
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous nanosheet wrapping for live imaging of suspension cells†
Hong
Zhang‡
a,
Takuto
Aoki‡
b,
Kanae
Hatano
c,
Kazuya
Kabayama
c,
Masaru
Nakagawa
d,
Koichi
Fukase
c and
Yosuke
Okamura
 *ab
*ab
aMicro/Nano Technology Center, Tokai University, 4-1-1 Kitakaname, Hiratsuka, Kanagawa 259-1292, Japan. E-mail: y.okamura@tokai-u.jp
bCourse of Applied Science, Graduate School of Engineering, Tokai University, 4-1-1 Kitakaname, Hiratsuka, Kanagawa 259-1292, Japan
cDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka, 560-0043, Japan
dInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
First published on 13th September 2018
Abstract
In the field of cell imaging, it is still a practical challenge to obtain the high quality live imaging of suspension cells, mainly due to undesirable cell movement in the imaging field during observation. This study describes a porous nanosheet wrapping method to noninvasively immobilize suspension cells for their live imaging. Perforated nanopores are fabricated on a nanosheet to enable the addition of external chemicals to cells, ranging from small molecules to macromolecules. Through several case studies, such as the live imaging of membrane staining of liposomes, transferrin endocytosis of B cells, and activation of platelets, it is verified that the confined space made by the nanosheet could provide a hydrodynamically stable environment for suspension cells, even if an aqueous stimulus is added through the nanopores in a static or a flowing condition. With this method, the live imaging of the whole activation process on a specific suspension cell in the imaging field is achieved, which is not feasible with the existing cell immobilization methods. This study suggests that the method of porous nanosheet wrapping will facilitate the visualization of the dynamic functions of suspension cells.
1. Introduction
Live imaging has now become a routine procedure for understanding dynamic cellular behaviors. Over the past decade, a series of super-resolution microscope technologies has been developed, which enable us to image cells with a higher resolution and at a faster speed.1 However, it is still challenging to obtain the live imaging of suspension cells and it is highly reliant on the specialized knowledge of each laboratory. Unlike adherent cells, suspension cells are cultured suspended in media and form little attachment to a substrate. This makes them locally unstable in an imaging field, especially when an aqueous external stimulus is added. Traditional methods for cell immobilization involve an adhesive coating to a substrate, using materials such as polylysine, collagen, fibronectin, antibody, a lipid-like anchor, etc.2–4 Embedding with agar, CyGEL, or Matrigel is also employed to fix cells.5–7 One can also seed suspension cells on well-designed chambers to stabilize their locations, such as an ibidi dish, microchip, or Live Cell Array chamber.8–10 Among these methods, suspension cells are forced into a particular location through a chemical or a physical interaction with the solid substrate, during which the cell behaviors may change to cope with the artificial environments. The requirement of sample immobilization and the dynamic nature of suspension cells always lead us to a dilemma, and a noninvasive imaging method for suspension cells is highly desirable.Inspired by the use of plastic food wrapping, we developed a nanosheet wrapping method to address the issue of sample shrinkage in tissue imaging.11 Herein, the term “nanosheet” refers to an ultrathin polymer film with thickness in the tens of nanometers, which exhibits a high level of transparency and flexibility.12 By means of a freestanding procedure, nanosheets can be detached from their prepared substrate and transferred to any solid or liquid surface with a strong self-adhesion. It is reasonable to suppose that a hydrodynamically stable environment can be obtained by wrapping suspension cells with a nanosheet, which would improve the accessibility of their imaging. In this study, we propose a porous nanosheet wrapping method for live imaging of suspension cells, where no chemical ligand or physical contact with cells is needed. Nanopores are fabricated on the nanosheets to enable the addition of other chemicals, and thus we could observe the cell responses to external stimuli in real time. To test the utility of this method, we demonstrate several case studies, such as the live imaging of membrane staining of liposomes, and the live imaging of transferrin endocytosis of B cells. Moreover, we employ porous nanosheet wrapping inside a flow chamber, and the platelet activation process is continuously tracked.
2. Materials and methods
2.1 Preparation of porous nanosheets
Porous nanosheets consisting of poly(lactic acid) (PLA) were prepared by a standard spin-casting and thermal nanoimprinting method. PLA was purchased from NatureWorks LLC (MN, USA) under the commercial name of Ingeo™ 4060D, which is synthesized from sugars primarily obtained from field corn. It is an amorphous polymer with an average D-lactide content of 12 wt%, a weight-average molecular weight (Mw) of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000, and a glass transition temperature of 55–60 °C.13 The PLA pellets were dissolved in toluene with a concentration of 20 mg mL−1, if not otherwise noted. Casting substrates were silicon wafers (100), deposited with a 200 nm thermally grown silicon oxide layer (KST World Co., Fukui, Japan), which were cut into a size of 35 × 35 mm2 before use. Spin-casting was performed on an MS-A100 spin coater (Mikasa Co., Ltd, Tokyo, Japan) at a rotation speed of 4000 rpm for 60 seconds. Poly(vinyl alcohol) (PVA, degree of polymerization: ca. 500, 86.5–89.0% hydrolyzed, Kanto Chemicals Co., Inc., Tokyo) was dissolved in water with a concentration of 10 mg mL−1. It was deposited onto the substrate prior to PLA to prepare a sacrificial layer.
000–200000, and a glass transition temperature of 55–60 °C.13 The PLA pellets were dissolved in toluene with a concentration of 20 mg mL−1, if not otherwise noted. Casting substrates were silicon wafers (100), deposited with a 200 nm thermally grown silicon oxide layer (KST World Co., Fukui, Japan), which were cut into a size of 35 × 35 mm2 before use. Spin-casting was performed on an MS-A100 spin coater (Mikasa Co., Ltd, Tokyo, Japan) at a rotation speed of 4000 rpm for 60 seconds. Poly(vinyl alcohol) (PVA, degree of polymerization: ca. 500, 86.5–89.0% hydrolyzed, Kanto Chemicals Co., Inc., Tokyo) was dissolved in water with a concentration of 10 mg mL−1. It was deposited onto the substrate prior to PLA to prepare a sacrificial layer.
After spin-casting, the substrate was pressed with a hard mold at 70 °C under 30 MPa for 60 seconds, using a thermal press AH-2003C (AS ONE Co., Osaka, Japan). The mold for nanoimprinting consisting of nickel with an annular array of cone structures was kindly donated by JVCKENWOOD Creative Media Co. Ltd (Kanagawa, Japan). The height and bottom diameter of each cone was ca. 0.85 and 0.80 μm, respectively, and the pitch distance was varied in the range from 1.5 to 6.0 μm. After cooling to room temperature, the substrate was immersed in water. The resulting nanosheets were subsequently released from the silicon substrate and floated to the surface of the water with the dissolution of the underlying PVA sacrificial layer. With the help of a homebuilt wire loop (diameter: ca. 28 mm), the nanosheets can be lifted out of the water and supported by the wire loop in the air. All the processes were conducted at room temperature (25 °C) and normal relative humidity (ca. 35% RH). Trypan blue (0.4 wt%, Mw = 873, Thermo Fisher Scientific Inc., MA, USA) was used as a tracer to visualize the permeability of the porous nanosheet. The images of a floating nanosheet, its wrapping of a sample of suspension cells, and the video of Trypan blue permeation were taken with a digital camera G7 X Mark II (Canon Inc., Tokyo).
2.2 Characterization of nanosheets
The thickness of the prepared nanosheets was determined by a scalpel scratch made on the films and measured using a stylus profilometer with Vision64 software (DektakXT, ver. 5.40 update 7, Bruker Co., MA). The stylus radius was 2 μm and the stylus force was set to be 3 mg, which gave a line resolution of 0.067 μm pt−1. Each sample was measured at nine arbitrary locations, and the thickness was obtained from three individual samples and shown as mean ± s.e.m. For characterization of the morphology of the nanosheets, floating films were supported on an Anodisc membrane (pore size: 0.1 μm, GE Healthcare, IL, USA), sputter-coated with platinum (3 mA, ca. 1 min), and observed using an S-4800 field emission scanning electron microscope (SEM, Hitachi High-Technologies Co., Tokyo) with an accelerating voltage of 3.0 kV. The diameter of the pores was manually measured from three individual SEM images by ImageJ software (NIH, USA) and shown as mean ± s.e.m.2.3 Imaging of membrane staining of liposomes
Egg phosphatidylcholine (EPC, COATSOME NC-50, Mw = ca. 773) and N-(carbonyl methoxypolyethyleneglycol)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt (DSPE-PEG, SUNBRIGHT DSPE-050CN, Mw of PEG = 5000) were purchased from NOF Co. (Tokyo). Liposomes comprising EPC/DSPE-PEG (100/1, mol mol−1) were prepared by a film hydration method.14 Specifically, EPC and DSPE-PEG were dissolved in chloroform and the mixture was dried to form a thin film using a rotary evaporator. HEPES buffer (1.0 wt%, pH = 7.4) was added and hydrated at 500 rpm for 5 hours. The suspended liposomes were purified by repeated cycles of centrifugation (3500 rpm, 10 min, and three times).N-(3-Triethylammoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide (FM 1-43, Mw = 612, excitation (Ex): 479 nm, emission (Em): 598 nm when bound to phospholipid membranes) dye was purchased from Thermo Fisher. To perform live imaging of liposome membrane staining with porous nanosheet wrapping in a static condition, FM 1-43 was dissolved in HEPES buffer as an external stimulus. 10 μL of liposome suspension was placed on a cover slip (diameter: 18 mm, thickness: 0.12–0.17 mm, Matsunami Glass Inc., Ltd, Osaka), which was wrapped with a porous nanosheet (pitch distance of 1.5 and 6.0 μm) and bonded to a 35 mm perforated bottom Petri dish (diameter of hole: 15 mm) with nail enamel. The Petri dish was firmly mounted on the microscope stage and 190 μL of FM 1-43 was slowly dripped onto the nanosheet to a final concentration of 10 μM. The motion of liposomes and the staining of the membrane were continuously recorded at room temperature for 15 min. As control groups, liposomes without nanosheet wrapping and those wrapped with a nonporous nanosheet were also observed.
2.4 Imaging of transferrin endocytosis of lymphocytes
Raji cells (a Burkitt's lymphoma B cell line) were obtained from the JCRB Cell Bank (Cell number: JCRB9012) and cultured in RPMI 1640 medium (Wako Pure Chemical, Ltd, Osaka) supplemented with 2 mM L-glutamine, 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin, and 10% fetal bovine serum at 37 °C (1.0 × 103 cells μL−1). Alexa Fluor 488 conjugated transferrin (Tf-488, human serum source glycoprotein, Mw = up to 80000, Ex = 488 nm, Em = 525 nm) was purchased from Thermo Fisher. First, 50 μL of B cell suspension was wrapped with nanosheet and mounted on the microscope stage using the same setup as described in the imaging of liposomes. Next, 140 μL of HEPES-buffered live cell imaging buffer (1.0% (w/v) BSA, 20 mM glucose, pH = 7.4) was first poured into the Petri dish to submerge the nanosheet, and 10 μL of Tf-488 dissolved in live cell imaging buffer was then added to a final concentration of 25 μg mL−1 as an external stimulus. The motion of B cells and their endocytosis of transferrin were continuously recorded at 37 °C for 60 min. As a control group, B cells wrapped with a nonporous nanosheet were also observed. Alexa Fluor 594 conjugated anti-human CD20 antibody (Ab-594, Mw = ca. 150000, Ex = 561 nm, Em = 595 nm) was purchased from BioLegend, Inc. (CA, USA); 10 μL of Ab-594 was dissolved in live cell imaging buffer and added to the nanosheet wrapped B cells with a final concentration of 0.5 μg mL−1 for testing.
2.5 Imaging of platelet activation under a flow condition
Blood was drawn from one healthy male volunteer and mixed with 10% (v/v) 3.8% (w/v) sodium citrate solution. An initial centrifugation step (120 × g, 15 min, 25 °C) yielded platelet rich plasma in the supernatant. The platelet count was adjusted to 3.5 × 105 platelets μL−1 using an automated hematology analyzer (pocH-100i, Sysmex Co., Hyogo, Japan). Thrombin receptor activator peptide (Ser-Phe-Leu-Leu-Arg-Asn-NH2 trifluoroacetate salt, TRAP) was purchased from Sigma-Aldrich Co. LLC (MO, USA). To perform live imaging of platelet activation with porous nanosheet wrapping under a flow condition, TRAP was dissolved in HEPES buffer to a concentration of 100 μM as an external stimulus. A cover slip (diameter: 25 mm) was coated with a nonporous PLA nanosheet and then blocked with BSA. Then, 10 μL of platelet suspension was placed on the cover slip and wrapped with a porous nanosheet with the BSA treated surface downwards. Once the sample was mounted on the microscope stage, TRAP solution was perfused into the chamber. Details about flow chamber design and particle image velocimetry analysis can be found in the ESI.† The motion and morphological change of platelets were continuously recorded at room temperature. As control groups, platelets without nanosheet wrapping and those wrapped with a nonporous nanosheet were also observed.This study was approved by the Tokai University Institutional Review Board for Human Research (Permit No. 16039). All the experiments related to live subjects were performed in compliance with the relevant laws and the institutional guidelines for human research and experiments. Regarding the studies involving human platelets, informed consent was obtained from the blood donor according to the institutional guidelines of Tokai University.
2.6 Microscopy and data analysis
In this study, all the imaging work was conducted in compliance with standard cell imaging protocols and technically supported by Nikon Instech Co., Ltd (Tokyo). Images of the membrane staining of liposomes and the particle image velocimetry were taken on an inverted confocal microscope (A1R+, Nikon) with a LU-N4 laser unit. A 60× water immersion objective lens (CFI Plan Apo VC60 × WI, numerical aperture (NA): 1.20, working distance (WD): 0.27 mm) was used. Laser lines at 488, 488, and 561 nm were used for FM 1-43, fluorescent nanoparticles, and Nile red, respectively. Correspondingly, fluorescence signals in the wavelength range of 570–620, 500–550, and 570–620 nm were detected via a photomultiplier tube. Images of the labeling of membrane antigen and the transferrin endocytosis of lymphocytes were taken on the same confocal microscope equipped with a controlled environmental incubator (INUBG2SF-TIZB, temperature: 37 °C, CO2 concentration: 5%, TOKAI HIT Co., Ltd, Shizuoka, Japan). A 60× oil immersion objective lens (CFI Apo 60 × H λS, NA: 1.40, WD: 0.14 mm) was used in this case. Laser lines at 488, and 561 nm were used for Tf-488 and Ab-594, respectively. The fluorescence signals in the wavelength range of 500–550, and 570–620 nm were detected. The cover slip was well focused to observe liposomes or B cells at this plane, and images and videos were captured over an area of 212 × 212 μm2 or 212 × 106 μm2 (160 × 80 μm2 for observing Tf-488 endocytosis with a scanner zoom of 1.33×) (512 × 512 pixels or 512 × 256 pixels, 0.414 μm pixel−1; 0.313 μm pixel−1 for the endocytosis of B cells). In the case of particle image velocimetry, image stacking was acquired from the location of the cover slip to a depth of 200 μm with a 1.0 μm z-step, over an area of 212 × 212 μm2 (1024 × 1024 pixels, 0.207 μm pixel−1). Images of platelet activation were taken on an inverted microscope (Eclipse Ti-E, Nikon) with a 100× oil immersion objective lens (CFI Apo TIRF 100× H, NA: 1.49, WD: 0.12 mm). The cover slip was well focused to observe platelets at this plane, and images and videos were captured over an area of 140 × 93 μm2 (1636 × 1088 pixels, 0.0855 μm pixel−1). Each microscope was installed on an anti-vibration table to avoid any effect from the ambient environment.All the images and videos were generated through NIS-Elements AR software (ver. 4.60, Nikon). The height of the porous nanosheet, namely the distance between the nanosheet and the cover slip (shown as mean ± s.e.m.) and the flow speed in particle image velocimetry were manually measured by ImageJ on three individual tests, where at least ten particles at each height were arbitrarily chosen. The velocity of the platelets during the imaging of platelet activation was measured by the same method used for nanoparticles, and shown as mean ± s.e.m. For a better visualization, images were trimmed to a size of 106 × 106 μm2 for the liposomes and B cell results (80 × 80 μm2 for the endocytosis of B cells), and a size of 70 × 46.5 μm2 for the platelet results in all the figures. Videos were processed and compressed using Camtasia Studio software (ver. 6.0, TechSmith Co., MI, USA).
3. Results and discussion
PLA, as a biocompatible polymer, was chosen to prepare the nanosheets using spin-casting. The thickness of the nanosheet is adjustable (Fig. S1, ESI†) and was set to 60 ± 5 nm. In our previous study, a 60 nm PLA nanosheet was proven to be mechanically strong (stress in bulging test: ca. 7 kPa).12 In addition, PLA provides a bio-innate surface to which bovine serum albumin (BSA) can stably adsorb to prevent nonspecific adhesion and activation of both adherent and suspension cells.15,16 Thermal nanoimprinting was applied to fabricate perforated pores on the nanosheets.17 The diameter of the pores was designed to be 840 ± 65 nm, smaller than that of ordinary cells (Fig. 1A and B). To obtain a freestanding nanosheet, a water-soluble polymer layer consisting of PVA was coated before PLA casting, and the PLA nanosheet can then float to the surface of water upon dissolution of the PVA underling layer (Fig. 1C). We found that Trypan blue, as a model for an aqueous stimulus, can freely penetrate the pores on these nanosheets (Video S1, ESI†).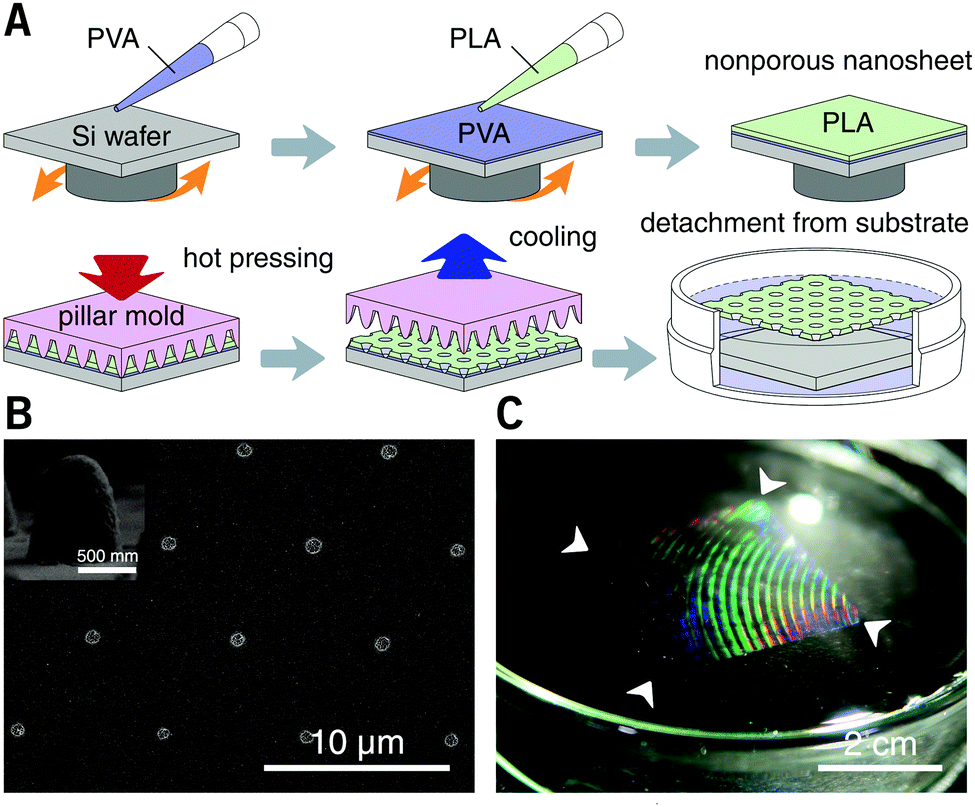 | ||
| Fig. 1 Preparation of porous nanosheets and nanosheet freestanding process. (A) Schematic of nanosheet preparation. PLA is spin-cast on a PVA pre-coated silicon wafer to form a thin film, and thermal nanoimprinting is used to fabricate perforated nanopores. (B) SEM image of a porous nanosheet with a pitch distance of 6.0 μm. Inset shows a cross-sectional view of the cone structure of the nickel mold used for nanoimprinting. (C) Photo of a freestanding porous nanosheet floating on the surface of water. Arrows show corners of the nanosheet, and the interference colors imply the existence of a structured surface. | ||
The nanosheet wrapping was accomplished with the help of a wire loop, and the margin of the nanosheet automatically adhered to the surface of the cover slip (Fig. 2A and B). It should be noted that the pitch distance between two adjacent pores is controllable as well, i.e., distances of 1.5, 3.0, and 6.0 μm could be obtained (Fig. S2, ESI†). Herein, a porous nanosheet with a pitch distance of 6.0 μm was found to be sufficiently robust for handling, and the following tests were performed on it, if not otherwise specified. The porous nanosheet wrapping was conducted in either a static condition or a flow condition, where an external stimulus was delivered to cells by diffusion or perfusion, respectively (Fig. 2C).
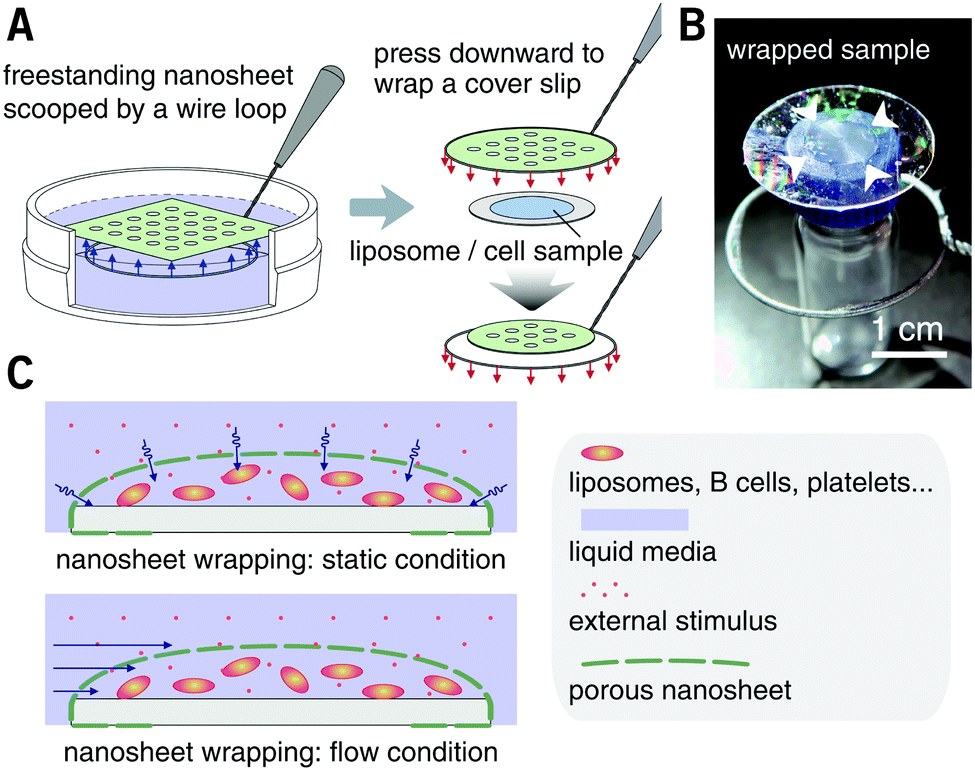 | ||
| Fig. 2 Porous nanosheet wrapping process and external stimuli addition. (A) Schematic of nanosheet wrapping process. The liposome or suspension cell sample is dripped on a cover slip in advance. The porous nanosheet is transferred from the surface of water to wrap the cover slip with the help of a wire loop. (B) Photo of a porous nanosheet wrapped sample. The diameter of the cover slip and the wire loop is 25 and 28 mm, respectively. Arrows show location of the wrapped sample (10 μL of liposome suspension). (C) Schematic of hydrodynamically stable environment obtained by wrapping suspension cells with a nanosheet, and the external stimuli penetrate the pores under a static or a flow condition. | ||
First, the membrane staining of liposomes was performed. EPC giant liposomes, as a model for suspension cells, were prepared by a film hydration method with a diameter of 7.2 ± 3.6 μm, similar to that of ordinary suspension cells. FM 1-43 is intensely fluorescent when it inserts into phospholipid membranes.18 By directly adding 190 μL of FM 1-43 into 10 μL of liposome suspension, it is natural that liposomes were dispersed in the turbulence and thereby moved out of the focal plane even after 15 minutes of observation (Fig. 3A and Video S2, ESI†). After being wrapped with a nanosheet, however, except for some disturbance in the initial few seconds, liposomes were kept locally stable, save for Brownian motion in the imaging field (Fig. 3B and Video S3, ESI†). This indicates that the nanosheet separated the liposome suspension and the aqueous stimulus. It acts as a barrier to prevent the mixing of two liquids and resists the surge pressure from above. The membrane of liposomes cannot be stained with a nonporous nanosheet, but as the nanosheet used here was porous, the liposomes could be immobilized and stained, owing to the passage of FM 1-43 through the pores of nanosheet. The staining process of the liposome membrane was well imaged (Fig. 3C and Video S4, ESI†). It should be noted that the time required for staining was shortened from ca. 7 to ca. 4 min if we applied a porous nanosheet with a minimum pitch distance that we fabricated in this study of 1.5 μm (Fig. 3D and Video S5, ESI†). The cell response time upon stimulus was expected to be adjustable by changing the permeation rate of the stimulus, which is always critical for imaging work with living cells.
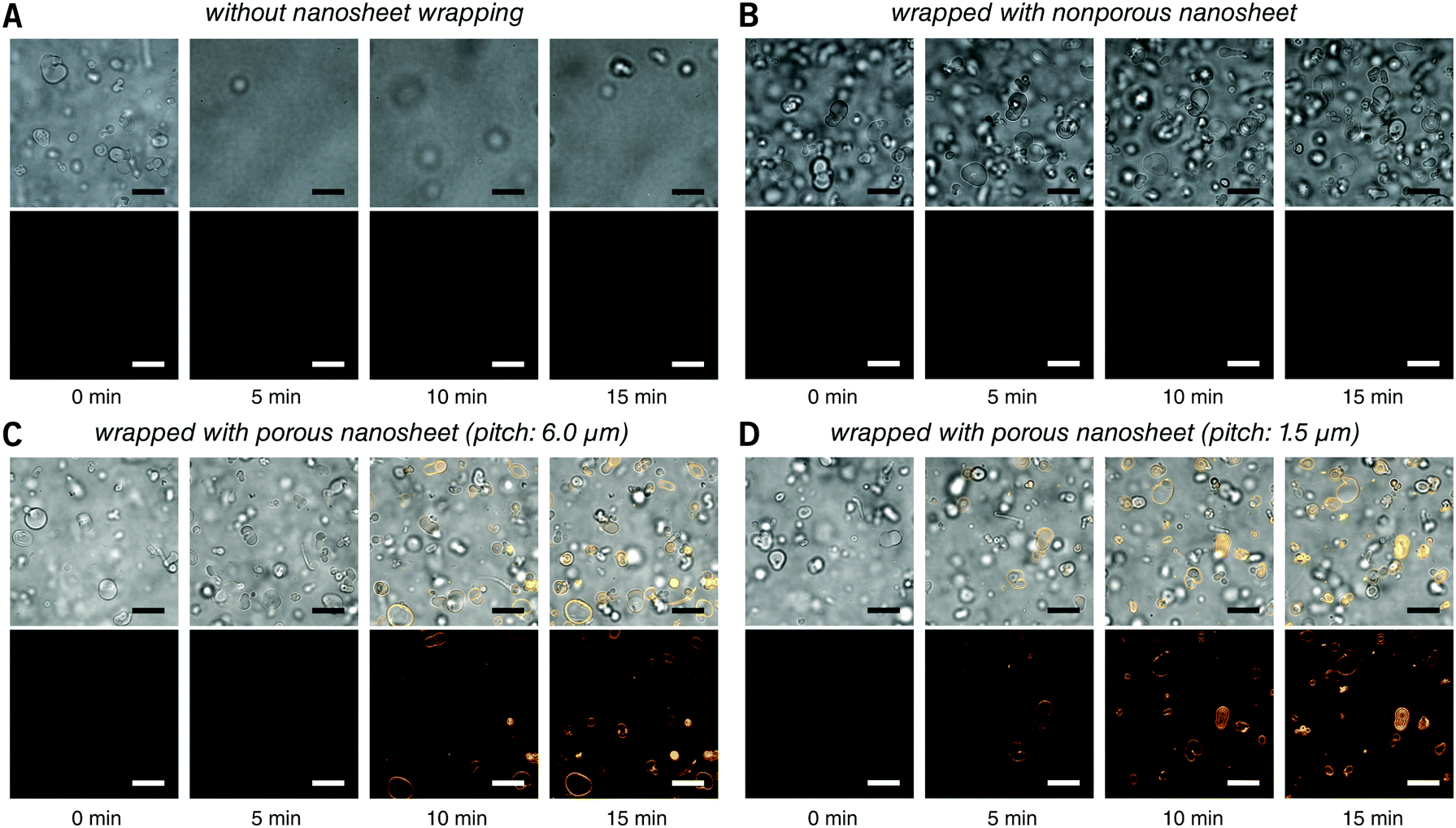 | ||
| Fig. 3 Real time observation of membrane staining of liposomes. (A) Directly dripped FM 1-43 in liposome suspension without nanosheet wrapping. (B) Liposomes wrapped with a nonporous nanosheet. (C) Liposomes wrapped with a porous nanosheet with a pitch distance of 6.0 μm; and (D) liposomes wrapped with a porous nanosheet with a pitch distance of 1.5 μm. Lower: FM 1-43 red fluorescence; upper: merged with brightfield image. The time for taking the snapshots is labeled below, and the original videos can be found as Videos S2–S5 (ESI†). Fluorescence images were individually adjusted to improve brightness and contrast. Scale bars: 20 μm in all panels. | ||
Raji cells, a B lymphocyte, were next selected as a typical suspension cell line for live imaging testing with porous nanosheet wrapping in a static condition. It is known that transferrin (Tf) can be endocytosed through transferrin receptors on the surface of cells.19 Herein, 50 μL of B cells was wrapped with a porous nanosheet with a pitch distance of 6.0 μm, and 150 μL of Fluor 488 conjugated Tf (Tf-488) was dripped in portions. During a 60 min continuous observation, B cells kept their viability in a suspension state, and the increasing amount of Tf-488 transported into the B cells was clearly visualized after ca. 20 min (Fig. 4 and Video S6, ESI†). By wrapping with a nonporous nanosheet, the B cells were locally stable but no fluorescence could be detected inside the B cells (Video S7, ESI†), which agrees with our results on liposomes. Furthermore, labeling of membrane antigen CD20, a B cell marker, was live imaged. Alexa Fluor 594 conjugated anti-CD20 antibody (Ab-594) was added to the porous nanosheet wrapped B cells and observed for 2 hours. Nanosheet wrapping induced cell morphology or function abnormality was not identified during the whole observation process and the specific binding of Ab-594 to CD20 was timely and in situ recorded from ca. 5 min (Video S8, ESI†). Taken together, our results show that the porous nanosheet wrapping is capable of immobilizing suspension cells and delivering various chemical stimuli, ranging from small molecules to macromolecules, such as proteins, nucleic acids, and polysaccharides.
 | ||
| Fig. 4 Live imaging of the transferrin endocytosis of B cells with the porous nanosheet wrapping method in a static condition. Lower: Tf-488 fluorescence; upper: differential interference contrast image. The time for taking the snapshots is labeled below, and the original video can be found as Video S6 (ESI†). Fluorescence images were individually adjusted to improve brightness and contrast. Scale bars: 15 μm in all panels. | ||
Blood cells, also a typical class of suspension cells, flow in the circulatory system. Compared to a static diffusion situation, imaging of blood cell responses to flowing external stimuli is more realistic. Here, a flow chamber was homebuilt, and a nanosheet wrapped cover slip was mounted (Fig. S3, ESI†).20 The nanosheet separates the flow channel into the outer space and the nanosheet wrapped space. By wrapping 10 μL of liquid, the height of the nanosheet was measured to be 135 ± 10 μm, which is much larger than the size of ordinary cells (Fig. 5A). As mentioned above, PLA is an essentially biocompatible and noninvasive material for suspension cells.12,15,16 In the case of nanosheet wrapping, there is no direct contact between nanosheet and cells, and thus we believe that the effect from the PLA nanosheet, if any, would be negligible. In this setup, a flow rate of 0.5 mL min−1 gave a shear rate of 35 s−1 outside.21 To investigate the effect of nanosheet wrapping on the flow in the chamber, particle image velocimetry was performed. Fluorescent nanoparticles (Φ = 200 nm) were perfused, and the flow speed in the height direction is shown in Fig. 5B. This profile presented a parabolic flow, which tends to have a zero velocity at the boundary of the cover slip or nanosheet. While the fluid flows into the porous nanosheet wrapped space, its velocity is much reduced by the nanosheet barrier. We expected that a hydrodynamically stable environment can be obtained in the nanosheet wrapped space, and the motion of suspension cells near the cover slip would be largely restricted.
 | ||
| Fig. 5 Particle image velocimetry on a porous nanosheet wrapping 10 μL of liquid in a flow chamber. (A) Image stacking is acquired from the surface of the cover slip with a 1.0 μm z-step. Arrow shows the direction of flowing nanoparticles, v, and the height of the porous nanosheet is shown as mean ± s.e.m. Representative slices are shown on right. Green: nanoparticles; red: Nile red loaded nanosheet. (B) Flow speed of nanoparticles in the height direction. The speed versus the normalized height was obtained from three individual tests (plotted by mean values). Inset is the magnified view of the results in the nanosheet wrapped space. Scale bars, 10 μm (A). | ||
This setup was then applied to visualize the morphological change of platelets from resting to activation state in real time. In a traditional method, the cover slip is coated with ligands, such as von Willebrand factor or fibrinogen, to activate platelets under a flow.22,23 Because such a capture process is performed at random, the real time imaging of a specific cell is difficult to achieve. In this study, however, the cover slip and the bottom of the porous nanosheet employed for wrapping were treated with BSA blocking to keep the platelets resting, and their activation was triggered only by a flowing stimulus. Here, TRAP, a ligation of protease-activated receptor, was used as an external activator.24 Wrapped with a nonporous nanosheet, platelets were locally stable but remained in a resting state (Fig. 6A and Video S9, ESI†), indicating that the activation of platelets cannot be triggered by contacting the nanosheet. Wrapped with a porous nanosheet, a continuous process of morphological changes of the platelets from their original disc shape, rounding, elaboration of filopods, to platelet aggregation was clearly tracked within 60 seconds (Fig. 6B and Video S10, ESI†). While the platelets moved slowly with a velocity of 0.8 ± 0.1 μm s−1 along the flow direction, most were still retained in the imaging field (140 × 93 μm2). Without nanosheet wrapping, however, the platelets were propelled with a high velocity of 15.3 ± 3.7 μm s−1 and left the imaging field quickly (Video S11, ESI†). This setup enables us to demonstrate a real time visualization of the activation of a specific cell, and even a cell–cell interaction process, which is not feasible with the existing cell immobilization methods. It should be noted that although the porous nanosheet wrapping was performed herein on a homebuilt flow chamber, any commercial one on which a cover slip can be mounted should work.
 | ||
| Fig. 6 Live imaging of platelets with the porous nanosheet wrapping method in a flow condition. (A and B) The activation and aggregation of platelets wrapped with a nonporous and a porous nanosheet under flowing TRAP, respectively. The cover slip and the bottom of the porous nanosheet were treated with BSA blocking in advance. Images were taken in situ with brightfield during the perfusion of TRAP. Insets are the magnified view of the corresponding regions as highlighted in the boxes. The time for taking the snapshots is labeled below, and the original videos can be found as Videos S9 and S10 (ESI†). Scale bars: 20 μm (A and B) and 3 μm (B inset). | ||
4. Conclusions
We describe herein a porous nanosheet wrapping method to noninvasively immobilize suspension cells, which can dramatically improve their live imaging upon external stimuli. This method makes use of the unique characteristics of an ultrathin polymer film and does not require any special device or expertise. In fact, the suspension cells mounted with this method can keep their dynamic nature, whereas their velocity is much restricted when an aqueous stimulus is added. Although still in an early stage, we expect that this simple and versatile method will facilitate visual investigations of the dynamic functions of suspension cells and will be of immense value in biology and medicine.Conflicts of interest
Y. O., T. A. and H. Z. have filed a Japanese patent application (No. 2016-225434) based on the porous nanosheet wrapping method.Acknowledgements
The authors wish to thank JVCKENWOOD Creative Media Co. Ltd for providing nickel molds for nanoimprinting, and Technical Service Coordination Office, Imaging Center for Advanced Research at Tokai University for technical assistance. This work is supported in part by a Cooperative Research Program of “Network Joint Research Center for Materials and Devices” (No. 2015096) (Y. O.), a Grant-in-Aid for Scientific Research on Innovative Areas “Nanomedicine Molecular Science” (No. 2306) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) (Y. O.), and a MEXT-Supported Program for the Strategic Research Foundation at Private Universities (Y. O.).References
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993 CrossRef CAS PubMed.
- D. Mazia, G. Schatten and W. Sale, J. Cell Biol., 1975, 66, 198 CrossRef CAS PubMed.
- K. Kato, K. Umezawa, D. P. Funeriu, M. Miyake, J. Miyake and T. Nagamune, Biotechniques, 2003, 35, 1014 CrossRef CAS PubMed.
- A. Revzin, K. Sekine, A. Sin, R. G. Tompkins and M. Toner, Lab Chip, 2005, 5, 30 RSC.
- Z. Z. Wang, P. Au, T. Chen, Y. Shao, L. M. Daheron, H. Bai, M. Arzigian, D. Fukumura, R. K. Jain and D. T. Scadden, Nat. Biotechnol., 2007, 25, 317 CrossRef CAS PubMed.
- D. H. Huberts, S. S. Lee, J. Gonzalez, G. E. Janssens, I. A. Vizcarra and M. Heinemann, Nat. Protoc., 2013, 8, 1019 CrossRef CAS PubMed.
- M. Fritzsche, R. A. Fernandes, V. T. Chang, H. Colin-York, M. P. Clausen, J. H. Felce, S. Galiani, C. Erlenkamper, A. M. Santos, J. M. Heddleston, I. Pedroza-Pacheco, D. Waithe, J. B. de la Serna, B. C. Lagerholm, T. L. Liu, T. L. Chew, E. Betzig, S. J. Davis and C. Eggeling, Sci. Adv., 2017, 3, e1603032 CrossRef PubMed.
- T. Tokuyama, S. I. Fujii, K. Sato, M. Abo and A. Okubo, Anal. Chem., 2005, 77, 3309 CrossRef CAS PubMed.
- M. Deutsch, A. Deutsch, O. Shirihai, I. Hurevich, E. Afrimzon, Y. Shafran and N. Zurgil, Lab Chip, 2006, 6, 995 RSC.
- A. R. Barr, J. V. Kilmartin and F. Gergely, J. Cell Biol., 2010, 189, 23 CrossRef CAS PubMed.
- H. Zhang, A. Masuda, R. Kawakami, K. Yarinome, R. Saito, Y. Nagase, T. Nemoto and Y. Okamura, Adv. Mater., 2017, 29, 1703139 CrossRef PubMed.
- Y. Okamura, K. Kabata, M. Kinoshita, D. Saitoh and S. Takeoka, Adv. Mater., 2009, 21, 4388 CrossRef CAS PubMed.
- Z. Duan, N. L. Thomas and W. Huang, J. Membr. Sci., 2013, 445, 112 CrossRef CAS.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238 CrossRef CAS PubMed.
- D. Niwa, T. Fujie, T. Lang, N. Goda and S. Takeoka, J. Biomater. Appl., 2012, 27, 131 CrossRef PubMed.
- Y. Okamura, R. Schmidt, I. Raschke, M. Hintze, S. Takeoka, A. Egner and T. Lang, Biophys. J., 2011, 100, 1855 CrossRef CAS PubMed.
- S. Matsui, H. Hiroshima, Y. Hirai and M. Nakagawa, Microelectron. Eng., 2015, 133, 134 CrossRef CAS.
- A. J. Cochilla, J. K. Angleson and W. J. Betz, Annu. Rev. Neurosci., 1999, 22, 1 CrossRef CAS PubMed.
- J. van Renswoude, K. R. Bridges, J. B. Harford and R. D. Klausner, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 6186 CrossRef CAS.
- K. Kawakami, Y. Harada, M. Sakasita, H. Nagai, M. Handa and Y. Ikeda, ASAIO J., 1993, 39, M558 CAS.
- S. M. Slack and V. T. Turitto, Thromb. Haemostasis, 1994, 72, 777 CAS.
- B. M. Cooke, S. Usami, I. Perry and G. B. Nash, Microvasc. Res., 1993, 45, 33 CrossRef CAS PubMed.
- B. van Aelst, H. B. Feys, R. Devloo, P. Vandekerckhove and V. Compernolle, J. Vis. Exp., 2016, 109, e53823 Search PubMed.
- R. R. Vassallo, T. Kieber-Emmons, K. Cichowski and L. F. Brass, J. Biol. Chem., 1992, 267, 6081 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S3, and captions for Videos S1–S11. Videos about Trypan blue penetration, membrane staining of liposomes, transferrin endocytosis and membrane antigen labeling of B cells, and activation of platelets: Videos S1–S11. See DOI: 10.1039/c8tb01943f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |