
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unraveling the molecular mechanisms of thermo-responsive properties of silk-elastin-like proteins by integrating multiscale modeling and experiment†
Jingjie
Yeo
 ab,
Wenwen
Huang
c,
Anna
Tarakanova
a,
Yong-Wei
Zhang
b,
David L.
Kaplan
c and
Markus J.
Buehler
*a
ab,
Wenwen
Huang
c,
Anna
Tarakanova
a,
Yong-Wei
Zhang
b,
David L.
Kaplan
c and
Markus J.
Buehler
*a
aLaboratory for Atomistic and Molecular Mechanics (LAMM), Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: mbuehler@mit.edu
bInstitute of High Performance Computing, A*STAR, 1 Fusionopolis Way, Singapore 138632, Singapore
cDepartment of Biomedical Engineering, Tufts University, 4 Colby Street, Medford, MA 02155, USA
First published on 3rd May 2018
Abstract
Adaptive hydrogels tailor-made from silk-elastin-like proteins (SELPs) possess excellent biocompatibility and biodegradability with properties that are tunable and responsive to multiple simultaneous external stimuli. To unravel the molecular mechanisms of their physical response to external stimuli in tandem with experiments, here we predict and measure the variation in structural properties as a function of temperature through coarse-grained (CG) modeling of individual and crosslinked SE8Y and S4E8Y molecules, which have ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 and 4:8 of silk to elastin blocks respectively. Extensive structural reshuffling in single SE8Y molecules led to the increased compactness of the structure, whereas S4E8Y molecules did not experience any significant changes as they already adopted very compact structures at low temperatures. Crosslinking of SE8Y molecules at high concentrations impeded their structural transition at high temperatures that drastically reduced the degree of deswelling through extensive suppression of the structural shuffling and the trapping of the molecules in high potential energy states due to inter-molecular constraints. This integrative experimental and computational understanding of the thermal response in single molecules of SELPs and their crosslinked networks should lead to further improvements in the properties of SELP hydrogels through predictive designs and their wider applications in biomaterials and tissue engineering.
:8 and 4:8 of silk to elastin blocks respectively. Extensive structural reshuffling in single SE8Y molecules led to the increased compactness of the structure, whereas S4E8Y molecules did not experience any significant changes as they already adopted very compact structures at low temperatures. Crosslinking of SE8Y molecules at high concentrations impeded their structural transition at high temperatures that drastically reduced the degree of deswelling through extensive suppression of the structural shuffling and the trapping of the molecules in high potential energy states due to inter-molecular constraints. This integrative experimental and computational understanding of the thermal response in single molecules of SELPs and their crosslinked networks should lead to further improvements in the properties of SELP hydrogels through predictive designs and their wider applications in biomaterials and tissue engineering.
Introduction
Hydrogels are colloidal suspensions of one or more polymers dispersed in fluids such as water. Owing to their high compliance and hydrophilicity, hydrogels have been widely applied in biomedical applications such as drug delivery, tissue engineering, and implants.1 However, significant advances in biomaterial applications demand far greater versatility than traditional static hydrogels manufactured from synthetic polymers. For instance, microneedles derived from silk are capable of imbibing bodily fluids for timed-release of drugs.2 These advances have driven the development of nature-inspired hydrogels whose properties are tunable and responsive to multiple simultaneous external stimuli. Stimuli-responsive hydrogels are also increasingly prepared from bio-inspired composites which provide excellent biocompatibility and biodegradability.3Protein-based biopolymers manufactured through genetic engineering are one such category of stimuli-responsive hydrogels that are designed through bio-inspiration. Hydrogels derived from silk-elastin-like proteins (SELPs) are notable examples. SELPs are derived from tandemly repeating units of elastin-like (GXGVP) and silk-like (GAGAGS) domains where the elastin-like domains are elastic with dynamic features4 and the silk-like domains convey mechanical stiffness through the formation of β-sheet secondary structures. An example of the dynamic changes observable in SELPs is the reversible transition in structure, from a soluble protein to a contracted aggregate, around a transition temperature, Tt.5 The stimuli-response of SELPs can be tuned further by changing the “X” residue located in the elastin-like domain to allow for sensitivity towards changes in pH, ionic strength, and phosphorylation to just name a few options.6 Therefore, the naming convention of SELPs helps to broadly clarify the multitude of possible combinations. For instance, if the S:E ratio is 1:8 and tyrosine (Y) is the “X” amino acid in the elastin block, the resulting molecule is termed as SE8Y.
Numerous experimental studies have detailed SELPs’ macromolecular structures derived from processes such as self-assembly through aggregation,7–9 electrospinning,10 and wet-spinning.11 In contrast, the precise mechanisms behind SELPs’ physical transitions on the molecular level in response to external stimuli have only begun to be unravelled recently through computational methods.5,12 The primary means were through fully atomistic molecular dynamics (FAMD) simulations of SELP molecules with high molecular weight, coupled with advanced large-scale replica exchange methods.13 Prior to these numerical studies, only the structural transitions of elastin-like peptides (ELPs) due to temperature have been studied, largely through much smaller scale sampling or low molecular weights using MD.14–17 However, even with advanced sampling methods such as Replica Exchange MD (REMD),13 analysing the properties of ELPs and SELPs with high molecular weight still requires significant computational resources and the data produced are frequently noisy.5,12,18 Additionally, although many properties of the SELP hydrogel are tunable, it was shown that the initial concentration of the SELP molecules strongly influences the swelling ratio of the corresponding hydrogel.5 For example, increasing the initial concentration of SE8Y from 2% to 10% results in a significant decrease in the degree of swelling by 60%. There is no clear understanding of why this phenomenon occurs, particularly the sub-nanoscale mechanisms for the aggregation of SELP molecules, as the concentration increases.
Therefore, this study addressed two significant problems. First, we showed that coarse-grained MD (CGMD) of individual SELP molecules, SE8Y with 644 residues and S4E8Y with 576 residues, enabled the rapid characterization of their thermal response due to potential energy landscapes that were significantly less rugged. There was a clear transition temperature beyond which the SE8Y molecule collapsed into a tightly-packed globular structure from an extended state, while the absence of a transition in S4E8Y was captured as well. This CG model was validated against both experimental and FAMD studies with excellent agreement. Second, through the combination of both experimental studies and the efficient conformational sampling of this CG model, we provided molecular evidence that the clustering and crosslinking of SELP molecules impeded their structural transition at high temperatures, leading to drastic reductions in the degree of deswelling. The molecules were trapped in states with high potential energy due to this aggregation. This understanding of the thermal response in single molecules of SELPs and their crosslinked networks should lead to further improvements in the properties of SELP hydrogels through predictive design and their wider applications in biomaterials and tissue engineering.
Results and discussions
Multiscale characterization of SELPs’ structural transition
To characterize the thermal response of single molecular SELPs, SE8Y and S4E8Y SELPs were synthesized with seamless cloning strategies and purified through inverse temperature transition cycling as described previously7,19 and outlined in the Experimental section. Transitions in these SELPs were determined through differential scanning calorimetry (DSC). The DSC data (Fig. 1a) showed a clear structural transition temperature, Tt, at 294 K for SE8Y, characterized by the endotherm during heating and the exotherm during cooling. In contrast, there were no such transitions for S4E8Y (Fig. 1b) as neither an endotherm nor exotherm were present. The structural transition in SE8Y was also evident by determining the hydrodynamic radius, RH, with dynamics light scattering (DLS) at 277 K and 330 K (Fig. 1c). On the one hand, there was a clear reduction in RH of ∼70% beyond the structural transition temperature of the single SE8Y molecule, indicating that it underwent a structural collapse. On the other hand, the RH of S4E8Y did not show a clear reduction (Fig. 1d), which further confirmed the absence of any structural transition at high temperatures. Peaks that were larger than 10 nm only indicated that some SELP proteins had aggregated.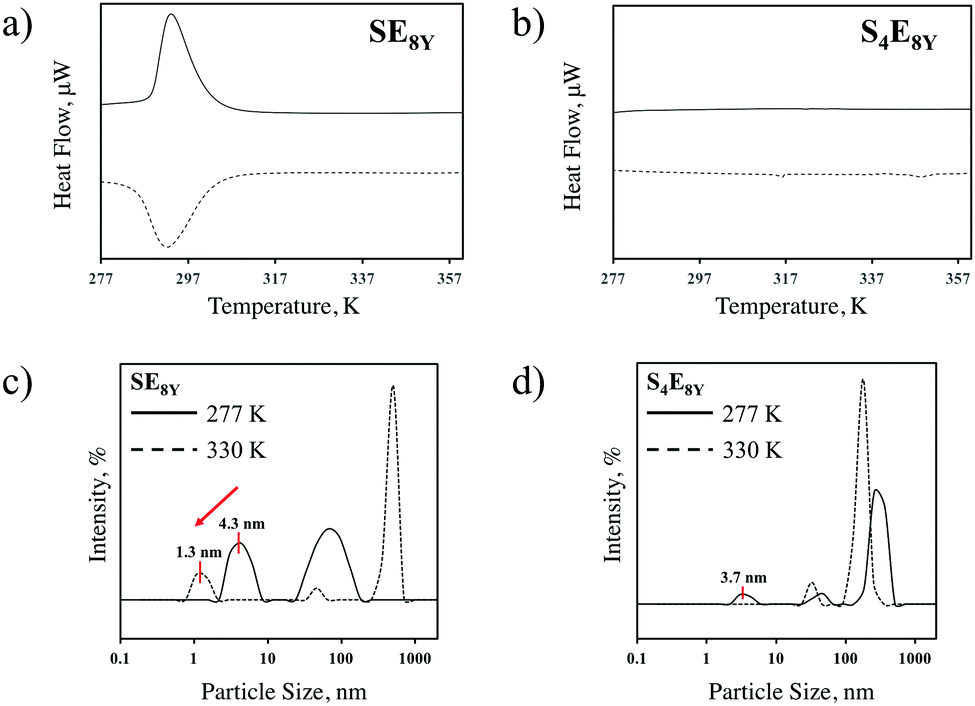 | ||
| Fig. 1 Comparison of the DSC heating curves for (a) SE8Y and (b) S4E8Y, showing a clear structural transition at 294 K for SE8Y, whereas there was no distinct transition for S4E8Y. The hydrodynamic radius from DLS data for (c) SE8Y shows a reduction in size from 4.3 nm to 1.3 nm, whereas (d) S4E8Y shows no transition. In (c) and (d), peaks that are larger than 10 nm are a result of protein aggregation. | ||
In order to understand the molecular mechanisms underlying these differences in structural response to changes in temperature, the structural transition characteristics of the SELPs were characterized through FAMD and CGMD across the temperature range of 277 K to 330 K, as outlined in the Experimental section and the ESI.†
The structural collapse beyond the transition temperature observed experimentally was also identified in both FAMD and CGMD, where the SE8Y molecule became significantly more compact at high temperatures (Fig. 2a and b) compared to the considerably more extended structure at 277 K. This compactness was characterized by two fundamental metrics of protein geometry: the radius of gyration (Rg) and the solvent-accessible surface area (SASA) of the SELPs. From the FAMD results (Fig. 2b), the SE8Y molecule showed a rather gradual decrease of less 10% in the molecule's Rg across the simulated temperature range. In contrast, the Rg and the SASA (Fig. 2c and d) determined by CGMD simulations predicted a clearly distinguishable structural transition at a transition temperature, Tt, of 296 K. This Tt is in remarkable agreement with our experimental value of 294 K from DSC. Between the temperatures of 277 K and 330 K, the difference in Rg was more than 20%, while the SASA was reduced by almost 10%.
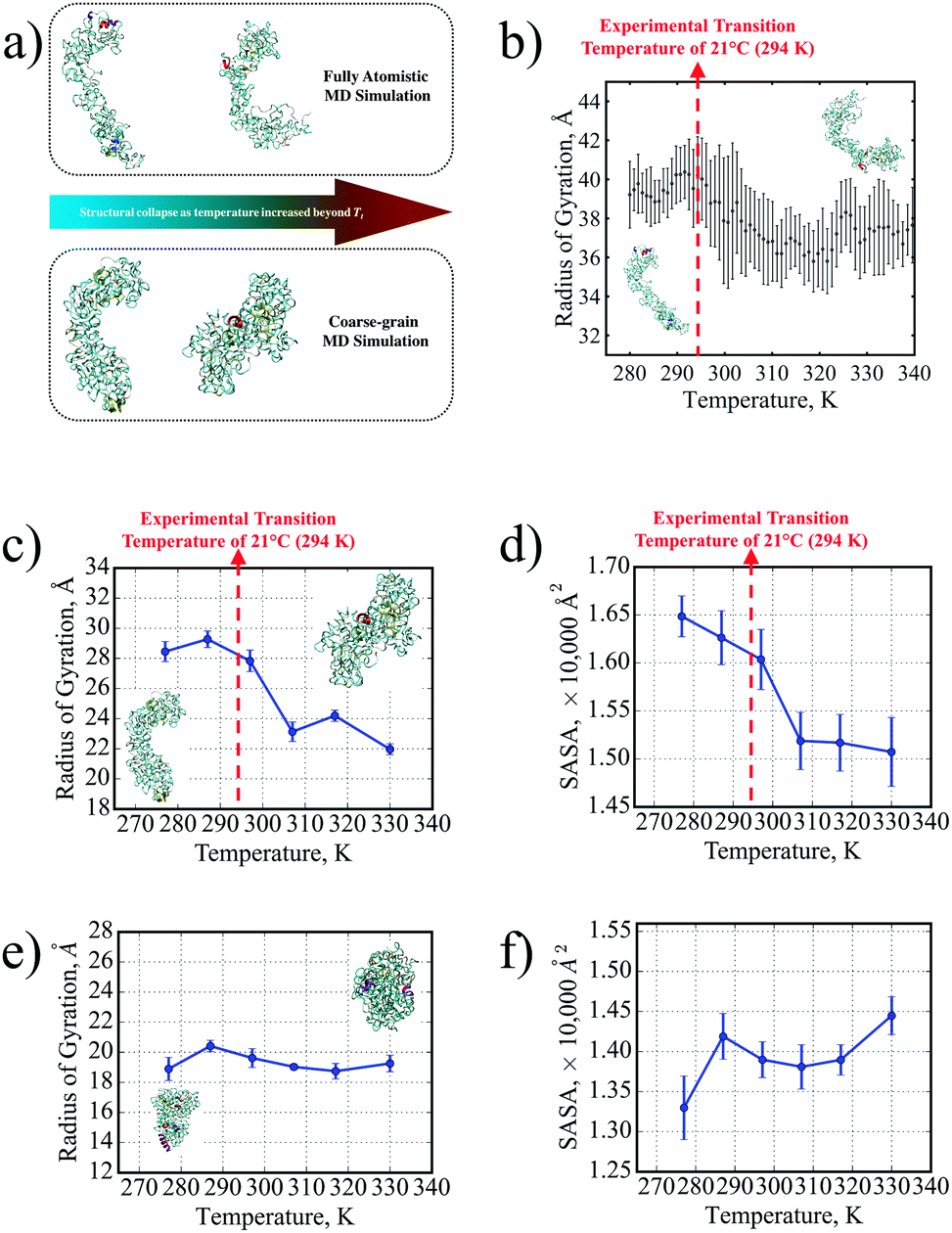 | ||
| Fig. 2 (a) A distinct structural transition temperature for SE8Y was captured by both FAMD and CGMD in terms of (b) the radius of gyration from FAMD and corresponding transitions in (c) the radius of gyration and (d) the SASA from CGMD. This is contrasted with the absence of distinct transitions in S4E8Y in terms of the (e) radius of gyration and (f) SASA as well. Insets in both (c) and (e) show the respective SELP molecule's conformations at 277 K (left) and 330 K (right). | ||
Moreover, this transition was suppressed for S4E8Y (Fig. 2c and d) with no distinct trends in either the Rg or the SASA across the entire temperature range. This result correlates with the absence of a structural transition indicated by our DSC and DLS experimental data. Even at the lowest temperature of 277 K, S4E8Y already adopted a very compact structure as the Rg was more than 30% smaller than that of SE8Y while the SASA was 20% lower. The fact that the thermo-responsive structural transition was clearly captured in SE8Y with a distinct transition temperature, while the absence of any transitions was also captured for S4E8Y, clearly demonstrates that the PLUM potential can adequately capture the dynamic properties of SELPs, in excellent agreement with our experimental studies. Therefore, the PLUM model is validated for further studies here on SELP hydrogel crosslinking, as well as for future work on various thermo-responsive properties in other SELP constructs.
The molecular mechanisms of the structural collapse can be observed in the contact maps of the SELP molecules (Fig. 3 and Fig. S3, ESI†) that illustrate the distance matrix between all possible pairs of α-carbon atoms in all residues. For SE8Y, the contact maps showed a gradual increase in the number of contacts made between the α-carbon atoms in each amino acid and α-carbon atoms at further neighbours (Fig. 3a and b). This was characterized by the increase in the yellow populated region away from the diagonal of the matrix. This implied that any particular residue i within the SELP molecule established increasing numbers of contacts with residues that were more than i ± 3 neighbours away (i.e. more than 3 residues away from i) as the temperature increased. This structural reshuffling led to the increasing compactness of the structure, thereby reducing the Rg and the SASA in SE8Y. In contrast, the compact structure of S4E8Y was a result of the large number of contact with far neighbours even at the lowest temperature of 277 K (Fig. 3c). This is the reason for the low initial Rg and SASA of S4E8Y. Moreover, structural reshuffling at the higher temperature of 330 K only resulted in a decrease in number of contacts with far neighbours while increasing near neighbour contacts as indicated by a localized redistribution of the yellow regions (Fig. 3d), thereby accounting for the lack of a clear structural transition from a looser structure to a more compact conformation observed in S4E8Y. Therefore, these results clearly demonstrate that having higher ratios of silk to elastin blocks greatly reduces the thermal responsiveness that is imbued by the presence of greater amounts of elastin domains with more dynamic structures.
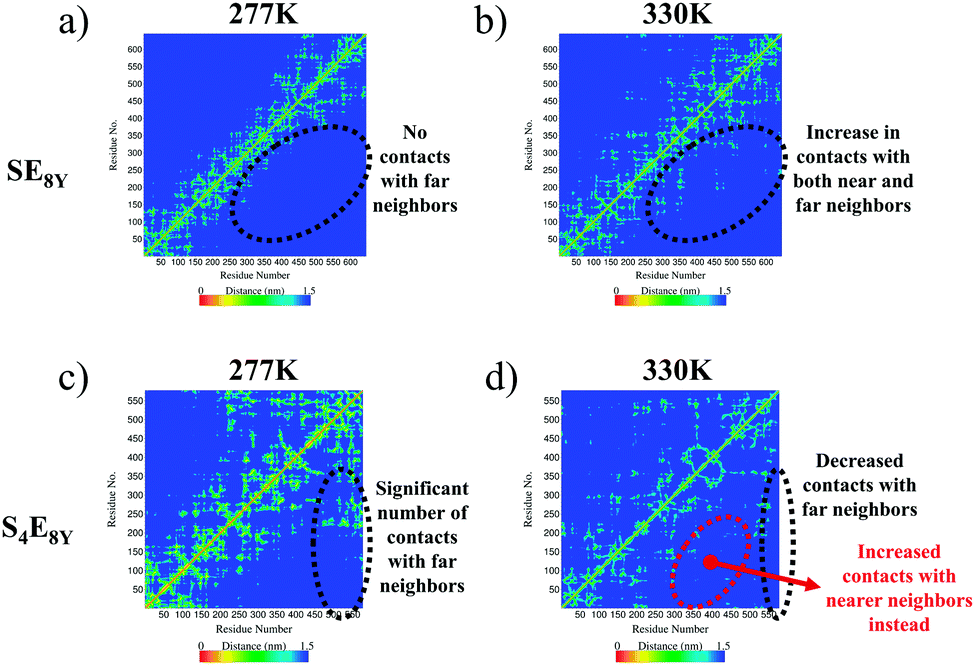 | ||
| Fig. 3 Contact maps showing the matrices of contact distances between the α-carbon atoms of each residue in (a), (b) SE8Y and (c), (d) S4E8Y at the temperatures of 277 K and 330 K, respectively. | ||
Concentration dependence of crosslinked SELPs’ structural transition
Having characterized the structural dynamics at the level of a single molecule, the SE8Y molecules were subsequently enzymatically crosslinked into hydrogels at 2% concentration with horseradish peroxidase (HRP). As HRP is a tyrosine-specific crosslinker, the phenolic sidechains of tyrosine residues form radicals in the presence of HRP and crosslinks are established when these radicals react with one another.Heating the hydrogels caused a shrinkage clearly observable when comparing the hydrogel size at 277 K and at 330 K (Fig. 4a, top row). In excellent correlation with the absence of a structural transition temperature in a single S4E8Y molecule, the S4E8Y hydrogel showed no response to changes in the temperature (Fig. 4a, bottom row). More crucially, the deswelling process was heavily influenced by the concentration of the SELP molecules. As shown in Fig. 4b, the SE8Y hydrogel synthesized at a significantly higher concentration of 20% showed a diminished deswelling response in comparison with the hydrogel synthesized from a concentration that was ten times lower. With a concentration of 2%, the deswelling ratio was more than 95%, whereas at a concentration of 20%, this ratio was significantly reduced to only 60%.
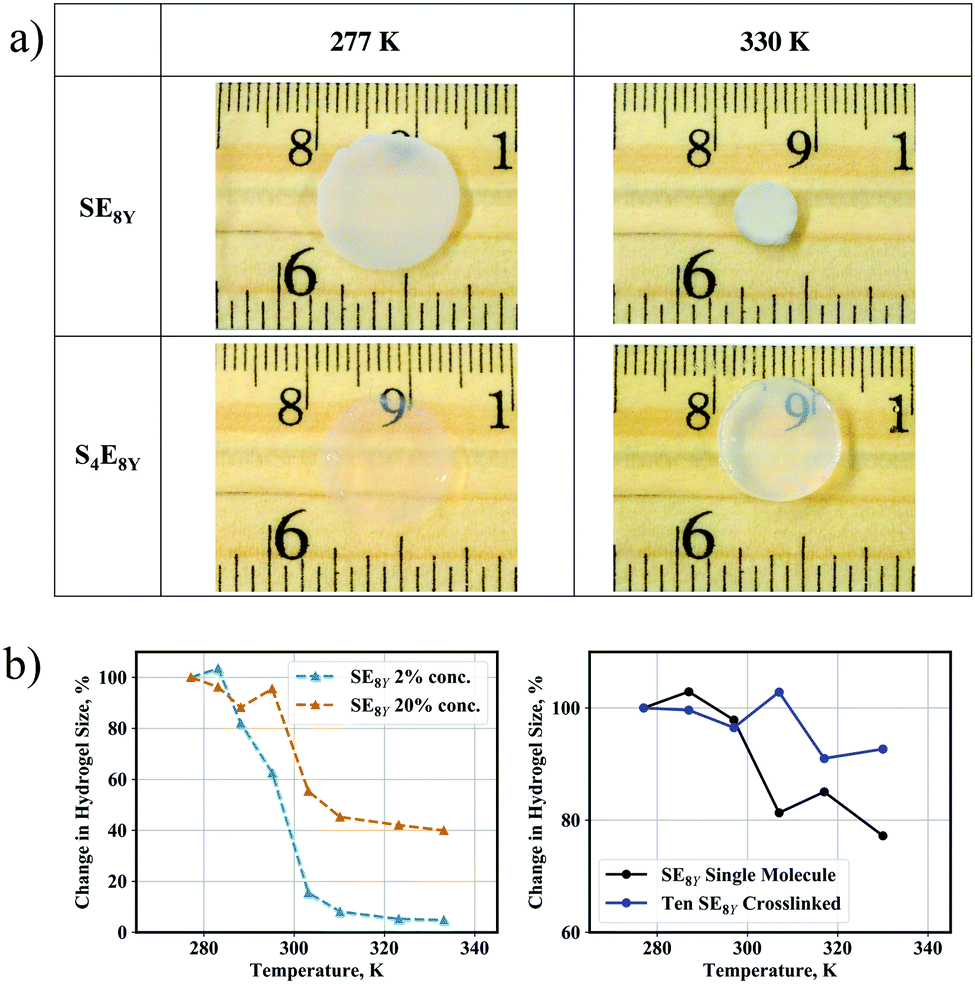 | ||
| Fig. 4 (a) Deswelling of SE8Y and S4E8Y hydrogels at 277 K and 330 K. (b) Variations in the size of experimental (left) and simulated (right) hydrogels that have different concentrations of SE8Y as the temperature increased, where the crosslinked CG model showed significantly reduced deswelling above the transition temperature (P < 0.0001), in good correlation with the trends from our experiments. | ||
To determine the molecular mechanisms behind this suppressed transition, CG crosslinked models consisting of SE8Y molecules were generated and this process is described in detail in the experimental methods. An example of the model is also illustrated in Fig. S2 (ESI†). By performing the simulations in the same temperature range of 277 K to 330 K, suppressed structural transitions were also observed in the crosslinked CG model, in good agreement with the trend observed in our experiments. A single SE8Y molecule showed a reduction in its radius of gyration from 28.4 Å at the temperature of 277 K to 22.0 Å at 330 K, which was a decrease of 23% (Fig. 4b and Fig. S4a, ESI†). However, the average radius of gyration for ten crosslinked molecules only showed a minimal reduction: from 30.8 Å at 277 K to 28.6 Å at 330 K, which was a reduction of merely 7%. Differences in the thermal response between our experimental and simulation data arise from the fact that the CG models are unable to capture the aggregation of multiple SELP clusters into even larger clusters and the crosslink density cannot be precisely determined and modelled. These aggregations will significantly increase the amount of deswelling as observed in our experimental SELP hydrogel. However, the trends of reduced deswelling with when SELP concentration increased ten-fold are in good agreement.
To understand the discrepancy in the thermal response at varying concentrations, the radius of gyration of each crosslinked molecule was analysed at the two temperatures of 277 K and 330 K (Fig. S4b and c, ESI†). Almost every molecule experienced inhibition in their thermal response such that the shrinkage was significantly reduced after the Tt, suggesting that the diminished response was a collective phenomenon rather than a response of molecules individually. Above the structural transition temperature corresponding to that of a single SE8Y molecule (i.e. 296 K), the inability of individual molecules to collapse was due to inter-molecular constraints preventing significant conformational changes in the protein backbones.
This was readily apparent from the contact maps at 277 K and 330 K (Fig. 5a and b) where there was extensive suppression of the shuffling to increase the number of contacts with far neighbours above the transition temperature, as observed from the lack of increase in yellow regions away from the main diagonal axis. This was in stark contrast to the distinct increase observed in a single, non-crosslinked SE8Y molecule (Fig. 2c and d) as discussed above.
 | ||
| Fig. 5 (a and b) Contact maps showing the matrices of contact distances between the α-carbon atoms of each residue in one of the crosslinked SE8Y molecules at the temperatures of 277 K and 330 K, respectively. Minimal variations in contact maps of the same chain (a) at 277 K and (b) at 330 K were observed, indicating that the chains had very minor variations in compactness beyond the transition temperature. (c) Change in potential energy of the SE8Y molecules with increasing temperature, where molecular clustering caused each molecule to be trapped in states of much higher potential energy above the transition temperature, hence they were unable to deswell. | ||
To characterize the extent that inter-molecular constraints influenced structural perturbations, the potential energy of each SE8Y molecule was determined (Fig. 5c) and averaged over all molecules in the crosslinked system. It can be observed that the collective inter-molecular constraints in crosslinked hydrogels caused the molecules to be trapped in a state of higher average potential energy. Below the transition temperature, the energetic behaviours of both systems were largely similar, showing slight increases compared to the baseline at 277 K. Above the transition temperature, the two curves diverged dramatically, where the increase at the highest temperature of 330 K was 30% and 50% from the baseline for single and crosslinked systems, respectively.
Conclusions
Tunable hydrogels that are responsive to multiple simultaneous external stimuli can be customized from SELPs, with the additional benefits of biocompatibility and biodegradability. Integrative experimental and computational approaches can help to optimize the design of these SELP hydrogels, especially with a bottom-up characterization of their molecular properties. This process can be significantly aided by advanced algorithms for modelling SELPs’ nanoscale characteristics. To demonstrate this, we harnessed potential energy landscapes that are less rugged to speedily characterize and uncover the fundamental molecular mechanisms underlying SELPs’ sequence- and concentration-dependent physical response to external stimuli. CGMD simulations of individual and crosslinked SE8Y and S4E8Y molecules were performed together with experimental synthesis and characterizations. Experimental DSC and DLS data showed a distinct structural transition temperature, Tt, at 294 K for single SE8Y molecules, beyond which the hydrodynamics radius, RH decreased by 70%. Such a transition was not found for S4E8Y molecules. Similarly, CG modelling of single SE8Y molecules found that the radius of gyration, Rg, and the SASA decreased beyond a Tt of 296 K by 20% and 10% respectively, collapsing into a tightly-packed globular structure from an extended state. The Tt and the corresponding structural changes were in close agreement with our experimental and FAMD data. Good agreement between CGMD and experimental data was also obtained in the observed absence of structural transitions in single S4E8Y molecules even at high temperatures of 330 K. Contact maps for both SELP sequences showed that structural reshuffling was very extensive in SE8Y molecules, leading to the increased compactness of the structure. In contrast, S4E8Y molecules did not experience significant changes due to their compact structures even at low temperatures, where the Rg was more than 30% smaller than that of SE8Y while the SASA was 20% lower at the temperature of 277 K. These results extensively validated the ability of the PLUM potential to accurately capture the single molecular temperature transitions of SELPs. Combined experimental studies and coarse-grained modelling of crosslinked SE8Y hydrogels demonstrated that the clustering and crosslinking of SE8Y molecules at high concentrations impeded their structural transition at high temperatures, leading to drastic reductions in the degree of deswelling. Experimentally, the deswelling ratio was more than 95% beyond the Tt at a concentration of 2%, reducing to only 60% at a concentration of 20%. The average Rg for the CG hydrogel model also showed minimal deswelling of 7%. Contact maps showed extensive suppression of the shuffling and did not allow for increased number of contacts with far neighbours above the Tt. By analysing the potential energies, this suppressed transition was attributed to the trapping of the molecules in high potential energy states due to inter-molecular constraints as a result of dense packing. Trends in average potential energy per molecule of both systems were largely similar below Tt, showing slight increases compared to the baseline at 277 K. Above the transition temperature, the potential energy increased by 30% and 50% from the baseline for single and crosslinked systems, respectively, at the highest temperature of 330 K. Through this detailed characterization of the effects of SELP sequences and concentration on the thermo-responsiveness of SELP hydrogels, we demonstrated the strong predictive capabilities that integrative experimental and computational approaches can provide for the design, optimization, and customization of SELP hydrogels for advanced applications in biomaterials and tissue engineering. Furthermore, this multiscale approach can potentially be applied to enhance the development of other protein-based materials.Materials and methods
Synthesis of SELP polymers
SELP genes and expression plasmids were constructed using our previously established procedures.7 The purity of the proteins was monitored via SDS-PAGE, and the molecular weights of the proteins were determined by MALDI-TOF (Bruker Corporation, Billerica, MA).Preparation of enzymatically cross-linked SELP hydrogels
To obtain crosslinked SELP hydrogels according to previously reported procedures,20 the lyophilized SELP powder was dissolved in deionized water at 4 °C for 4 h to form a SELP stock solution. Horseradish peroxidase (HRP) type VI lyophilized powder (Sigma-Aldrich, St. Louis, MO) was mixed with deionized water to form a 40 mg mL−1 HRP stock solution with a concentration of 10000 U mL−1. In accordance with our previous studies,5 to fabricate a 10% SELP hydrogel, 6 μL of HRP stock solution was added to 100 μL 10% SELP stock solution, and then the crosslinking reaction of SELP was initiated by adding 0.2 μL of 30 wt% H2O2 solution to the SELP and HRP mixture with a final H2O2 concentration of 18 × 10−3 M. The reaction mixture was mixed by gentle pipetting prior to gelation. The enzymatically cross-linked SELP hydrogels were formed by incubation at 4 °C overnight. To fabricate 2% and 20% SELP hydrogels, the same SELP:HRP:H2O2 ratio and crosslinking reaction protocol was used for SELP gelation. An inversion test was used to qualitatively characterize the gelation time for each SELP hydrogel. The formed SELP hydrogels were dialyzed in DI water for 24 h to remove the residual, unreacted HRP.
Differential scanning calorimetry
DSC measurements were performed with Nano DSC II Model 6100 (Calorimetry Sciences Corp., Lindon, UT) to capture the inverse transition temperature of SELP hydrogels. The 2% SELP hydrogels were equilibrated at the initial temperature for 10 min, and then heated/cooled in the sample chamber at a rate of 2 °C min−1. The same volume of solvent was placed in the reference chamber during each scan. The baseline scans were taken with the solvent under the same condition and subtracted from the sample scans.Dynamic light scattering
Dynamic light scattering (DLS) was carried out on a DynaPro Titan instrument (Wyatt Technology, Santa Barbara, CA) equipped with a temperature controller. Cuvettes with 1 mm path length were used. All samples were filtered through 0.2 μm Millex® syringe filters (EMD Millipore, Darmstadt, Germany) before measurement. SELP solutions (0.2 mg mL−1) were stabilized at each temperature for 10 minutes prior to measurement. To obtain the hydrodynamic radii, the intensity autocorrelation functions were analyzed using the Dynamics software (Wyatt Technology, Santa Barbara, CA).Swelling and deswelling properties
The swelling ratio of the SELP hydrogels at 4 °C was determined by the ratio of the weight of the hydrogel equilibrated in deionized water to the weight of corresponding as-prepared gel. For deswelling studies, the swollen samples equilibrated in deionized water at 4 °C were transferred to 4 °C buffer solution, and the deswelling ratio was determined by the ratio of the weight of the hydrogel in buffer to the weight of the hydrogel equilibrated in deionized water. To investigate the stimuli responsive properties, the hydrogels were equilibrated at 4, 10, 15, 22, 37, 50, and 60 °C in deionized water and buffers. The equilibrium deswelling ratio upon environmental stimulus is defined as the ratio of the weight of the hydrogel under the specific stimuli to the weight of the hydrogel equilibrated in deionized water at 4 °C.Fully atomistic molecular simulation setup
Both FAMD and CGMD simulations were performed in order to characterize phenomena on different time scales, as laid out in Fig. S5 (ESI†). The FAMD simulations followed our established procedures.5,12,18 Using CHARMM version 35b1,21 extended straight chain conformations of SE8Y and S4E8Y were constructed from elastin and silk blocks. The SE8Y chain was a 14-mer alternating silk-elastin chain that had the sequence of [(GVGVP)4(GYGVP)(GVGVP)3(GAGAGS)]14, while the S4E8Y chain had the sequence of [(GVGVP)4(GYGVP)(GVGVP)3(GAGAGS)4]9. The same SELP sequences were studied in both simulations and experiments. Energy minimization through the steepest descent algorithm was performed to prevent steric clashes.Subsequently, REMD simulations were carried out in the canonical (NVT) ensemble with the CHARMM19 all-atom energy function.21 The implicit solvent was implemented as the EEF1 force field with a Gaussian effective solvent energy function.22 REMD improves conformational sampling by integrating Monte Carlo exchanges into a classical molecular dynamics simulation scheme. This scheme helps the sampled protein to escape local free energy minima in the replicas with high temperatures, thus allowing wider sampling of the conformational space. Full details of the force field and the REMD methodology are laid out in the ESI.† Twenty-four temperature replicas were created and their temperatures were exponentially distributed between 280 and 480 K.23 Replica exchanges were attempted every 0.5 ps with 2 fs time step to achieve a 15% exchange acceptance rate with the Metropolis criterion. K-means clustering was performed on the ensemble of structures from the last 1000 exchanges at the lowest temperature replica. Clustering was based on mutual similarity by root-mean-square deviation (RMSD of 3 Å) and performed in the MMTSB tool set.24
The lowest-energy representative structure in the most populated cluster was selected for further refinement with REMD in explicit solvent to correct for local structural approximations. These simulations were implemented in GROMACS version 5.0125 with the CHARMM27 force field.26 The representative protein was solvated in a rectangular water box with fully periodic boundary conditions. The entire model system contained approximately 200000 atoms. After energy minimization through the steepest descent algorithm, the solvent was equilibrated while the protein was fixed. This was done in two equilibration stages of 100 ps each with a time step of 1 fs. The NVT ensemble was implemented in the first stage to stabilize the system's temperature. The NPT ensemble was implemented in the second stage to stabilize the system's pressure. Subsequently, the restraints on the protein were removed and equilibrated for an additional 100 ps in the NPT ensemble. The Berendsen thermostat27 and the Parrinello–Rahman barostat28 were used for temperature and pressure coupling, respectively. Covalent bonds with hydrogen atoms were constrained with the LINCS algorithm.29 The short range electrostatic interactions and Lennard-Jones interactions were evaluated with a cutoff of 10 Å. Particle-mesh Ewald summation30 was used to calculate long range electrostatic interactions with a grid spacing of 1.6 Å and a fourth order interpolation.
Finally, REMD was performed with 120 replicas in explicit solvent, starting from representative structures derived from implicit solvent REMD, at temperatures exponentially distributed from 280 K to 400 K.23 A 2 fs time step was used, each replica was simulated for 20 ns, and exchanges were attempted after 2 ps equilibration runs. Exchange acceptance ratios, based on the Metropolis criterion, were between 20–30%, which signifies adequate sampling. K-means clustering was performed on the ensemble of structures in the final 2 ns to group structures into clusters according to a RMSD of 12 Å for the low-temperature replica at 280 K and for the high-temperature replica at 330 K. Representative structures with the lowest potential energy were chosen from most populated clusters. Analysis of representative structures was carried out using the MMTSB script package.24
Coarse-grain molecular simulation setup
Following the extensive FAMD simulations, the representative structure of a single SELP molecule at 280 K was coarse-grained using the scheme from the PLUM potential.31 In this CG model, heavy atoms in each amino acid of the protein backbone are fully represented while their corresponding sidechains are coalesced into a single CG bead. This leads to an intermediate-resolution CG model of four beads per amino acid with implicit water solvent interactions. The full details of the PLUM potential are laid out in the ESI.† There are multiple benefits of using this approach. First, the rugged potential energy landscape encountered in AAMD is smoothened due to significantly reduced interactions in CG simulations. Conformational sampling is greatly enhanced, where the estimated speedup factor was estimated to be three orders of magnitude.32 For instance, the time to fold the protein (AAQAA)3 was experimentally determined to be on the order of 100–500 ns at room temperature, and yet CG simulations with the PLUM potential were able to achieve the same protein folding from an extended conformation within 500 ps of simulation time.32 Second, computational resources are drastically diminished due to greatly enhanced sampling together with significant reductions in the number of interactions that have to be computed.Using the PLUM potential implemented in the open-source CGMD software package Extensible Simulation Package for Research on Soft matter (ESPResSo),33,34 the transition temperature of this CG model was validated against experimental and FAMD data by simulating a single SELP molecule in the temperature range of 277 to 330 K with a simulation time of 5 ns each. Using a conservative estimate based on the reported speedup factor,32 5 ns of simulation time corresponds to more than 1 μs of real sampling time of an already extensively equilibrated SELP structure from FAMD. A time step of 1 fs was used and non-bonded interactions were slowly uncapped over 1 ps to minimize structural perturbations upon starting the simulation.
To simulate dityrosine-crosslinked clusters of SE8Y molecules, tyrosine residues that were exposed on the surface of a single SE8Y molecule were determined first in PyMol.35 A surface exposed residue was defined as being within 0.35 nm from the surface of the molecule. Ten SE8Y molecules were then placed one after another in an iterative fashion using Packmol36 and PyMol35 scripts, such that randomly selected surface tyrosine sidechain beads were able to be packed at less than 10 Å to each other without creating overlaps in any of the other beads (see Fig. S2, ESI†). These sidechain beads were then crosslinked by the addition of a harmonic spring potential to mimic bonding. The transition temperature of these crosslinked clusters was determined again in the same manner as for a single SE8Y molecule.
Molecular dynamics data analysis tools
For all the FAMD and CGMD simulations above, radius of gyration, solvent accessible surface area, and contact maps were analyzed using the open-source GROningen MAchine for Chemical Simulations (GROMACS) analysis tools,25 PyMol,35 in-house Bash and Python scripts, and script libraries such as MDTraj.37 All visualization of molecular models was performed using combinations of Visual Molecular Dynamics (VMD),38 PyMol,35 and in-house TCL, Python, Bash, and Matlab scripts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the US Department of Defense, Office of Naval Research (N00014-16-1-233) and the National Institutes of Health (U01 EB014976). J. Y. and Y-W. Z. acknowledge support from Singapore's Agency for Science, Technology and Research (A1786a0031). Computational simulations were performed on the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation grant number ACI-1053575, the MIT Engaging Cluster, and Singapore's A*STAR Computational Resource Centre and National Supercomputing Centre.Notes and references
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- K. Tsioris, W. K. Raja, E. M. Pritchard, B. Panilaitis, D. L. Kaplan and F. G. Omenetto, Adv. Funct. Mater., 2012, 22, 330–335 CrossRef.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- Z. Megeed, J. Cappello and H. Ghandehari, Adv. Drug Delivery Rev., 2002, 54, 1075–1091 CrossRef PubMed.
- W. Huang, A. Tarakanova, N. Dinjaski, Q. Wang, X. Xia, Y. Chen, J. Y. Wong, M. J. Buehler and D. L. Kaplan, Adv. Funct. Mater., 2016, 26, 4113–4123 CrossRef PubMed.
- Q. Wang, X. Xia, W. Huang, Y. Lin, Q. Xu and D. L. Kaplan, Adv. Funct. Mater., 2014, 24, 4303–4310 CrossRef PubMed.
- X.-X. Xia, Q. Xu, X. Hu, G. Qin and D. L. Kaplan, Biomacromolecules, 2011, 12, 3844–3850 CrossRef PubMed.
- Y. Lin, X. Xia, M. Wang, Q. Wang, B. An, H. Tao, Q. Xu, F. Omenetto and D. L. Kaplan, Langmuir, 2014, 30, 4406–4414 CrossRef PubMed.
- W. Hwang, B.-H. Kim, R. Dandu, J. Cappello, H. Ghandehari and J. Seog, Langmuir, 2009, 25, 12682–12686 CrossRef PubMed.
- Y. Ner, J. A. Stuart, G. Whited and G. A. Sotzing, Polymer, 2009, 50, 5828–5836 CrossRef.
- W. Qiu, W. Teng, J. Cappello and X. Wu, Biomacromolecules, 2009, 10, 602–608 CrossRef PubMed.
- A. Tarakanova, W. Huang, Z. Qin, D. L. Kaplan and M. J. Buehler, ACS Biomater. Sci. Eng., 2017, 3, 2889–2899 CrossRef.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef.
- G. C. Yeo, A. Tarakanova, C. Baldock, S. G. Wise, M. J. Buehler and A. S. Weiss, Sci. Adv., 2016, 2, e1501145 CrossRef PubMed.
- N. K. Li, F. G. Quiroz, C. K. Hall, A. Chilkoti and Y. G. Yingling, Biomacromolecules, 2014, 15, 3522–3530 CrossRef PubMed.
- B. Li, D. O. V. Alonso and V. Daggett, J. Mol. Biol., 2001, 305, 581–592 CrossRef PubMed.
- J. Huang, C. Sun, O. Mitchell, N. Ng, Z. N. Wang and G. S. Boutis, J. Chem. Phys., 2012, 136, 085101 CrossRef PubMed.
- A. Tarakanova, W. Huang, A. S. Weiss, D. L. Kaplan and M. J. Buehler, Biomaterials, 2017, 127, 49–60 CrossRef PubMed.
- W. W. Huang, A. Tarakanova, N. Dinjaski, Q. Wang, X. X. Xia, Y. Chen, J. Y. Wong, M. J. Buehler and D. L. Kaplan, Adv. Funct. Mater., 2016, 26, 4113–4123 CrossRef PubMed.
- B. P. Partlow, C. W. Hanna, J. Rnjak-Kovacina, J. E. Moreau, M. B. Applegate, K. A. Burke, B. Marelli, A. N. Mitropoulos, F. G. Omenetto and D. L. Kaplan, Adv. Funct. Mater., 2014, 24, 4615–4624 CrossRef PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef.
- T. Lazaridis and M. Karplus, Proteins: Struct., Funct., Bioinf., 1999, 35, 133–152 CrossRef.
- A. Patriksson and D. van der Spoel, Phys. Chem. Chem. Phys., 2008, 10, 2073–2077 RSC.
- M. Feig, J. Karanicolas and C. L. Brooks Iii, J. Mol. Graphics Modell., 2004, 22, 377–395 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef.
- T. Bereau and M. Deserno, J. Chem. Phys., 2009, 130, 235106 CrossRef PubMed.
- T. Bereau, Unconstrained Structure Formation in Coarse-Grained Protein Simulations, PhD thesis, Carnegie Mellon University, 2011 Search PubMed.
- H. J. Limbach, A. Arnold, B. A. Mann and C. Holm, Comput. Phys. Commun., 2006, 174, 704–727 CrossRef.
- A. Arnold, O. Lenz, S. Kesselheim, R. Weeber, F. Fahrenberger, D. Roehm, P. Košovan and C. Holm, in Meshfree Methods for Partial Differential Equations VI, ed. M. Griebel and M. A. Schweitzer, Springer Berlin Heidelberg, 2013, ch. 1, vol. 89, pp. 1–23 Search PubMed.
- W. L. DeLano, The PyMOL molecular graphics system, 2002, http://pymol.org Search PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- R. T. McGibbon, K. A. Beauchamp, M. P. Harrigan, C. Klein, J. M. Swails, C. X. Hernández, C. R. Schwantes, L.-P. Wang, T. J. Lane and V. S. Pande, Biophys. J., 2015, 109, 1528–1532 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb00819a |
| This journal is © The Royal Society of Chemistry 2018 |