
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTurn-on fluorescence detection of protein by molecularly imprinted hydrogels based on supramolecular assembly of peptide multi-functional blocks†
Edmondo
Battista
 a,
Pasqualina L.
Scognamiglio
b,
Nunzia
Di Luise
b,
Umberto
Raucci
c,
Greta
Donati
c,
Nadia
Rega
ac,
Paolo A.
Netti
abd and
Filippo
Causa
*abd
a,
Pasqualina L.
Scognamiglio
b,
Nunzia
Di Luise
b,
Umberto
Raucci
c,
Greta
Donati
c,
Nadia
Rega
ac,
Paolo A.
Netti
abd and
Filippo
Causa
*abd
aInterdisciplinary Research Centre on Biomaterials (CRIB) Università degli studi di Napoli “Federico II”, Piazzale Tecchio 80, 80125, Napoli, Italy. E-mail: causa@unina.it
bCenter for Advanced Biomaterials for HealthCare@CRIB, Istituto Italiano di Tecnologia, Largo Barsanti e Matteucci 53, 80125 Napoli, Italy
cDipartimento di Scienze Chimiche, Università degli studi di Napoli “Federico II”, Complesso Universitario di M.S.Angelo, via Cintia, I-80126, Napoli, Italy
dDipartimento di Ingegneria Chimica, dei Materiali e della Produzione Industriale (DICMAPI), Università degli studi di Napoli “Federico II”, Piazzale Tecchio 80, 80125, Napoli, Italy
First published on 29th January 2018
Abstract
Synthetic receptors for biomacromolecules lack the supramolecular self-assembly behavior typical of biological systems. Here we propose a new method for the preparation of protein imprinted polymers based on the specific interaction of a peptide multi-functional block with a protein target. This peptide block contains a protein-binding peptide domain, a polymerizable moiety at the C-terminus and an environment-sensitive fluorescent molecule at the N-terminus. The method relies on a preliminary step consisting of peptide/protein supramolecular assembly, followed by copolymerization with the most common acrylate monomers (acrylamide, acrylic acid and bis-acrylamide) to produce a protein imprinted hydrogel polymer. Such a peptide block can function as an active assistant recognition element to improve affinity, and guarantees its effective polymerization at the protein/cavity interface, allowing for proper placement of a dye. As a proof of concept, we chose Bovine Serum Albumin (BSA) as the protein target and built the peptide block around a BSA binding dodecapeptide, with an allyl group as the polymerizable moiety and a dansyl molecule as the responsive dye. Compared to conventional approaches these hydrogels showed higher affinity (more than 45%) and imprinted sensitivity (about twenty fold) to the target, with a great BSA selectivity with respect to ovalbumin (α = 1.25) and lysozyme (α = 6.02). Upon protein binding, computational and experimental observations showed a blue shift of the emission peak (down to 440 nm) and an increase of fluorescence emission (twofold) and average lifetime (Δτ = 4.3 ns). Such an approach generates recognition cavities with controlled chemical information and represents an a priori method for self-responsive materials. Provided a specific peptide and minimal optimization conditions are used, such a method could be easily implemented for any protein target.
Introduction
The precision of nature is inspiring chemists in developing wholly synthetic receptors, adapting concepts of supramolecular chemistry to appropriately locate key residues involved in the interaction with targets.1,2 To this end, as an alternative to natural receptors,3–5 Molecularly Imprinted Polymers (MIPs) are emerging as a platform of materials, being highly robust and relatively inexpensive, and useful for the development of selective sorbents in a diverse range of applications such as separations, sensing, catalysis and drug discovery.6–11 Among the different imprinted polymer strategies, the bulk imprinting of hydrogels results in a successful approach, suitable for biological macromolecules.12 Hydrogels provide a highly hydrated bioinspired environment with tunable mechanical properties and high chemical flexibility for the straightforward production of engineered analyte-specific HydroMIPs (hydrogel molecularly imprinted polymers).13–15 Moreover, unlike with small molecule templates,16,17 the imprinting of biomacromolecules (>1500 Da) still poses challenges.7,18,19 Indeed, one of the major limitations for proteins and oligonucleotides (DNA/RNA) is their complex and flexible three-dimensional structure that is easily modified upon environmental changes (i.e. temperature, ionic strength or pH) and exposure to different patterns of interactions, impairing their natural selectivity.20 Nevertheless, successful examples of imprinted hydrogels for proteins were obtained through a configurational biomimetic approach,21 metal ion-mediated imprinting22 or making use of assistant recognition polymer chains (ARPCs).23 However, these approaches are based on labor-intensive monomer pre-polymer screening toward specific templates with limited applicability to all biomacromolecules. Moreover, further issues arise with the development of MIPs for biosensing applications. Several efforts have been made to realize optically active MIPs, in particular, environmentally sensitive dyes have been incorporated within the site of interaction,24,25 showing changes in their optical properties (i.e. wavelength of emission, intensity and/or lifetime) with respect to physical or chemical changes in their surroundings.26,27 Along these lines, Takeuchi's group introduced approaches for a posteriori in-cavity modification of protein-imprinted hydrogels to obtain multi-functional materials (on/off switching of molecular recognition, signal transduction of binding events and photoresponsive and catalytic activities).28,29 The resulting procedures are rather complicated as they often require the synthesis of small organic molecules, the modification of sacrificial proteins and some specific cleavage procedures after the polymer synthesis. Thus the need to chemically access the cavity might be achieved by an a priori easy approach that exploits the assembly of structurally and chemically complementary biomolecules to drive the generation of recognitive cavities with well-oriented chemical information.Peptides have been widely used as probes in protein–protein or peptide–protein interactions for drug delivery, enzyme catalysis and biosensors.30,31 Although peptides possess low binding affinities, the great advantage of their use lies in the easy and versatile cost-effective chemical synthesis with high yield, high stability and access to non-native chemistries. The use of peptides in macromolecular imprinting approaches has not been reported so far. Few examples describe a rational design of peptides for the development of ATP synthetic receptors.32,33 Peptides offer the possibility to create molecular scaffolds with tailored block units with multiple functions, for example recognition, signal transduction, catalysis and tethering.34–36
Herein we introduce a novel approach for protein imprinting based on the in-cavity incorporation of a peptide multi-functional block, working as an Active Assistant Recognition Element (AARE), and at the same time capable of driving the proper orientation of the target at the polymer interface, participating in the polymer network to control the cavity chemistry and reporting the binding event. In detail, we introduced a pre-polymerization phase where the AAREs interact with the protein (peptide/protein supramolecular assembly), co-polymerize with acrylate monomers and retain the fluorophore in an appropriate location to obtain a Hybrid Peptide–Polymer Imprint (HyPPI) (Scheme 1).
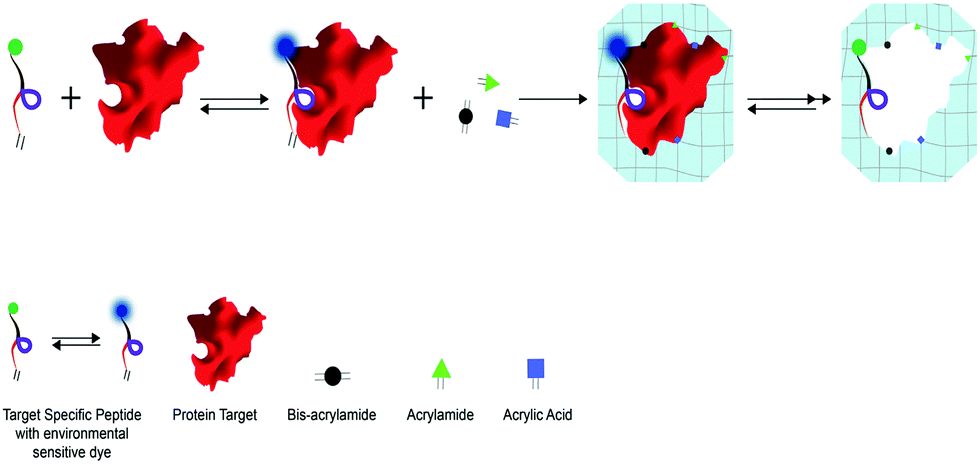 | ||
| Scheme 1 HyPPI approach: the affine peptide for a given target is used as a molecular scaffold to guide the specific interaction and support an environment-sensitive fluorophore (dansyl). The HyPPI approach relies on two pre-polymerization steps where (i) a peptide block/template supramolecular complex is formed and subsequently (ii) the bimolecular complex is conditioned with the mixture of monomers to form weak interactions and then co-polymerized to obtain the hydrogel network. In this way, a hybrid recognition cavity is formed between the polymer and the multi-functional peptide block, capable of directing the template and of locating the functionalities at precise 3D positions. | ||
In this study, we rationally designed and characterized a peptide block with three different units addressing: (i) the protein recognition, (ii) the polymerization phase and (iii) the fluorescence reporting. As a proof of concept we chose Bovine Serum Albumin as the protein target, a BSA-binding dodecapeptide as a binder, an allyl group as the polymerizable unit and a dansyl molecule as the environment-sensitive unit. Then, the effect of AARE inclusion in HyPPIs was evaluated in terms of the binding and specificity properties in comparison with conventional polyacrylamide HydroMIP. Finally, the optical sensing capabilities of the responsive materials were studied by quantum mechanical calculations and experimental analysis in terms of fluorescence emission and lifetime.
Result and discussion
Rational design and characterization of AARE
Herein, the AARE is based on a Serum Albumin peptide (SAp) specifically interacting with the BSA protein (Fig. 1a). | ||
| Fig. 1 AARE design – (a) AARE structure: the design of the AARE incorporates the insertion of a polymerizable group and a reporter dye in the flanking parts of the binding region. Specifically, at the C-terminus the polymerizable unit was introduced as Fmoc-Asp(OAll)-OH, an aspartic acid with an allyl group on the side chain; whereas at the N-terminus a dansyl derivative was introduced as Fmoc-Lys(dansyl)-OH, spaced by two β-Ala residues, to allow free motion of the dye. (b) Upper panel: HOMO and LUMO contour plots computed for the dansyl molecule in a water solution. Lower panel: HOMO and LUMO contour plots computed for the AARE in a water solution. (c) and (d) ITC analysis and fluorescence experiment results of the interaction of AARE with BSA, respectively. | ||
Previously selected by Dennis et al.37 through phage display screening, SAp (Table S2, ESI†) recognizes BSA with a one to one stoichiometry and a dissociation constant in a low micromolar range, as confirmed by ITC (KD = 21.7 ± 3.3 μM) and SPR (KD = 22 ± 1 μM for fitting kinetic parameters, whereas KD = 18.2 ± 8.4 μM from the fit of the binding isotherm) experiments (Fig. S7(a and b) and S8, ESI†). Here, SAp was used as a scaffold for the specific introduction of functionalities in order to ensure the polymerization at the protein/cavity interface, and to allow for suitable location of the responsive probe. Specifically, we considered the conjugation of SAp with the dansyl dye, in order to exploit its capability to recognize the change of polarity when passing from an aqueous solution environment to in proximity to the protein (Fig. 1a). Quantum mechanical calculations performed in a water solution suggest that the photophysical signatures of the dansyl dye remain unaffected by its conjugation into the peptide structure, Fig. 1b (see the ESI† for the computational details). From an experimental point of view, the AARE is still able to interact with BSA, retaining the same affinity and stoichiometry as the native peptide sequence (KD = 13.18 μM and the stoichiometry n ≈ 1) (Fig. 1c). It is strongly sensitive to the local polarity of its surrounding environment (Fig. 1d). With an increase in the BSA concentration, both an increase of the fluorescence emission intensity and a blue shift of the emission peak are observed (Fig. 1d). Specifically, the wavelength of the maximum emission shifts from 540 nm to 500 nm, whereas the fluorescence intensity increases up to a BSA concentration of 1 μM (equimolar to the peptide concentration). The control experiment with a n-AARE sequence (i.e. non affine AARE peptide) is reported in Fig. S9 (ESI†). We tried to interpret the effect of the BSA protein on the AARE fluorescence using quantum mechanical calculations. The protein environment is usually characterized by a low dielectric constant (ε ≈ 2.5).38 The protein polarity effect on the dansyl emission energy can be therefore compared to that of a low polar solvent, which can be in turn modeled implicitly.39 In this way it is possible to take into account the effect of the protein polarity, although an atomistic description of the residues is lost. Here, the emission energy of the dansyl molecule was calculated both in cyclohexane (ε = 2.02), representative of the local environment encountered by the dansyl in the presence of the BSA protein, and in water, simulating the local environment in its absence (Fig. S10, ESI†). Calculated values show a nice quantitative agreement with the experimental ones obtained from the spectrophotometric titration with the BSA protein. Specifically, the emission energy is computed to be 523 and 494 nm in water and cyclohexane solution, respectively, thus reproducing 30 out of the 40 nm total shift in the emission maximum. In both the solvents the emission is from an intramolecular charge transfer (ICT) state initially generated by a HOMO–LUMO transition, with the charge transfer involving the dimethylamino and the naphthyl groups. This ICT state is destabilized with respect to the ground state when passing from a polar to a non-polar environment, causing a blue shift in the emission band. We can therefore attribute the blue shift of the fluorescence band to the change of the local dielectric constant felt by the dansyl dye upon the formation of the BSA–AARE complex. Regarding the increase of the fluorescence emission in the presence of BSA, we can hypothesize, also on the basis of previous studies, that decay processes quenching the fluorescence yield (when the dansyl tag is embedded in a highly polar environment) become less important with the formation of the BSA–AARE complex.40 In order to deepen the understanding of this key aspect, the fluorescence decay kinetics in solution were also analyzed. The AARE and the control, n-AARE, showed a bi-exponential decay with a τ average of around 8.7 ns and 8.2 ns, respectively (Table S3, ESI†). Upon titration with BSA, the AARE τ average value increases up to 12.5 ns, at saturation point. Specifically, the two components τ1 and τ2 reached respective values of around 4.6 and 16.6 ns, while no significant variations were observed in the case of n-AARE (Table S3, ESI†). It is worth noting that the τ2 value increases significantly with the BSA concentration and this can be easily attributed to the ICT state, which is sensitive to an apolar environment. On the basis of the τ2 values, we estimated the non-radiative decay rates for the ICT state by adopting a general protocol recently formulated by us41,42 and briefly reported in the ESI.† Values of 0.0500 and 0.0319 ns−1 were respectively computed for the non-radiative decay rates in water and BSA with a ratio equal to 1. These data clearly suggest that the non-radiative decay pathways are less important in an apolar environment, leading to an enhancement of dansyl fluorescence emission. Based on these investigations, we can conclude that the dansyl dye comes into contact with a low polarity region of the protein after the AARE–BSA complex formation. This in turn enables the enhancement of the quantum yield upon BSA binding, and consequently also of the lifetime and the fluorescence intensity values.
Setup for HydroMIP preparation
Prior to the integration of the AAREs inside the recognition cavity, we set up a basic formulation of polyacrylamide HydroMIP for the preparation of BSA imprinted hydrogels. Hydrogels for imprinting are not very highly crosslinked and therefore mechanical integrity, as well as the capability to swell and collapse in different environments, has to be carefully taken into account to guarantee their use in imprinting processes. Along these lines, a preliminary study was carried out to find the best formulation in terms of effective re-binding, imprinting factor and selectivity in monomers and crosslinking concentrations preserving mechanical integrity. The BSA protein is employed as a template molecule. The polymer matrix formed here was based on the co-polymerization of acrylamide (AAm) and acrylic acid (AAc) with bis-acrylamide (Bis) as the cross-linking agent, according to a T% and C% variation scheme (Table S1, ESI†). This is one of the most commonly used recipes for macromolecular imprinting under mild polymerization conditions in order to avoid thermal unfolding of proteins.20–22 Soon after the polymerization the bulk hydrogel was comminuted into particles with an average size of 150 μm (Fig. S3, ESI†) and washed several times with different solutions to remove the templating agent, BSA, with an efficiency at least of 79.3% (Fig. S1, ESI†). The results reported in the radar chart (Fig. 2) allow the different features to be appreciated at a glance. Thus, the preparation 15%T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10%C better satisfied all considered parameters with a high imprinting ratio and selectivity (with respect to an interfering protein, lysozyme) and one of the highest capabilities to bind the template protein.
:10%C better satisfied all considered parameters with a high imprinting ratio and selectivity (with respect to an interfering protein, lysozyme) and one of the highest capabilities to bind the template protein.
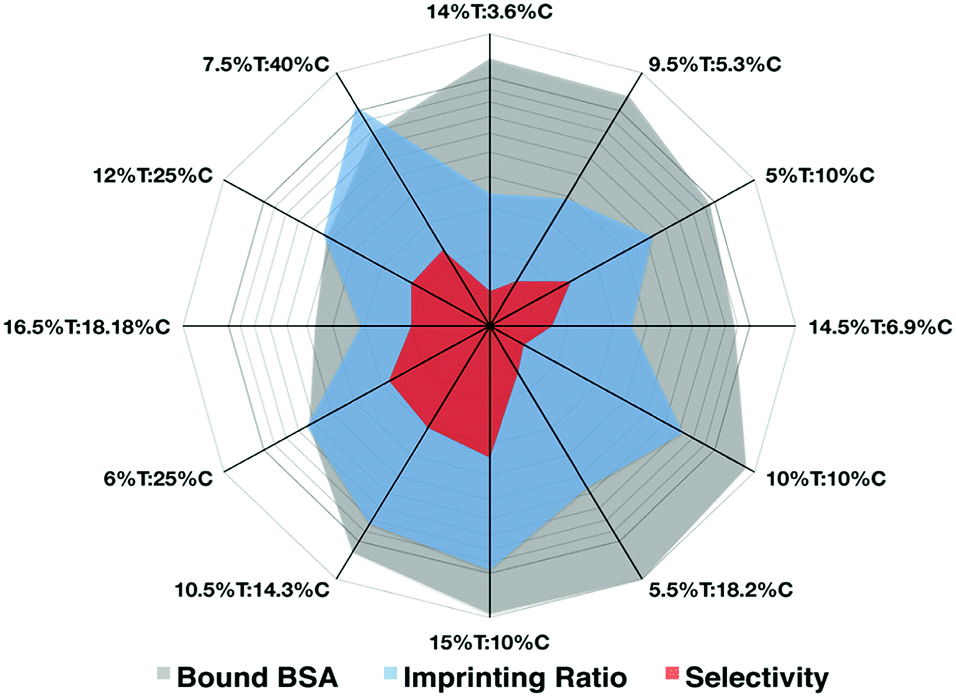 | ||
| Fig. 2 In the radar plot the three parameters considered for the imprinting recipe expressed as T% and C% are highlighted (plotted in log scale). (The raw data are reported in Table S1, ESI†). | ||
HyPPIs: imprinting and binding features with the fluorescent BSA
For the preparation of HyPPI, the monomers and all synthetic conditions selected for the HydroMIP setup were used to incorporate an AARE inside the recognition cavity. For the optimization experiments with HyPPI, the AARE sequences were synthesized without a dansyl reporter and are defined as SAp-Oall (the Serum Albumin peptide with an allyl group), (Scheme 1). SAp-Oall is added to the template in 1:1 stoichiometry during the pre-polymerization step and results in a very low quantity with respect to the monomers (molar ratio Sap-Oall/monomers = 1/105), and so a significant influence on the imprinting process is not expected. The adsorption features of the fluorescein-conjugated template (BSA–FITC) were analyzed via confocal microfluorimetry both in HydroMIP and in HyPPI, and compared to evaluate the contribution of SAp-Oall in the recognition properties. The microscopic images of the imprinted polymer microparticles after being soaked in BSA solutions of different concentrations are shown in Fig. 3a.
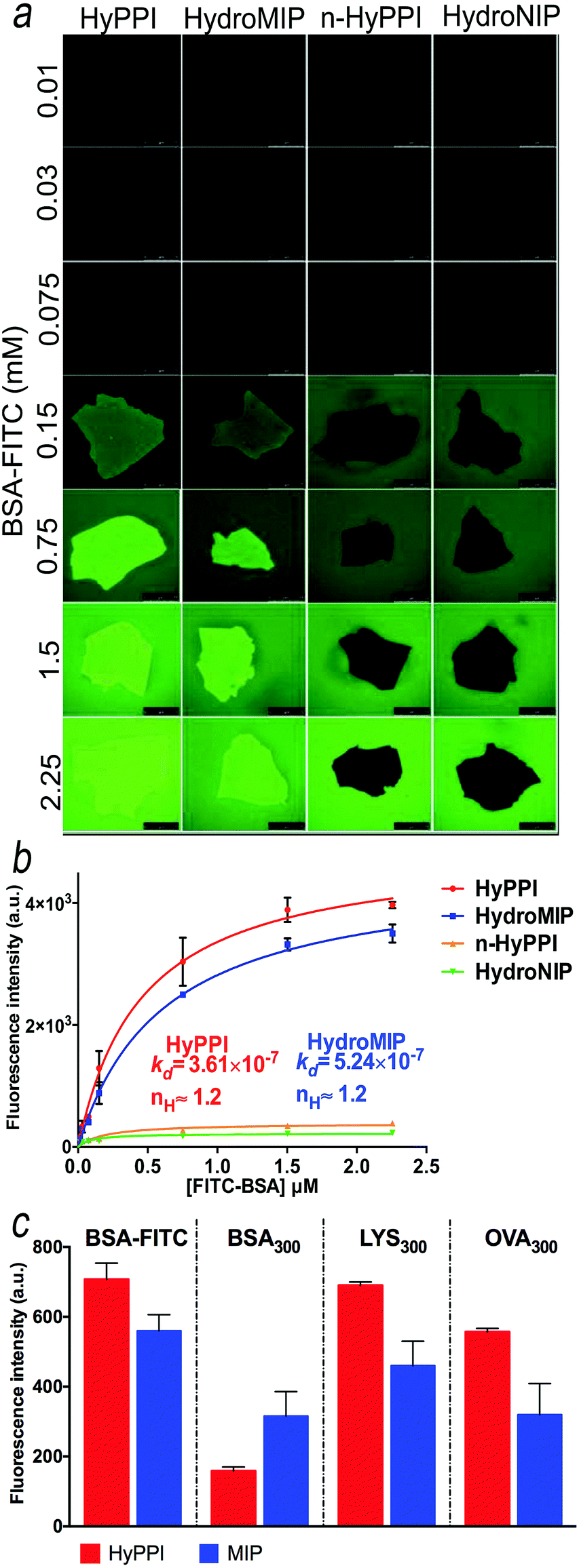 | ||
| Fig. 3 Direct and competitive adsorption assays – (a) micrographs of the binding of BSA–FITC on imprinted materials; (b) the plot of fluorescence intensity (taken from a ROI inside the polymer) versus the concentration of BSA–FITC. HyPPI and HydroMIP curves, after subtraction of the non-specific binding (e.g. n-HyPPI and HydroNIP, respectively), were fitted by Hill's equation (data not shown, KD values are expressed as mol L−1). Scale bars are 50 μm; (c) comparison of the competition adsorption ability of BSA–FITC on HydroMIP and HyPPI in the presence of a 300-fold excess of BSA, LYS and OVA. | ||
To study the affinity of HydroMIP and HyPPI (and related not-imprinted controls), the fluorescence emission of the labeled template versus its concentration (in the range of 0.01 to 2.25 μM) is plotted (Fig. 3b) and fitted with Hill's equation, allowing estimation of the dissociation binding constants in a low micromolar range. Notably, the Hill coefficient (nH), equal to 1.2, is indicative of the site cooperativity during the recognition process for both HyPPI and HydroMIP, an effect that is already reported for protein imprinted polymers due to the multiple interactions and heterogeneous distribution of binding sites.43,44 However, considering the ratio between the affinity constants (KD) determined from the curve-fitting in Fig. 3b (KD HyPPI = 3.61 × 10−7 M and KD HydroMIP = 5.24 × 10−7 M), a 45.15% improvement of HyPPI performance is demonstrated.
Furthermore, in order to probe the role of the peptide in a selective recognition process, selectivity experiments were conducted using binary mixtures of proteins, more representative of the environments in which these polymers would actually be used.45,46 Specifically, we carried out competitive binding experiments to evaluate the re-binding efficiency of BSA–FITC (at a fixed concentration) in the presence of one of these three different competitive proteins (at different molar ratios) using microfluorimetry: the same BSA (not-fluorescent) and another two interfering proteins with different molecular weights and isoelectric points. Specifically, we used lysozyme (LYS), a small (with a molecular weight of 14 kDa) surface-charged protein, and ovalbumin (OVA), a 44 kDa protein containing a high percentage of homologous sequences with BSA. Moreover, OVA was unable to interact with SAps during the phage display screening.37 As shown in Fig. 3c and in Fig. S11 and S12 (ESI†), HyPPI exhibited a higher selectivity than HydroMIP. Specifically, in the presence of a 300-fold excess of BSA, the fluorescence intensity of BSA–FITC halves in the case of HydroMIP. Under the same conditions, the fluorescence intensity of BSA–FITC in HyPPI is 75% lower than that of the control, confirming a more selective cavity. In the case of LYS, no statistical differences were recorded. LYS is positively charged at a pH below its pI value, which would lead to remarkable electrostatic interactions between the protein and the formed polymers. However, in the presence of LYS, BSA adsorption is not affected because of the specific interactions occurring between the BSA and HyPPI cavities. In contrast, in the presence of an excess of OVA, the HyPPI still adsorbs BSA–FITC even if in a lower amount (85%), and the same is true in the case of HydroMIP although the amount of BSA–FITC adsorbed is even less (about 50%). All these results are evidence that HydroMIPs are able to impart selectivity on the basis of size and charge as in the case of LYS, while for homologous sequences (BSA and OVA), a lower selectivity is reported.44 Instead, for HyPPIs a combination of a memory effect in size, conformation, chemical properties and multiple complementary interactions played an important role in BSA recognition. As a result, the imprinted polymers preferentially bind BSA with respect to homologous proteins. This can be ascribed to the AARE which guides the recognition of the template and confers to HyPPIs important discriminating features.
Dansyl-HyPPI: fluorescence transduction of BSA binding
Dansyl-HyPPI (i.e. hybrid dansyl-peptide–polymer imprint) was synthesized to obtain highly selective and responsive imprinted materials. In this case we would exploit the capability of AAREs to effectively orientate the dye and to directly report the binding event. Specifically, the AARE was introduced in the synthesis of the hybrid cavities by replacing SAp-Oall under the same polymerization conditions. The responsiveness of dansyl-HyPPIs to template rebinding is directly evaluated by interrogating tiny amounts of material (1 mg) excited at 330 nm with a spectrofluorometer. It is worth noting that high fluorescence intensity in the imprinted (Fig. 4a inset) and not imprinted (Fig. S13, ESI†) hydrogels is recorded, and their emission wavelengths shifted to a more blue region (440 nm and 410 nm, respectively) compared to that of the AARE (540 nm). This behavior indicates a strong hydrophobic interaction between dansyl molecules and polymer chains in both cases. However, after BSA incubation, the fluorescence intensity of dansyl-HyPPI considerably increases with a dose-dependent behavior, as shown in Fig. 4a. This suggests that the sensitivity of dansyl to the presence of BSA is still retained after its co-polymerization in the hydrogel network and that the reporter is properly located and oriented within the cavity for effective sensing. A linear dynamic range of 0.20–2.65 μM was explored. As a reference sample, we considered the dansyl-HyPPI_negativeCTRL (where n-AARE is used in the imprinting process) and in this case no increase of fluorescence intensity and no shift of the wavelength of maximum emission were observed upon BSA adsorption (Fig. 4a). The KD determined by fitting the dansyl-HyPPI data with the standard Hill's equation was 1.8 ± 0.5 μM. The resulting limit of detection was about 0.56 μM. The sensitivity factor (Sf) of these hydrogels for BSA was calculated by Sf = ΔF/F0, in which F0 is the fluorescence intensity of the background signal.47 The Sf values of dansyl-HyPPI and dansyl-HyPPI_negativeCTRL for BSA were 0.65 and 0.03, respectively. The imprinted sensitivity (IS), from the ratio of Sf(dansyl-HyPPI)/Sf(dansyl-HyPPI_negativeCTRL), was equal to 21 and confirmed the high responsiveness of the material to the target, attributable to the suitable location of the specific peptide inside the cavity. Furthermore the selectivity of the dansyl-HyPPI was evaluated by comparing the optical response of the BSA imprinted polymers to that of other proteins. Specifically, we carried out direct adsorption experiments to evaluate the re-binding efficiency of OVA and LYS (exploring a concentration range between 0 and 2.25 μM) to the dansyl-HyPPIs.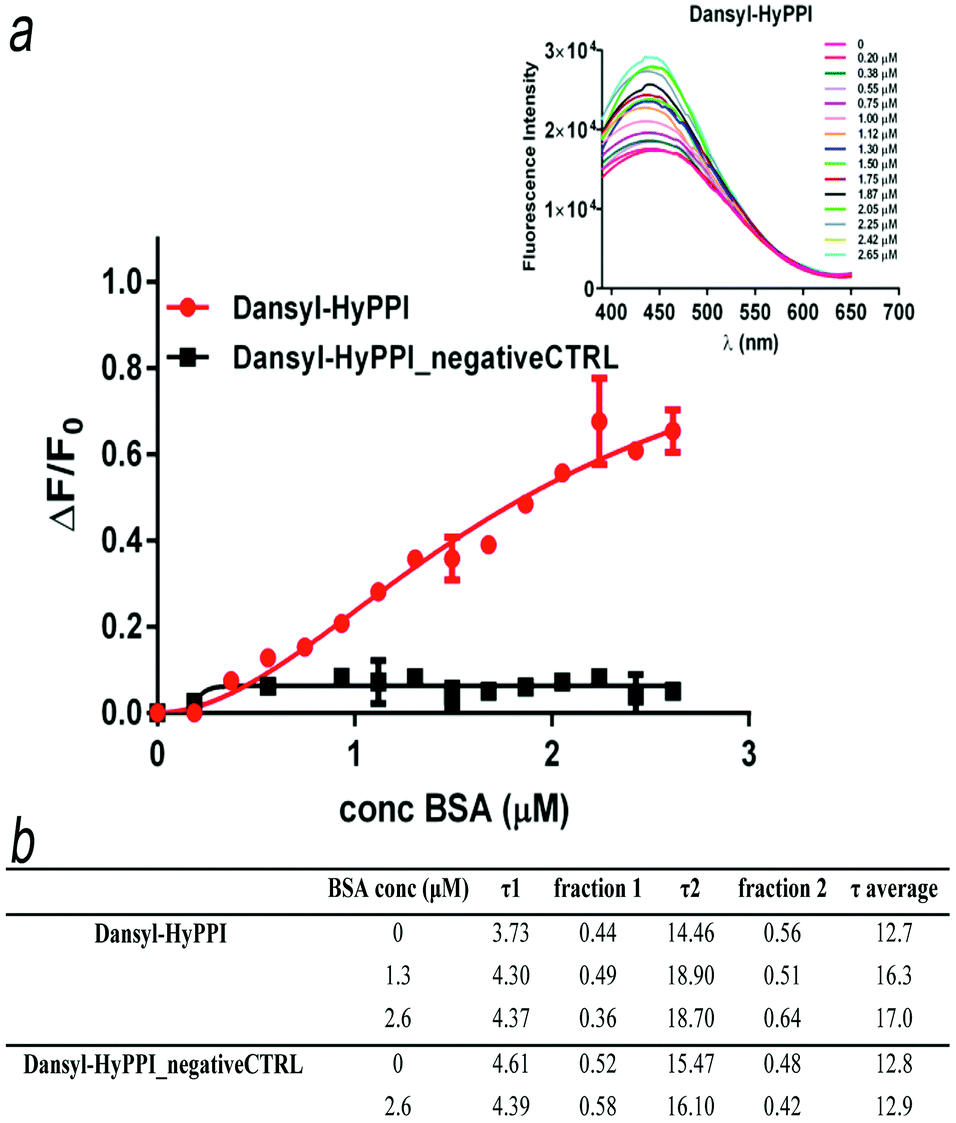 | ||
| Fig. 4 “Turn on” fluorescence – (a) ΔF/F0 plots for dansyl-HyPPI and dansyl-HyPPI_negativeCTRL titrated with BSA solutions ranging from 0 to 2.65 μM. The inset represents an overlay of spectra from the BSA titration of dansyl-HyPPI; (b) lifetime values of AAREs co-polymerized in the polymer matrix. | ||
No enhancements in fluorescence intensity and no shift of the maximum wavelength of the band were observed upon LYS adsorption (Fig. 5a).
 | ||
| Fig. 5 The fluorescence response of the dansyl-HyPPI to (a) lysozyme and (b) ovalbumin titrations. (c) A plot of ΔF/F0 as a function of both protein concentrations. | ||
Instead, the binding of the competitor OVA to dansyl-HyPPI was considerably weaker, and a plateau was not reached, showing a lower affinity and binding capacity than that of BSA (Fig. 5b). In Fig. 5c, a plot of ΔF/F0 as a function of both protein concentrations is reported. The selectivity (α) of the imprinted hydrogel was evaluated using the ratio of sensitivity of BSA to other proteins, α = Sf(BSA)/Sf(protein). The α(BSA/OVA) value was 1.25 and α(BSA/LYS) was 6.02 for the dansyl-HyPPI, confirming the good selectivity for imprinted molecules of these new imprinted hydrogels.
Furthermore, in the analysis of the dansyl-HyPPIs lifetimes without BSA, two components were identified, corresponding to τ1 = 3.73 ns and τ2 = 14.46 ns. The increased value of τ2 with respect to that evaluated in solution (Fig. 4b) again reflects the different microenvironment experienced by the dansyl electronic states once in the cavities. After protein incubation, a considerable increase in the τ2 values was observed at high BSA concentrations (up to 18.70 ns). As a consequence, the average lifetime for dansyl-HyPPI changes from 12.7 ns to 17.0 ns, at saturation concentrations. As result, it is possible to analytically quantify the presence of the protein by following the τ2 signal. In contrast, the corresponding dansyl-HyPPI_negativeCTRL showed no sensitivity toward protein titration. Dansyl HyPPI non-radiative decay constants, calculated according to quantum mechanical analysis, were 0.0326 and 0.025 ns−1 for BSA concentrations of zero and 2.6 μM, respectively. These values are smaller than those obtained for the AARE peptide in solution, demonstrating that fluorescence quenching mechanisms are more inhibited in the presence of the polymer, as previously indicated by experimental data (Fig. 4a inset and Fig. S13, ESI†).47 Here a fluorescence “turn-on” mechanism inside imprinted hydrogels was successfully implemented in a straightforward way by exploiting a supramolecular complex between the target template and the specific peptide derivatized with a reporter unit consisting of a solvatochromic dye. As result a clear relationship between the protein concentrations and lifetime average/emission intensity has been demonstrated, giving such a material efficient self-reporting properties.
Conclusions
We have reported a facile method to generate a protein imprinted polymer with chemically-controlled recognition cavities. Such an approach allows a priori in-cavity modification, through the integration of peptide multi-functional blocks rationally designed to include three different units: a peptide binder that plays a role in BSA recognition; an allyl moiety useful for its inclusion into polymer networks and a dansyl derivative responsible for the fluorescence reporting. Such an approach relies on an easy modification of conventional imprinting methods introducing a pre-polymerization step to allow for supramolecular assembly, in order to appropriately place the peptide inside the cavity. As a proof of concept of such an approach, we introduced a single peptide for the BSA protein.The assembly of a peptide block with the BSA protein drives the suitable in-cavity location of all functional units for both improved selective recognition and effective sensing of the protein. Specifically, the as obtained hybrid peptide–polymer protein imprinted materials showed enhanced binding capability and a remarkable selectivity in the presence of interfering proteins (OVA and LYS). Such a material also proved its ability to self-report the protein-binding event, acting as a fluorescence biosensor. Indeed, optical response variations (in terms of fluorescence intensity and lifetime) upon the rise in BSA concentration are ascribable to an effective interaction between the protein and environment-sensitive dansyl molecule, after defined spatial positioning inside the cavity.
Along these lines, it is possible to realize multi-point, noncovalent interactions through the use of a mixture of affine sequences during the pre-polymerization phase, which could target different protein epitopes and predictably further increase the protein affinity and selectivity.
For the development of effective synthetic receptors, the arrangement in 3D space of the different units, acting as functional sites, is an important factor and can benefit a highly cooperative combination of interactions. In this regard, the use of peptide molecules as scaffolds in polymer imprinting is useful to restrict the molecular orientation of active sites. Because of their easy chemical synthesis, peptide molecules can implement many types of reactive groups for a plethora of chemical cross-linking and transduction strategies. The proposed approach provides the basis for a new and general strategy, applicable to any target, for sensing, chromatography or catalysis purposes.
Experimental section
Materials
Acrylamide (AAm), acrylic acid (AAc), methylene bis-acrylamide (BIS), bovine serum albumin (BSA), sodium chloride (NaCl), BSA–fluorescein isothiocyanate conjugate (FITC–BSA), lysozyme (Lys), ovalbumin (OVA), fluorescein isothiocyanate (FITC), disodium carbonate (Na2CO3), potassium persulfate (KPS) and N,N,N′,N′-tetramethylethylenediamine (TEMED) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phosphate buffer saline (PBS) was provided as tablets by MP Biomedicals and was always used at pH 7.4 and 0.01 M. Reagents for peptide synthesis (Fmoc-protected amino acids, resins and activation and deprotection reagents) were from Novabiochem and InBios. Solvents for peptide synthesis and HPLC analyses were from Romil.Polymer synthesis and characterization
The setup of the recipe and the relative amounts of the monomers (acrylamide and bis-acrylamide) have been optimized according to two key parameters: T% and C% (respectively total monomer % and cross-linker %) (see the ESI,† and Table S1). Specifically, we alternately varied the amount of AAm and AAc between 3–9% and 1.5–4.5% (expressed in w/v%), respectively, while varying the cross-linker amount between 0.5–3% to obtain different preparations tagged as T% and C% (see the ESI†). In addition, taking into account the reactivity ratio of the monomers, the ratio of AAm to AAc was fixed at AAm/AAc 2:1. The optimized HydroMIP formulation was 9% AAm, 4.5% AAc and 1.5% BIS (expressed in %w/v) added to a BSA solution at a monomer/template molar ratio (M/T) fixed at 10000:1. AAm, AAc and BIS were mixed and neutralized (pH ∼ 7) with 1M NaOH and then added to a buffered BSA solution, under gentle stirring for 30 min. Polymerization was initiated by purging the solution with N2 for 3 min and adding potassium persulfate (KPS, 0.6 w/v% monomers) to generate free radicals, and was catalyzed by N,N,N′,N′-tetramethylethylenediamine (TEMED, 0.8 w/v% monomers), under vigorous magnetic stirring and a continuous nitrogen stream (for 30 min at RT).12,21 The synthesis of HyPPIs was carried out with the same recipe of HydroMIPs using a BSA/SAp-Oall complex (or BSA/AARE complex) instead of the BSA solution, in a1:1 molar ratio as provided by ITC (Isothermal Titration Calorimetry) analysis. In any case, the polymerization was terminated by transferring the monolithic gel in a beaker, adding 90 mL of PBS and homogenizing for 15 min in an ice bath to produce particles. The resulting microparticles were collected by centrifugation and washed repeatedly to remove: (i) the protein templates, (ii) the adsorbed oligomers and (iii) the unreacted monomers (Fig. S1, ESI†). The chemical, morphological and dimensional characterizations of the particles are reported in Fig. S2–S4 in the ESI.† Control polymers, defined as HydroNIPs (hydrogel non-imprinted polymers) and n-HyPPIs (non Hybrid Peptide–Polymer Imprints) were prepared exactly under the same conditions without BSA.
Active Assistant Recognition Elements (AAREs) design, synthesis and binding characterization
The Active Assistant Recognition Elements (AAREs) are peptides designed and prepared by a solid phase method following the Fmoc strategy on a Rink amide resin as reported elsewhere47 (Table S2, ESI†). The allyl group at the C-terminus was incorporated as Fmoc-Asp(Oallyl)-OH (N-α-(9-fluorenylmethyloxycarbonyl)-L-aspartic acid β-allyl ester) and spaced from the active core sequence by Fmoc-βAla-OH (N-(9-fluorenylmethyloxycarbonyl)-β-alanine). At the N-terminus, after the sequential addition of two residues of Fmoc-βAla-OH, the dansyl group was introduced using the modified amino acid Fmoc-Lys(dansyl)-OH (N-α-(9-fluorenylmethyloxycarbonyl)-N-ε-dansyl-L-lysine). Following this, the peptide purity and identity were confirmed by liquid chromatography mass-spectrometry (HPLC-MS) analyses on an Agilent 6530 Accurate-Mass Q-TOF LC/MS spectrometer, as shown in Fig. S5 (ESI†). The identity of the AARE was confirmed by NMR experiments. H NMR assignment of the AARE was carried out by means of TOCSY and NOESY experiments (Fig. S6, ESI†). The binding characterization between BSA and the peptides was performed through ITC, SPR (Surface Plasmon Resonance) and fluorescence/lifetime experiments (Fig. S7–S9, ESI†).Computational analysis of fluorescence features by TD-DFT
The investigation of the ground and excited state potential energy surfaces of a dansyl-peptide model system was carried out through density functional theory (DFT) and its time dependent version (TD-DFT), respectively. Solvent effects were described by the polarizable continuum model in its conductor-like version (CPCM), considering water (ε = 78.36) and cyclohexane (ε = 2.02) as solvents.48 The B3LYP/6-31+G(d,p)/CPCM and TD-B3LYP/6-31+G(d,p)/CPCM levels of theory were adopted to locate the ground and excited state minimum energy structures. All calculations were performed with the Gaussian09 suite of programs.49Microfluorimetry for binding and selectivity studies
Batch re-binding tests were performed using microfluorimetry on a confocal laser-scanning microscope.50 The albumin fluorescein isothiocyanate conjugate (BSA–FITC), in the 0.01–2.25 μM range, was used to directly visualize the adsorption in the polymer matrix (5 mg) suspended in 0.5 mL of PBS buffer. Selectivity studies were carried out by performing competition assays through the addition of binary protein mixtures of BSA–FITC, and alternately BSA, lysozyme (LYS) and ovalbumin (OVA), to a suspension including polymer gel particles. All captured images were analyzed with public domain image-processing ImageJ software. For details see the ESI.†Steady-state and lifetime fluorescence studies for BSA/dansyl-HyPPIs interactions
Fluorescence emission spectra and lifetime values of dansyl-HyPPIs and those in complex with BSA were collected in a quartz cell using a spectrofluorometer (FluoroMax-4 Horiba Scientific). Selectivity experiments were carried out through lysozyme or ovalbumin adsorption, exploring the 0–2 μM range. For all details see the ESI.†Statistical analysis
All data are presented as means ± standard deviation (S.D.). The significant differences between groups were evaluated with Tukey's method after analysis of variance (ANOVA). In all statistical analyses, **p < 0.05 was considered to be statistically significant.Conflicts of interest
The authors declare no competing conflicts of interest.Acknowledgements
The authors would like to acknowledge Dr Luca Raiola for NMR spectra acquisition and interpretation.References
- C. S. Mahon and D. A. Fulton, Nat. Chem., 2014, 6, 665–672 CrossRef CAS PubMed.
- R. Xing, S. Wang, Z. Bie, H. He and Z. Liu, Nat. Protoc., 2017, 12, 964–987 CrossRef CAS PubMed.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- D. Cai, L. Ren, H. Zhao, C. Xu, L. Zhang, Y. Yu, H. Wang, Y. Lan, M. F. Roberts, J. H. Chuang, M. J. Naughton, Z. Ren and T. C. Chiles, Nat. Nanotechnol., 2010, 5, 597–601 CrossRef CAS PubMed.
- M. Yoshikawa, K. Tharpa and Ş.-O. Dima, Chem. Rev., 2016, 116, 11500–11528 CrossRef CAS PubMed.
- J. Q. Liu and G. Wulff, J. Am. Chem. Soc., 2008, 130, 8044–8054 CrossRef CAS PubMed.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- Z. Zhang, X. Zhang, B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 5412–5419 CrossRef CAS PubMed.
- J. Orozco, A. Cortés, G. Cheng, S. Sattayasamitsathit, W. Gao, X. Feng, Y. Shen and J. Wang, J. Am. Chem. Soc., 2013, 135, 5336–5339 CrossRef CAS PubMed.
- I. E. Tothill, Semin. Cell Dev. Biol., 2009, 20, 55–62 CrossRef CAS PubMed.
- D. R. Kryscio and N. A. Peppas, Acta Biomater., 2012, 8, 461–473 CrossRef CAS PubMed.
- S. M. Reddy, Q. T. Phan, H. El-Sharif, L. Govada, D. Stevenson and N. E. Chayen, Biomacromolecules, 2012, 13, 3959–3965 CrossRef CAS PubMed.
- F. Causa, A. Aliberti, A. M. Cusano, E. Battista and P. A. Netti, J. Am. Chem. Soc., 2015, 137, 1758–1761 CrossRef CAS PubMed.
- H. F. El-Sharif, D. M. Hawkins, D. Stevenson and S. M. Reddy, Phys. Chem. Chem. Phys., 2014, 16, 15483 RSC.
- R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 829–835 CrossRef CAS PubMed.
- S. Shinde, Z. El-Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. Gjorloff Wingren and B. Sellergren, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef CAS PubMed.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC.
- M. Cieplak and W. Kutner, Trends Biotechnol., 2016, 34, 922–941 CrossRef CAS PubMed.
- D. R. Kryscio, M. Q. Fleming and N. A. Peppas, Macromol. Biosci., 2012, 12, 1137–1144 CrossRef CAS PubMed.
- N. M. Bergmann and N. A. Peppas, Ind. Eng. Chem. Res., 2008, 47, 9099–9107 CrossRef CAS.
- H. F. El-Sharif, H. Yapati, S. Kalluru and S. M. Reddy, Acta Biomater., 2015, 28, 121–127 CrossRef CAS PubMed.
- J. Gao, H. Tian, Y. Wang, Q. Yang, D. Liu, Y. Wang and H. Mi, Biomaterials, 2012, 33, 3344–3352 CrossRef CAS PubMed.
- Z. Tao, E. C. Tehan, R. M. Bukowski, Y. Tang, E. L. Shughart, W. G. Holthoff, A. N. Cartwright, A. H. Titus and F. V. Bright, Anal. Chim. Acta, 2006, 564, 59–65 CrossRef CAS PubMed.
- Y. Inoue, A. Kuwahara, K. Ohmori, H. Sunayama, T. Ooya and T. Takeuchi, Biosens. Bioelectron., 2013, 48, 113–119 CrossRef CAS PubMed.
- O. Hayashida, N. Ogawa and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 13698–13705 CrossRef CAS PubMed.
- A. T. Krueger and B. Imperiali, ChemBioChem, 2013, 14, 788–799 CrossRef CAS PubMed.
- Y. Suga, H. Sunayama, T. Ooya and T. Takeuchi, Chem. Commun., 2013, 49, 8450 RSC.
- H. Sunayama, T. Ooya and T. Takeuchi, Chem. Commun., 2014, 50, 1347–1349 RSC.
- S. Pavan and F. Berti, Anal. Bioanal. Chem., 2011, 402, 3055–3070 CrossRef PubMed.
- M. Sainlos and B. Imperiali, Nat. Protoc., 2007, 2, 3201–3209 CrossRef CAS PubMed.
- J. Matsui, J. Nagano, D. Miyoshi, K. Tamaki and N. Sugimoto, Biosens. Bioelectron., 2009, 25, 563–567 CrossRef CAS PubMed.
- A. Takata, K. Usui and J. Matsui, Mol. Imprinting, 2015, 3, 65–70 CAS.
- M. Morell and J. Puiggalí, Polymers, 2013, 5, 188–224 CrossRef.
- A. S. Carlini, L. Adamiak and N. C. Gianneschi, Macromolecules, 2016, 49, 4379–4394 CrossRef CAS PubMed.
- J. Kopecek and J. Yang, Angew. Chem., Int. Ed. Engl., 2012, 51, 7396–7417 CrossRef CAS PubMed.
- M. S. Dennis, M. Zhang, Y. G. Meng, M. Kadkhodayan, D. Kirchhofer, D. Combs and L. A. Damico, J. Biol. Chem., 2002, 277, 35035–35043 CrossRef CAS PubMed.
- M. K. Gilson and B. H. Honig, Biopolymers, 1986, 25, 2097–2119 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato and W. W. Parson, Biochim. Biophys. Acta, 2006, 1764, 1647–1676 CrossRef CAS PubMed.
- Y. Hu, K. Horie, H. Ushiki, F. Tsunomori and T. Yamashita, Macromolecules, 1992, 25, 7324–7329 CrossRef CAS.
- M. Savarese, A. Aliberti, I. De Santo, E. Battista, F. Causa, P. A. Netti and N. Rega, J. Phys. Chem. A, 2012, 116, 7491–7497 CrossRef CAS PubMed.
- M. Savarese, U. Raucci, P. A. Netti, C. Adamo, I. Ciofini and N. Rega, Chem. Phys. Lett., 2014, 610–611, 148–152 CrossRef CAS.
- N. Lavignac, K. R. Brain and C. J. Allender, Biosens. Bioelectron., 2006, 22, 138–144 CrossRef CAS PubMed.
- H. F. El-Sharif, D. M. Hawkins, D. Stevenson and S. M. Reddy, Phys. Chem. Chem. Phys., 2014, 16, 15483–15489 RSC.
- J. Liu, Q. Deng, D. Tao, K. Yang, L. Zhang, Z. Liang and Y. Zhang, Sci. Rep., 2014, 4, 5487 CrossRef CAS PubMed.
- H. R. Culver, S. D. Steichen and N. A. Peppas, Biomacromolecules, 2016, 17, 4045–4053 CrossRef CAS PubMed.
- A. M. Cusano, F. Causa, R. D. Moglie, N. Falco, P. L. Scognamiglio, A. Aliberti, R. Vecchione, E. Battista, D. Marasco, M. Savarese, U. Raucci, N. Rega and P. A. Netti, J. R. Soc., Interface, 2014, 11, 20140718 CrossRef PubMed.
- S. Leone and D. Picone, PLoS One, 2016, 11, e0158372 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. M. Hawkins, A. Trache, E. A. Ellis, D. Stevenson, A. Holzenburg, G. A. Meininger and S. M. Reddy, Biomacromolecules, 2006, 7, 2560–2564 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb03107f |
| This journal is © The Royal Society of Chemistry 2018 |