethyne as an electron acceptor to construct a sky-blue thermally activated delayed fluorescence material, 1,2-bis(4-(10H-phenoxazin-10-yl)phenyl)ethyne (DPE-DPXZ), using 10H-phenoxazine (PXZ) as an electron donor fragment. A sky-blue organic light-emitting diode employing DPE-DPXZ as the emitter demonstrated an external quantum efficiency exceeding 10%. Furthermore, another alkyne derivative, 1,2-bis(4-(9,9-dimethylacridin-10(9H)-yl)phenyl)ethyne (DPE-DDMAc), in combination with 9,9-dimethyl-9,10-dihydroacridine (DMAc) as the donor was also studied to explore the singlet–triplet splitting energy governed by donor–acceptor alternation.
Introduction
The replacement of current light-emitting materials, such as traditional fluorophores with limited harvesting capabilities of only 25% electrogenerated singlet excitons and noble metals containing phosphorescent emitters with unity exciton utilization ratios, with pure aromatic compounds has recently become one of the most attractive fields for future highly efficient, low-cost, and large-scale display and illumination applications.1–6 The thermally activated delayed fluorescence (TADF, E-typed fluorescence) mechanism, which requires narrowing of the singlet–triplet splitting energy (ΔEST) for effective reverse intersystem crossing (RISC) with ambient thermal energy activation, is among the most promising candidates for realizing 100% internal quantum efficiency (IQE) organic light-emitting diodes (OLEDs).7–9 As this technique is still in its infancy, the pursuit of pure aromatic compounds with the capacity to harvest triplet excitons is ongoing.To achieve small ΔEST, frontier molecular orbitals (FMOs) closely related to the lowest energy transition should exhibit a small electronic wave function overlap integral (〈ϕi|ϕf〉, where ϕi denotes initial state orbitals and ϕf denotes final state orbitals), which is directly proportional to electron exchange energy (J). Small FMO overlap results in a small J value and produces an efficient RISC process, as described by the Boltzmann equation, kRISC ∝ exp(−ΔEST/kBT), in which kRISC, kB, and T denote the RISC rate constant, Boltzmann constant, and temperature, respectively.10–12 Following this guideline, molecular design strategies employing highly twisted intramolecular charge transfer (TICT) framework to break the π-conjugation and combining donors (D) and acceptors (A) with high triplet energies (T1) have become the most successful for TADF molecules. Numerous D–A compounds utilizing diverse chemical moieties have been reported. Emitters for OLEDs based on acceptors, such as sulfones, carbonyls, nitrogen heterocycles, benzonitrile, boron heterocycles, phosphorus oxide, and cyclophosphazene, have achieved excellent performances matching those of phosphorescent OLEDs and covering the full RGB and near-infrared (NIR) spectra.8,10,13–27
Alkyne groups have usually been used as “p-type” units or π bridges,28–30 due to their two electron-rich C–C π bonds, which have been considered as a cylinder of electrons wrapped around the σ bond. However, according to hybrid orbital theory, the more s character in a hybrid orbital, the more electronegative the hybridized carbon atom, because s-orbital electrons are closer to the nucleus than those in p orbitals. Therefore, sp hybridized carbons are more electronegative than sp2 hybridized carbons, which, in turn, are more electronegative than sp3 hybridized carbons. Accordingly, ethyne is a stronger acid than both ethylene and ethane (Fig. 1). This electronegativity increases with an increasing s character percentage in the orbital, which indicates the potential of alkyne groups as acceptor moieties. Moreover, the electron-accepting ability of the diphenylethyne (DPE) moiety originates from its moderate energy level compared to that of 9,9-dimethyl-9,10-dihydroacridine (DMAc) and 10H-phenoxazine (PXZ).
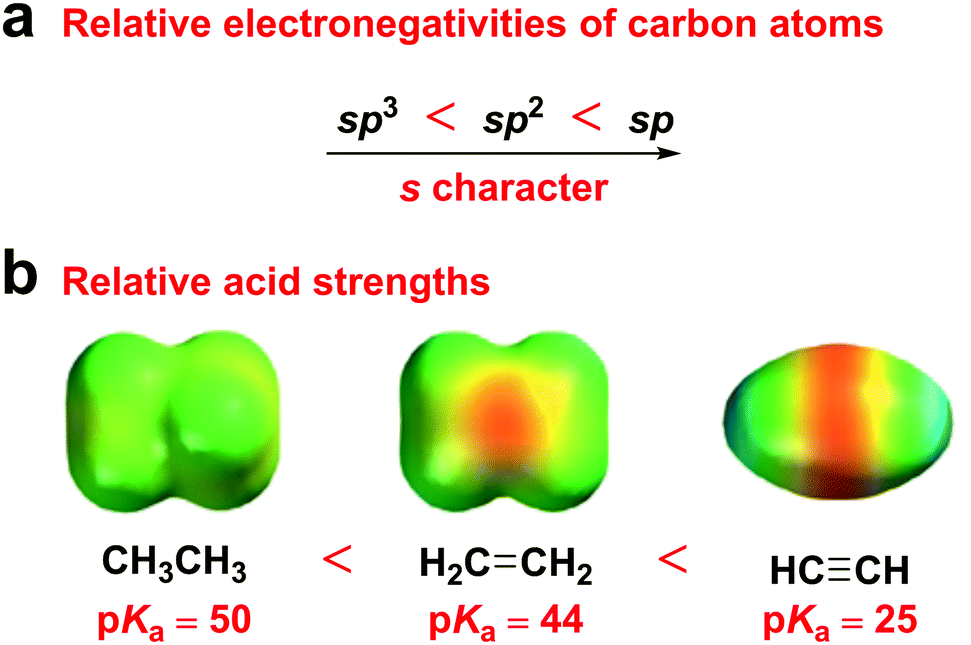 | ||
| Fig. 1 (a) Relative electronegativities of carbon atoms. (b) Relative acid strengths and electrostatic potential maps of ethane, ethylene, and ethyne. | ||
In this work, considering the strong electronegativity of sp-hybridized carbons contributed by the large s character (50%), we used alkyne unit diphenylethyne (DPE), which has seldom been considered as an electron-withdrawing group, as an electron acceptor for the first time. A sky-blue TADF emitter was serendipitously developed by combining DPE acceptor and PXZ donor moieties (Fig. 2). The lowest singlet (S1) and triplet (T1) state energies of the molecule were delicately governed by combining a moderately electron-donating donor and a weakly electron-withdrawing DPE moiety to guarantee a small ΔEST and large energy gap for blue emission. For the first time, a sky-blue OLED using DPE-DPXZ as the emitter exhibited an external quantum efficiency (EQE) surpassing 10% with an emission peak at 475 nm. Insight into the material design is thoroughly analysed in this article.
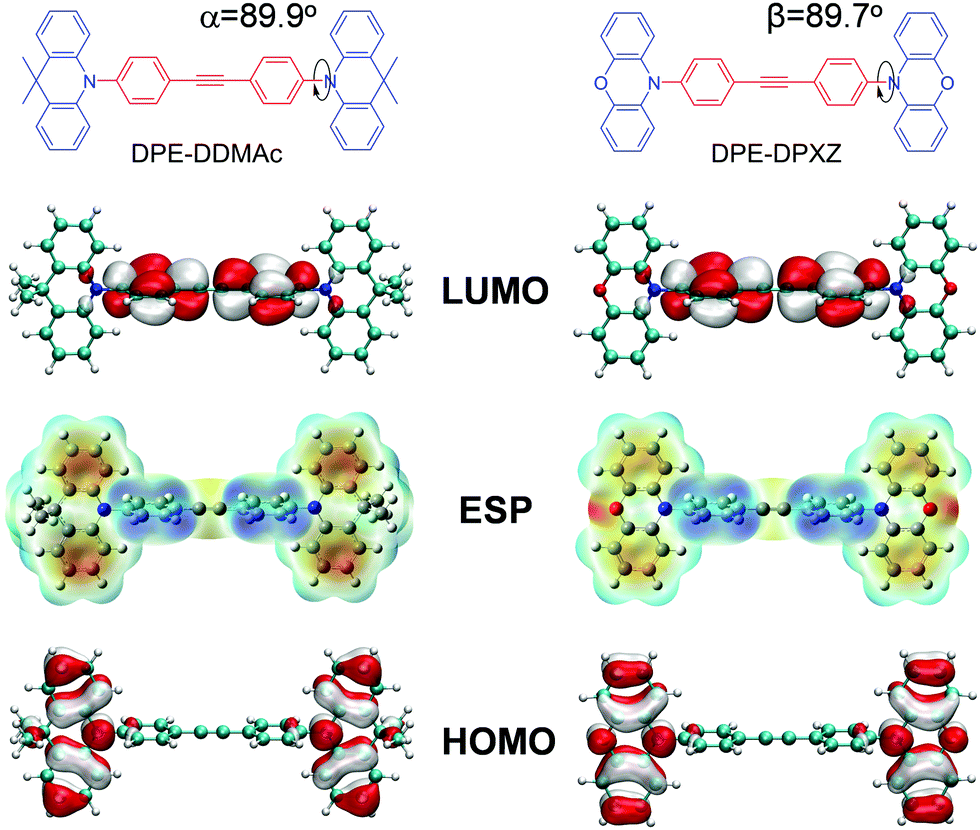 | ||
| Fig. 2 Molecular structures, frontier molecular orbitals, and electrostatic potential distributions of DPE-DDMAc and DPE-DPXZ calculated at the B3LYP/6-31G* level in the gas phase (α and β denote the dihedral angles between the electron donors and acceptor). | ||
Experimental
1H and 13C NMR spectra were recorded on a Bruker NMR spectrometer operating at 600 and 150 MHz, respectively. All data were recorded in deuterated chloroform (CDCl3) solution at room temperature. Mass spectra were recorded on a Waters Synapt G2 Mass Spectrometer. Thermogravimetric analysis (TGA) was performed on Netzsch TG 209 instrument under nitrogen flow at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were performed on Netzsch DSC 209 under nitrogen flow using several heating and cooling cycles with a heating rate of 10 °C min−1 and a cooling rate of 20 °C min−1. Cyclic voltammetry (CV) was carried out on the CHI-600D electrochemical workstation using a Pt working electrode, Pt wire as the counter electrode, and Ag/AgCl as the reference electrode at a scanning rate of 100 mV s−1 in a nitrogen-saturated solution of n-Bu4NPF6 (0.1 mol L−1) in anhydrous acetonitrile and dichloromethane (DCM). Single crystals of DPE-DDMAc were grown from chloroform/methanol mixtures. Processing of the intensity data was carried out using Rigaku R-AXIS, and structure solution and refinement were carried out using the SHELXTL suite of X-ray programs. The crystal structure was deposited at CCDC (deposition number, 1555726).†Ultraviolet-visible absorption spectra were recorded using a PerkinElmer Lambda 950-PKA UV/Vis spectrophotometer, while photoluminescence (PL) and phosphorescence spectra (77 K) were recorded using a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon). PL quantum yields (PLQYs) were measured using the integrating sphere of a Hamamatsu absolute PL quantum yield spectrometer (C11347). Transient PL decay at room temperature was measured using Quantaurus-Tau fluorescence lifetime measurement system (C11367-03, Hamamatsu Photonics Co., Japan). Transient PL decays of film samples at various temperatures were investigated under N2 atmosphere using a Quantaurus-Tau fluorescence lifetime measurement system (C11367-03, Hamamatsu Photonics Co., Japan) equipped with an Oxford Instruments nitrogen cryostat (Optistat DN).
Compound DPE-DPXZ was easily synthesized using a Buchwald–Hartwig C–N coupling reaction in high yield (>80%).31 For comparison, reference compound DPE-DDMAc was also synthesized following the same procedure, using 9,9-dimethyl-9,10-dihydroacridine (DMAc) as the donor. Molecular structures of the two alkyne compounds, DPE-DDMAc and DPE-DPXZ, are shown in Fig. 2, and full characterizations are included in the ESI†. The thermal properties of DPE-DDMAc and DPE-DPXZ were examined by TGA and DSC under a nitrogen atmosphere. These two compounds showed good thermal stability, with decomposition temperatures above 380 °C, making film deposition feasible using a vacuum thermal evaporation technique (Fig. S21, ESI†). Furthermore, no glass transition was found for DPE-DDMAc, while the glass transition temperature (Tg) of DPE-DPXZ was above 120 °C, which ensured their morphological stability.
Results and discussion
Computational simulations
To explore the molecular structure of these compounds, a density functional theory (DFT) simulation was conducted and the ground state geometries were initially optimized at the B3LYP/6-31G* level in the gas phase. Interestingly, the central DPE moiety can adopt two conformers (horizontal and vertical) for both optimized geometries, which were confirmed with no imaginary frequency (Fig. S10 and S13, ESI†). A triple bond consists of one σ bond and two π bonds, with the σ bond formed by sp–sp orbital overlap and π bonds formed by side-to-side overlap of unhybridized p orbitals (Fig. 3). As the two unhybridized p orbitals on each carbon are perpendicular to each other, conjugation of the DPE backbone can be well maintained in both horizontal and vertical conformers, ensuring moderate edge-to-edge overlap between the triple bond and adjacent phenyl rings. Given that the horizontal conformers of the two compounds were more energetically stable than the vertical conformers, we selected the former as the initial guess for the subsequent simulation, as supported by the single-crystal X-ray structure analysis of DPE-DDMAc. As shown in Fig. S17 (ESI†), DPE-DDMAc in the single-crystal structure was confirmed to be the horizontal conformer, which was consistent with the relationship between the energy levels of the two conformers, according to DFT calculations. | ||
| Fig. 3 Orbital hybridization in an acetylene bond. | ||
Geometries, FMO distributions, and electrostatic surface potential (ESP) analyses of the two molecules are presented in Fig. 2. Due to steric hindrance by the adjacent hydrogen atoms on the six-membered rings and DPE backbone, the defined donors and acceptor are almost orthogonal (α = 89.9° and β = 89.7°, respectively). As shown by the ESP distribution (Fig. 3, S11 and S14, ESI†), the negative site was induced by the central acetylene bond due to the large s character (50%) of the sp hybridization pattern, in which the peripheral electron cloud was tightly confined within the triple bond. In that case, the acetylene bond provided a moderate electron-withdrawing ability. Further evidence was found in the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions, which showed that the HOMO was dominantly located on the PXZ or DMAc moieties, while the LUMO was mainly located on DPE unit. This confirmed that DPE acted as the acceptor (Fig. 3 and Fig. S12, S15, ESI†).
Based on the optimized ground state conformers, a time-dependent DFT (TD-DFT) simulation was performed to gain insight into the excited-state paradigm of these two compounds.
Initially, the charge transfer amount (q) was calculated and the optimal Hartree–Fock percentage (OHF) was determined (Fig. S9, ESI,† and Table 1).32 This amendment can minimize so-called self-interaction errors or electron-correlation problems in the TD-DFT simulation, which could result in an underestimate or overestimate of the ICT transition energies.33–35 Taking this into account, the zero–zero lowest charge transfer singlet state energy (E0–0 (1CT)), lowest CT triplet state energy (E0–0 (3CT)), and lowest locally triplet excited state energy (E0–0 (3LE)) were calculated and compared (see ESI†).36 For both emitters E0–0 (3LE) was almost the same (≈2.55 eV) and fell below the corresponding E0–0 value (3CT), indicating that T1 presented LE character (Table 1). Further computational evaluation of the separated molecular fragments (DPE, DMAc, and PXZ) demonstrated that the T1 state could be dominated by the central DPE unit because the simulated values of E0–0 (3LE) for PXZ and DMAc moieties were much higher than that of DPE (2.61 eV). Given the slight bathochromic shift of E0–0 (3LE) in DPE-DDMAc and DPE-DPXZ compared with that of DPE the fragment, moderate electron interaction between the D and A could be anticipated to endow moderate FMO overlap for increased transition possibilities under optical excitation.37 Furthermore, considering the stronger electron-donating ability of PXZ compared to that of DMAc unit, the E0–0 (1CT) of DPE-DPXZ was 0.25 eV lower than that of DPE-DDMAc, causing a narrow calculated zero–zero band ΔEST (0.18 eV) (Table 1). From this perspective, DPE-DPXZ could possess TADF character and the potential for blue emission considering its large CT gap (2.88 eV).
| Compounds | q | OHF (%) | E VA (S1, OHF) (eV) | E 0–0 (1CT) (eV) | E 0–0 (3CT) (eV) | E 0–0 (3LE) (eV) | ΔEST (eV) | HOMO (eV) | LUMO (eV) | E g (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| a HOMO/LUMO levels and the energy gap (Eg) were calculated at the B3LYP/6-31G* level. b Ionization potential (IP) and electron affinity potential (EA) were calculated from CV results. EIP was estimated from (Eoxonset + 4.303) and EEA was roughly approximated from EIP – Eabsonset (Eabsonset is the optical absorption onset energy estimated in toluene solution). | ||||||||||
| DPE-DDMAc | 0.85 | 35.5 | 3.22 | 2.98 | 2.83 | 2.54 | 0.44 | −4.94a/−5.19b | −1.71a/−2.17b | 3.23 |
| DPE-DPXZ | 0.86 | 36.3 | 2.97 | 2.73 | 2.72 | 2.55 | 0.18 | −4.72a−/−4.98b | −1.84a/−2.15b | 2.88 |
| PXZ | — | — | — | — | — | 2.65 | — | −4.68a | −0.23a | 4.45 |
| DMAc | — | — | — | — | — | 2.98 | — | −4.88a | −0.06a | 4.82 |
| DPE | — | — | — | — | — | 2.61 | — | −5.69a | −1.25a | 4.44 |
Photophysical properties
Photophysical investigation validated our computational prediction and analysis. UV-vis absorption and PL spectra of dilute solutions in a variety of solvents (ranging from nonpolar to polar) are shown in Fig. 4. Small molar extinction coefficients (<5 mM cm−2) were observed for the two alkyne compounds due to the large dihedral angle between the donor and DPE units that possibly suppressed transition (Fig. 2). The PL spectra showed a clear dependence on solvent polarity. The toluene solution of DPE-DDMAc and DPE-DPXZ emitted deep-blue and pure-blue emissions centred at 428 and 468 nm, respectively. The emission peak of DPE-DDMAC was red-shifted to 501 nm in acetonitrile solution. The emission wavelength of DPE-DPXZ underwent a larger bathochromic shift from 468 to 551 nm as the polarity of the solvent increased. Solvatochromism effects indicated the ICT character fluorescence of two compounds, confirming the acceptor role of DPE. The emission of DPE-DPXZ was red-shifted compared to that of DPE-DDMAc due to PXZ having a stronger electron-donating ability than DMAc. | ||
| Fig. 4 Solvent-dependent UV-vis absorption and photoluminescence spectra of (a) DPE-DDMAc and (b) DPE-DPXZ, measured in diluted solutions (0.01 mM L−1). | ||
Furthermore, low-temperature fluorescence and phosphorescence spectra of two alkyne compounds and their corresponding molecular fragments were acquired in toluene solution. In good agreement with the TD-DFT analyses, the LE character phosphorescence of DPE-DDMAc and DPE-DPXZ was experimentally confirmed and dominated by the DPE unit. As shown in Fig. 5, the phosphorescence spectra of two emitters showed a dramatic departure from those of their corresponding donor sections. In contrast, there was little deviation from the mutual DPE fragment in the T1 emission. Moreover, the phosphorescence spectra of both compounds exhibited specific vibrational structures that were similar to that of DPE. These results showed that T1 of the investigated compounds was localized at the DPE centre.
 | ||
| Fig. 5 Normalized fluorescence and phosphorescence spectra of DPE-DDMAc, DPE-DPXZ, and their corresponding molecular fragments measured in toluene solution (0.01 mM L−1) at 77 K. | ||
The slight red-shift of T1 emissions in DPE-DDMAc and DPE-DPXZ could be explained by incomplete D–A electronic decoupling.38 For greater understanding, potential surface scans were conducted along dihedral angles between the electron donors and acceptors for the two alkyne compounds in the gas phase at the B3LYP/6-31G* level. The potential surface scan showed that these molecules could be less rigid given the rather smooth energy distribution in a small range of dihedral angle alternation (±20°) between the optimized structures (Fig. S16, ESI†). The steeper energy-rise trend when reducing the α angle indicated that the electronic interaction of DPE-DDMAc was weaker than that of DPE-DPXZ, explaining the greater red-shift of 3LE emission for DPE-DPXZ than DPE-DDMAc. Therefore, increased electronic decoupling shows potential for maintaining a high T1 state. Accordingly, for two alkyne compounds, displacement of the donor moieties with different electron-donating abilities affects not only the S1 energy level, but also slightly manages the T1 energy level, resulting in distinct exchange energies (Scheme 1 and Table S1, ESI†). As a result, the ΔEST of DPE-DDMAc and DPE-DPXZ were evaluated to be 0.55 and 0.40 eV, respectively, via low-temperature fluorescence/phosphorescence spectra.
 | ||
| Scheme 1 Schematic diagram of singlet–triplet splitting energy governed by donor–acceptor alternation. | ||
The HOMO energy levels of DPE-DDMAc and DPE-DPXZ were examined using cyclic voltammetry (Fig. S22, ESI†), and LUMO energy levels were further estimated from the optical absorption onset energy in toluene solution. The HOMO and LUMO energy levels were estimated to be −5.19 and −2.17 eV for DPE-DDMAc, and −4.98 and −2.15 eV for DPE-DPXZ, respectively (Table 1). Compared to DPE-DDMAc, the stronger electron-donating capability of PXZ made the HOMO energy level of DPE-DPXZ shallower by 0.21 eV. Meanwhile, there was a slight difference between the LUMO energy levels of the two alkyne compounds due to the LUMO dispersing over the same acceptor (DPE), according to DFT simulation.
To approximate the situation in OLED applications, DPE-DDMAc and DPE-DPXZ were evaluated in solid-state films. Given the high S1 and T1 energy levels of the investigated materials, bis(2-(diphenylphosphino)phenyl)etheroxide (DPEPO) with ultrahigh T1 (3.30 eV) was selected as the host and a low doping concentration (10 wt%) was adopted to avoid severe bimolecular interactions.39 When DPE-DDMAc was doped into DPEPO, a lifetime of only 6.70 ns was observed (Fig. S20, ESI†) due to the high ΔEST value making up-conversion of triplet excitons impossible. In good agreement with TD-DFT simulation results, a relatively small ΔEST of DPE-DPXZ was achieved by delicate tuning of the S1 and T1 states, which enabled RISC to take place to harvest triplet excitons, yielding delayed fluorescence (DF). As shown in Fig. 6, DPE-DPXZ initially decayed promptly with a lifetime of 5.58 ns, then followed a delayed decay with a lifetime of 542.70 μs, verifying its TADF character. Millisecond-level τDF was noted under vacuum conditions considering the ΔEST > 0.2 eV, meaning more chances for quenching processes by oxygen in air (Fig. 6). As mentioned, the small kTADF (in the order of 2) should be responsible for the long τDF because the reverse up-conversion process plays a rate-limiting role in controlling the excited-state lifetime of DF (τDF).40 To further verify the TADF nature of DPE-DPXZ, temperature-dependent transient PL spectra were measured from 77 to 350 K (Fig. 7). Following the temperature increase, the ratio of DF showed significant monotonic enhancement from 21.3% at 100 K to 66.9% at 300 K, demonstrating the occurrence of a thermally activated process. Conversely, the DF ratio dropped to 53.1% when the temperature was further increased to 350 K. This was explained by the significantly enhanced nonradiative (internal conversion) channel for depletion of long-lifetime excitons.15,41
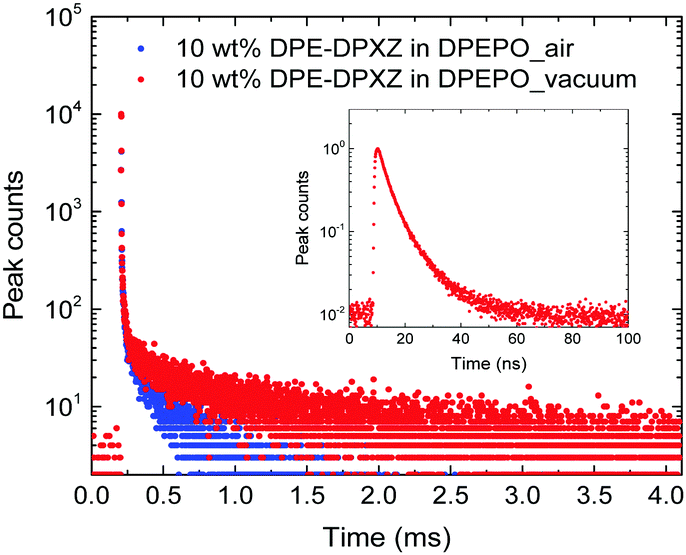 | ||
| Fig. 6 Transient PL decays of 10 wt% DPE-DPXZ in DPEPO measured under air and vacuum conditions using a fluorescence lifetime spectrometer (C11367-01, Hamamatsu Photonics) in TCC900 mode. Inset: Prompt component of the decay recorded in M9003-01 mode. | ||
 | ||
| Fig. 7 Temperature-dependent transient PL decays of 10 wt% DPE-DPXZ in DPEPO-doped film measured under vacuum conditions. Owing to the wide impulse response function (flash lamp with long decay signals), the delayed fluorescence ratio was determined and shown (inset) using the proportion of integrated area cutting from the fitting line (grey lines) and axis, and the overall integration area calculated from the curves and axis. | ||
OLED characterization
According to computer simulations and photophysical properties analysis, DPE-DPXZ was shown to possess TADF character, while DPE-DDMAc had no TADF character. Therefore, DPE-DPXZ was confirmed to be more appropriate for application in OLED devices. The three device architectures utilized were as follows: (I) ITO (95 nm)/TAPC (35 nm)/DPE-DPXZ (25 nm)/TmPyPB (40 nm)/LiF (0.8 nm)/Al (100 nm); (II) ITO (95 nm)/TAPC (35 nm)/10 wt% DPE-DPXZ in CBP (25 nm)/TmPyPB (40 nm)/LiF (0.8 nm)/Al (100 nm); (III) ITO (95 nm)/TAPC (35 nm)/DAcDB (10 nm)/10 wt% DPE-DPXZ in DPEPO (25 nm)/DPEPO (10 nm)/TmPyPB (40 nm)/LiF (0.8 nm)/Al (100 nm). As a severe concentration quenching effect was noted for the DPE-DPXZ neat film, device I exhibited very poor performance with a maximum EQE of 2.89% (Fig. 8), in agreement with photophysical properties analysis (Fig. S20, ESI†). Moreover, device I showed the highest onset voltage (Von) among the three groups of devices, demonstrating that it had the worst charge carrier balance, which was ascribed to an imbalance between the hole and electron injecting and transporting abilities of DPE-DPXZ. In device II, better performance was achieved with less bimolecular interaction causing a concentration quenching effect. A much lower Von and higher maximum brightness of the device were noted as more balanced charge carrier injection and transport were realized when DPE-DPXZ was doped into the 4,4′-bis(carbazol-9-yl)biphenyl (CBP) host. For device III, in which electron-blocking and hole-blocking layers were introduced, the EQE exceeded 10% and a blue emission at 475 nm was observed, which was quite close to the PL peaks of DPE-DPXZ in toluene solution and 10 wt% DPE-DPXZ in a DPEPO-doped film. The triplet excitons of DPE-DPXZ were better confined within the emissive layer, which resulted in a higher performance than the other two devices. Considering the relatively low photoluminescence quantum yield of the DPE-DPXZ doped film (30.0 ± 3.0%), the high EQE achieved might be ascribed to the enhanced light out-coupling efficiency. Rigid and bar-shaped TADF emitters tend to flatly orientate within the EML, which might result in enhanced light out-coupling efficiency and a high EQE (see ESI†).27,42–45 However, owing to the poor charge carrier mobility of DPEPO, the maximum brightness was low. At the same operation voltage, the current density of device II employing bipolar host CBP was much larger than that of device III. At a high operation current density, TADF-OLED suffered from severe singlet–triplet annihilation (STA) and triplet–triplet annihilation (TTA) owing to the large τDF and imbalance of charge carrier injection and transport.46,47 Accordingly, further strategies to reduce the ΔEST and device optimization could have potential for producing OLEDs based on DPE derivative compounds with better performance. Further work to this effect is already underway in our laboratory.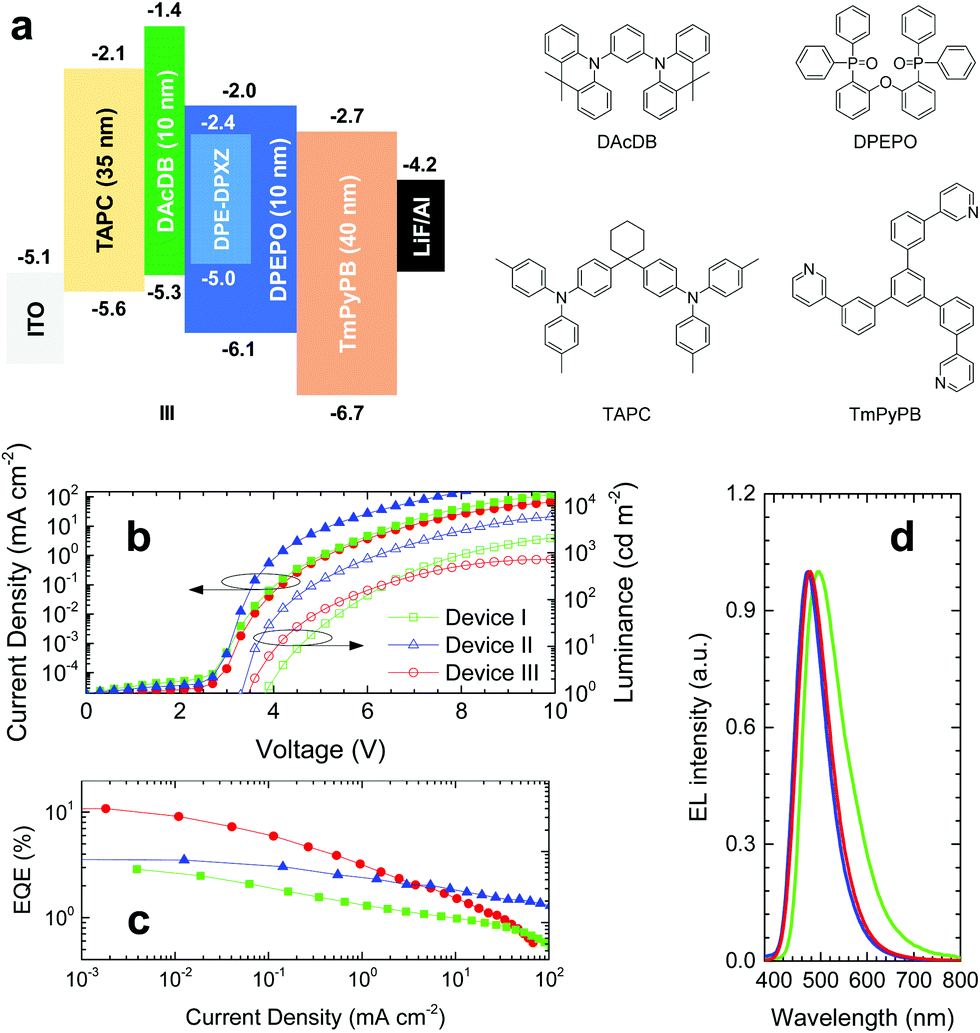 | ||
| Fig. 8 (a) Device architectures and materials used in OLEDs. (b) Current density–voltage–luminance (J–V–L), (c) external quantum efficiency–current density (EQE–J) characteristics of devices I, II, and III, and (d) electroluminescence (EL) spectra of corresponding devices at a current density of 0.1 mA cm−2. | ||
Conclusions
Herein, we proposed the first TADF emitter based on a DPE acceptor and applied it in an OLED device. The DPE moiety was confirmed to be a weak acceptor due to the special sp-hybridization of the C–C triple bond with high s character, which is a potential fragment for blue TADF material design. The 3LE state of the emitters was delicately governed by electronic interactions in D–A decoupling. Reduced interaction further elevated the 3LE state, which prompted reverse up-conversion for a more effective TADF process. Although a DF phenomenon was observed for DPE-DPXZ, further decreasing the ΔEST was critical for enhancing the RISC process, which is beneficial for highly efficient and low-efficiency roll-off OLED application operating at high current densities and brightness.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge financial support from the National Key R&D Program of China (2016YFB0401004), the National Natural Science Foundation of China (51625301, 51573059 and 91233116), the 973 Project (2015CB655003), and the Guangdong Provincial Department of Science and Technology (2016B090906003 and 2016TX03C175).Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- Y. G. Ma, H. Y. Zhang, J. C. Shen and C. M. Che, Synth. Met., 1998, 94, 245–248 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- S. Reineke, Nat. Photonics, 2014, 8, 269–270 CrossRef CAS.
- S. Reineke, Nat. Mater., 2015, 14, 459–462 CrossRef CAS PubMed.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS PubMed.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, Jr., S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- Q. S. Zhang, B. Li, S. P. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- G. Xie, X. Li, D. Chen, Z. Wang, X. Cai, D. Chen, Y. Li, K. Liu, Y. Cao and S.-J. Su, Adv. Mater., 2016, 28, 181–187 CrossRef CAS PubMed.
- X. Y. Cai, B. Gao, X. L. Li, Y. Cao and S. J. Su, Adv. Funct. Mater., 2016, 26, 8042–8052 CrossRef CAS.
- Y. C. Li, X. L. Li, D. J. Chen, X. Y. Cai, G. Z. Xie, Z. Z. He, Y. C. Wu, A. Lien, Y. Cao and S. J. Su, Adv. Funct. Mater., 2016, 26, 6904–6912 CrossRef CAS.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- L. S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- J. Lee, N. Aizawa, M. Numata, C. Adachi and T. Yasuda, Adv. Mater., 2017, 29, 1604856 CrossRef PubMed.
- S. Y. Lee, C. Adachi and T. Yasuda, Adv. Mater., 2016, 28, 4626–4631 CrossRef CAS PubMed.
- T. Nishimoto, T. Yasuda, S. Y. Lee, R. Kondo and C. Adachi, Mater. Horiz., 2014, 1, 264–269 RSC.
- D. D. Zhang, M. H. Cai, Y. G. Zhang, D. Q. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
- S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS PubMed.
- A. Abate, M. Planells, D. J. Hollman, V. Barthi, S. Chand, H. J. Snaith and N. Robertson, Phys. Chem. Chem. Phys., 2015, 17, 2335–2338 RSC.
- S. Kazim, F. J. Ramos, P. Gao, M. K. Nazeeruddin, M. Grätzel and S. Ahmad, Energy Environ. Sci., 2015, 8, 1816–1823 CAS.
- W. Shi, L. Wang, M. Umar, T. Awut, H. Mi, C. Tan and I. Nurulla, Polym. Int., 2009, 58, 800–806 CrossRef CAS.
- M. J. Mio, L. C. Kopel, J. B. Braun, T. L. Gadzikwa, K. L. Hull, R. G. Brisbois, C. J. Markworth and P. A. Grieco, Org. Lett., 2002, 4, 3199–3202 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- R. J. Magyar and S. Tretiak, J. Chem. Theory Comput., 2007, 3, 976–987 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS PubMed.
- M. Fagnoni, Angew. Chem., Int. Ed., 2010, 49, 6709–6710 CrossRef CAS.
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 128, 5833–5838 CrossRef.
- C. Han, Y. Zhao, H. Xu, J. Chen, Z. Deng, D. Ma, Q. Li and P. Yan, Chem. – Eur. J., 2011, 17, 5800–5803 CrossRef CAS PubMed.
- X. Y. Cai, X. L. Li, G. Z. Xie, Z. Z. He, K. Gao, K. K. Liu, D. C. Chen, Y. Cao and S. J. Su, Chem. Sci., 2016, 7, 4264–4275 RSC.
- J. Li, T. Nakagawa, J. MacDonald, Q. Zhang, H. Nomura, H. Miyazaki and C. Adachi, Adv. Mater., 2013, 25, 3319–3323 CrossRef CAS PubMed.
- T. Komino, Y. Sagara, H. Tanaka, Y. Oki, N. Nakamura, H. Fujimoto and C. Adachi, Appl. Phys. Lett., 2016, 108, 5 CrossRef.
- L. Zhao, T. Komino, M. Inoue, J. H. Kim, J. C. Ribierre and C. Adachi, Appl. Phys. Lett., 2015, 106, 063301 CrossRef.
- C. S. Oh, C.-K. Moon, J. M. Choi, J.-S. Huh, J.-J. Kim and J. Y. Lee, Org. Electron., 2017, 42, 337–342 CrossRef CAS.
- M. Liu, R. Komatsu, X. Y. Cai, K. Hotta, S. Sato, K. K. Liu, D. C. Chen, Y. Kato, H. Sasabe, S. Ohisa, Y. Suzuri, D. Yokoyama, S.-J. Su and J. Kido, Chem. Mater., 2017, 29, 8630 CrossRef CAS.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- T. Furukawa, H. Nakanotani, M. Inoue and C. Adachi, Sci. Rep., 2015, 5, 8429 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis routes, computation summary, photo-physical properties and OLED device characterization data. CCDC 1555726. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7tc02763j |
| ‡ The authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |