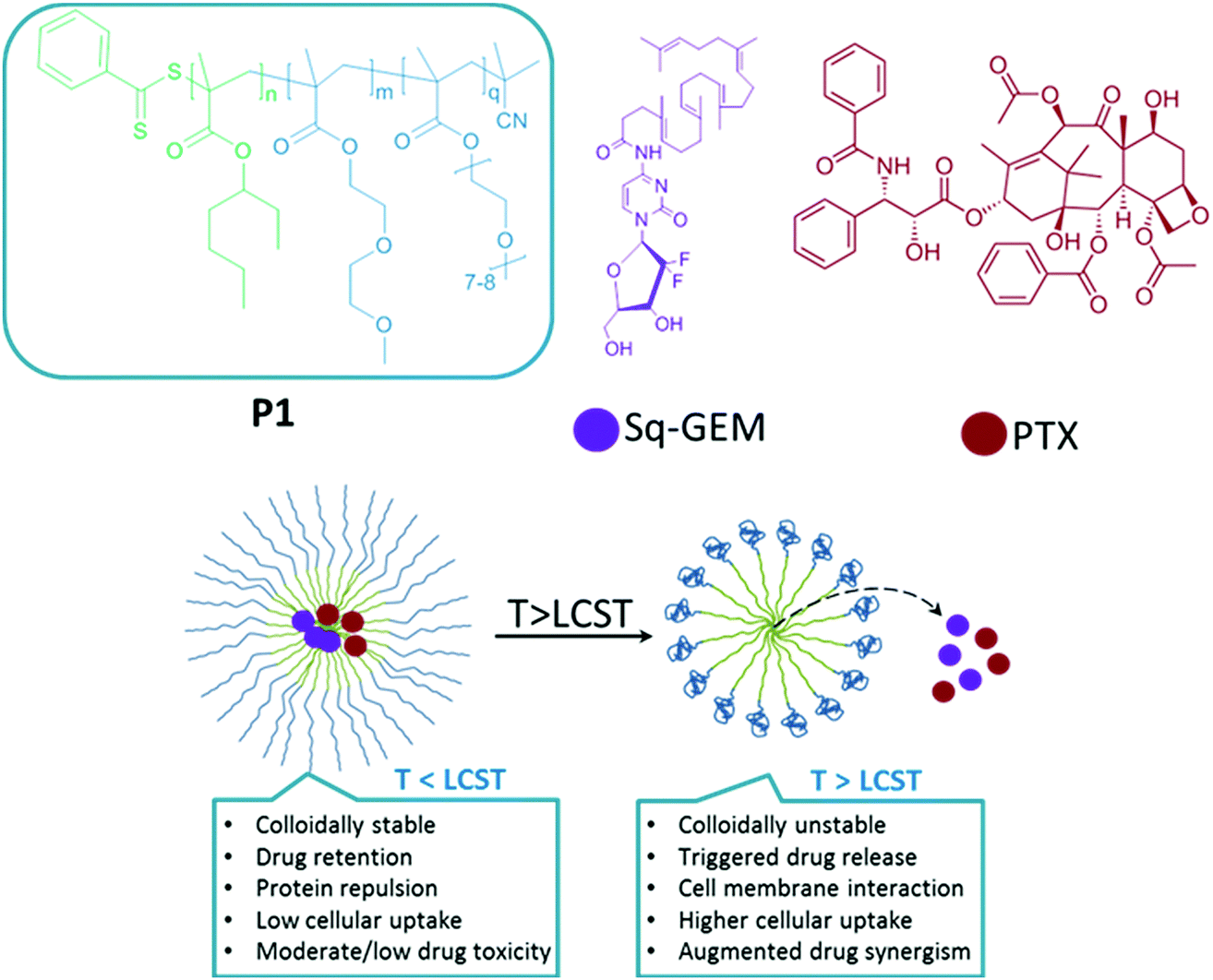
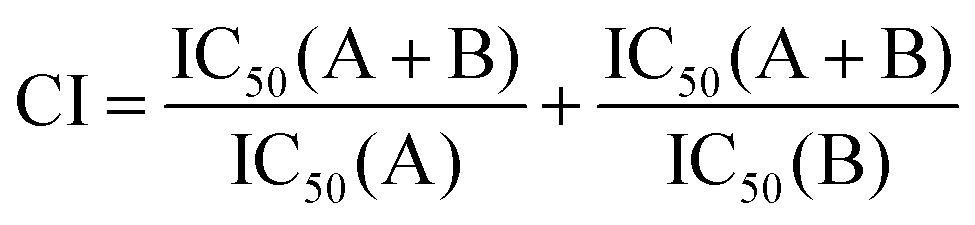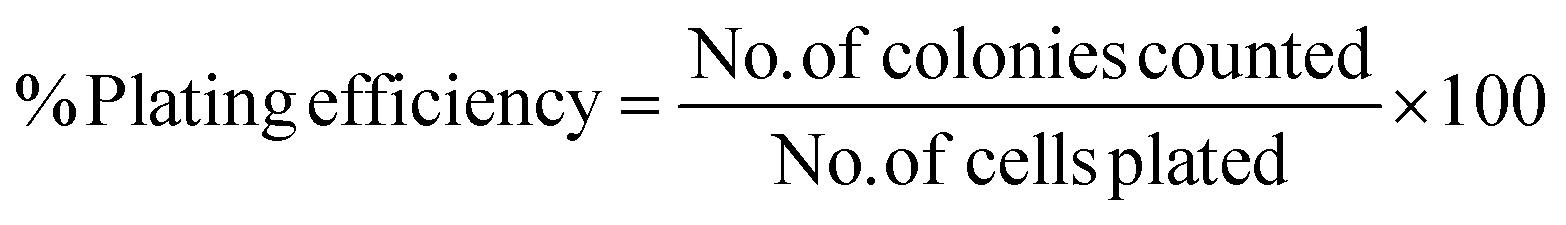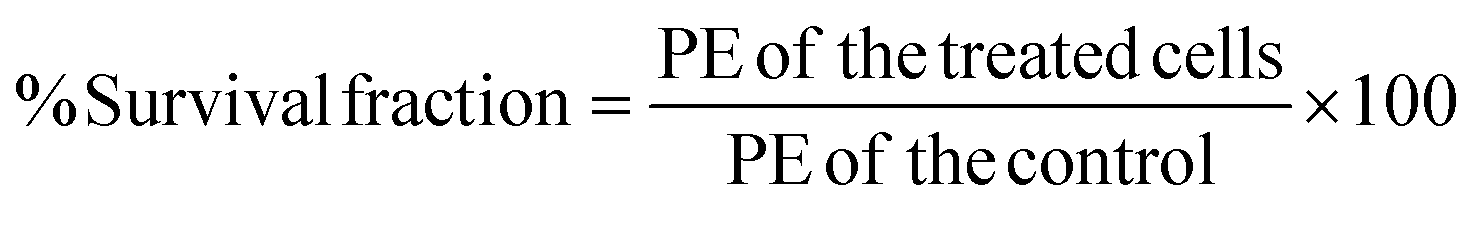Dual controlled delivery of squalenoyl-gemcitabine and paclitaxel using thermo-responsive polymeric micelles for pancreatic cancer†
Received
7th November 2017
, Accepted 9th March 2018
First published on 28th March 2018
Abstract
In this study we report the synthesis of a themroresponsive block copolymer by reversible addition fragmentation transfer polymerization comprising poly(2-ethylhexyl methacrylate)-b-poly[di(ethylene glycol)methyl ether methacrylate-co-oligo(ethylene glycol)methyl ether methacrylate] as hydrophobic and thermoresponsive blocks respectively. The polymer self-assembles into sub-50 micelles and can carry simultaneously two drug molecules, namely squalene-gemcitabine and paclitaxel. Both drugs can be released from the micellar compartment in a thermally controlled manner owing to the controllable disruption of the micellar corona above the lower critical solution temperature of the polymer. We demonstrate that the formulation augments synergistically the cytotoxicity of the two drugs in vitro against a model pancreatic cancer cell line. More importantly, it is shown that the polymer exerts a direct interaction with the cell membrane which further augments the cytotoxicity of the drug cargo in a thermally controlled manner.
Introduction
Pancreatic cancer is the 4th most lethal cancer in Europe with disappointing prognosis and survival rates for 1, 5, and 10 years patients not exceeding 21%, 3%, and 1%, respectively1,2 – nearly the same since the early 1970s. More than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 patients are diagnosed with pancreatic cancer in Europe annually which is now ranked as the 9th most common cancer in Europe and the 4th most common cause of cancer death in the Western world. Gemcitabine (2,2-difluoro 2-deoxycytidine) (GEM) is the front line drug for pancreatic cancer, with relatively moderate therapeutic potency due to poor pharmacokinetics, high dosage required for effective therapy, and the rapid development of drug resistance.3,4 GEM is a highly soluble molecule that is rapidly de-aminated (intra- and extra-cellularly) by cytidine deaminase (CDA) – which is abundant in blood plasma and the liver- and results in the non-cytotoxic metabolite 2-deoxy-2,2-difluorouridine.5 As a result, GEM has an extremely low plasma half-life (<60 minutes, in human subjects, data can be accessed here6) and hence, to compensate for the poor pharmacokinetics, GEM is administered at very high doses via 30 minute intravenous infusion sessions at >1000 mg m−2.
000 patients are diagnosed with pancreatic cancer in Europe annually which is now ranked as the 9th most common cancer in Europe and the 4th most common cause of cancer death in the Western world. Gemcitabine (2,2-difluoro 2-deoxycytidine) (GEM) is the front line drug for pancreatic cancer, with relatively moderate therapeutic potency due to poor pharmacokinetics, high dosage required for effective therapy, and the rapid development of drug resistance.3,4 GEM is a highly soluble molecule that is rapidly de-aminated (intra- and extra-cellularly) by cytidine deaminase (CDA) – which is abundant in blood plasma and the liver- and results in the non-cytotoxic metabolite 2-deoxy-2,2-difluorouridine.5 As a result, GEM has an extremely low plasma half-life (<60 minutes, in human subjects, data can be accessed here6) and hence, to compensate for the poor pharmacokinetics, GEM is administered at very high doses via 30 minute intravenous infusion sessions at >1000 mg m−2.
Recently, the combination of GEM with other chemotherapeutic agents has been explored as an aggressive means to maximize the potency of therapeutic protocols, with the most notable examples the FOLFIRINOX7 protocol, and the co-administration of GEM with paclitaxel (in the form of an albumin carrying PTX vehicle, nab-PTX). In a large scale phase III clinical study, it was found that the rate of survival was significantly higher in the nab-PTX–GEM group than in the GEM group—by 59% at 1 year (35% vs. 22%) and by more than 100% at 2 years (9% vs. 4%).8 Interestingly, nab-PTX was found to improve the tumor uptake of GEM in solid tumors and increased the levels of the non-deaminated active molecule by suppressing the activity of CDA through the activation of intracellular reactive oxygen species (ROS) secretion;9 in addition, PTX was found to increase the permeability of the tumors by decreasing the stiffness and loosening of the tumor outer stroma.10 These findings imply that the co-delivery of GEM with nab-PTX could further enhance the therapeutic efficiency by exerting augmented synergism between the two drugs.
Therefore, we hypothesized that the development of drug delivery systems that co-carry multiple GEM and PTX molecules in a single carrier could augment the synergistic activity of the two drugs and also, possibly, alleviate the pharmacokinetic limitations, such as the rapid deamination of GEM in blood plasma, and the absence of alternative formulations of nab-PTX, especially given the difficulty to formulate the PTX molecule in more compliant forms (it is currently administered in alcohol/castor oil mixtures).11
Of particular interest are thermoresponsive micelles comprising the use of polymeric building blocks that exhibit a so called lower critical solution temperature (LCST).12–14 These polymers exist in well hydrated form below the LCST, and dehydrate above the LCST which in turn induces a collapsing of the micellar compartment leading to triggered release of the molecular cargo. Therefore, in a therapeutic scenario, these micelles can (1) protect the drug molecules from premature hydrolysis/deactivation (as in the case of GEM) (2) carry multiple drug molecules (3) be accumulated at the tumor sites at higher rates compared to other tissues/organs owing to the enhanced permeation and retention effect (EPR) which is observed in certain animal models (and partly in pancreatic cancer).15–19
In principle, this concept could significantly improve the delivery of drugs with a narrow therapeutic index and more significantly elevate the maximum tolerated dose. Interestingly, this approach becomes even more relevant in the case of multi-drug therapies where cumulative toxicity is a severe limitation; surprisingly, the number of studies on thermoresponsive micelles used for multidrug therapeutics has only very recently emerged despite the apparent formulation advantages they potentially exert.20
In the present study, we report on a novel thermoresponsive block copolymer that self-assembles into well defined, sub-50 nm micelles that can co-carry two drug molecules, namely, PTX and a hydrophobic derivative of GEM, that is, squalene-GEM (Sq-GEM). It is demonstrated that the polymeric micelles, not only augment the synergism of the two drugs above the polymers’ LCST, but also exert an active interaction with the cell membranes, which evidently seems to accelerate the translocation of the drug molecules in the cytoplasm. To the best of our knowledge, this is one of the very first reports on drug synergism augmentation by thermoresponsive micelles accompanied by simultaneous cell membrane interaction.
Experimental
Materials
All solvents were of analytical or HPLC grade and were purchased from Sigma-Aldrich. Double distilled water was used throughout all the experiments. 2,2′-Azobis(2-methylpropionitrile) (AIBN), 2-butanone, 2-cyano-2-propyl benzodithioate (CTA/RAFT agent), 2-ethylhexyl methacrylate (EHMA), chloroform, diethyl ether, di(ethylene glycol)methyl ether methacrylate (DiEGMA), pyrene, fetal Bovine serum (FBS), hexane, oligo(ethylene glycol)methyl ether methacrylate (OEGMA300), phosphate buffered saline (PBS), penicillin–Streptomycin, L-glutamine, Dulbecco's modified eagle's medium-high glucose (DMEM), dimethyl sulfoxide (DMSO), dimethylformamide (DMF) thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. Paclitaxel (PTX), Taxus brevifolia was purchased from Fluorochem Ltd. Squalenoyl-gemcitabine (Sq-Gem) was synthesized by our previously published synthesis route.21
One pot synthesis of poly(DiEGMA-co-OEGMA300)-b-EHMA (P1) by RAFT polymerization
DiEGMA (6 mmol, 1.107 mL), OEGMA300 (4 mmol, 1.143 mL), AIBN (0.02 mmol, 3.3 mg) and CTA (0.08 mmol, 18 mg) were dissolved in 2-butanone (8 mL) in a 25 mL round bottom flask. The reaction mixture was sealed with rubber septum and purged with argon for 10 minutes. The polymerization started by placing the vessel in an oil bath at 75 °C and was left under magnetic stirring. After 24 hours, EHMA (4 mmol, 896 μL) was dissolved in 3 mL 2-butanone purged with Ar, and introduced to the reaction via a fine needle. The reaction was stopped after 24 hours by cooling down the reaction and exposing it to air. The polymer was precipitated in excess hexane, dried under reduced pressure, and collected as a pink viscous fluid (yield: 78%). The targeted DP for hydrophilic block and hydrophobic block were 120 and 50 respectively.
Synthesis of fluorescent polymer for cellular uptake assessment
A co-polymer of DiEGMA and OEGMA300 with molar ratio of 60:40 and degree of polymerization of 120 was synthesized. DiEGMA (6 mmol, 1.107 mL), OEGMA300 (4 mmol, 1.143 mL), AIBN (0.02 mmol, 3.3 mg) and CTA (0.08 mmol, 18 mg) were dissolved in 2-butanone (8 mL) in a 25 mL round bottom flask. The reaction mixture was sealed with rubber septum and purged with argon. The polymerization started by placing the vessel in an oil bath at 75 °C and was left under magnetic stirring. After 24 hours, the hydrophobic block with degree of polymerization of 50 was synthesized by introducing an argon-purged solution (3 mL, 2-butanone) with EHMA (3.95 mmol, 885 μL) and fluorescein O-methacrylate (FOMA) (0.05 mmol, 20 mg) to the reaction via a fine needle. The reaction was stopped after 24 hours by cooling down the reaction and exposing it to air. The polymers were washed twice in excess hexane in order to dissolve unreacted monomer and precipitate synthesized polymers. The precipitated polymers were dried under reduced pressure and collected as a pink viscous fluid (yield: 73%).
Nuclear magnetic resonance spectroscopy
Liquid state 1H NMR spectra of the polymers were recorded on a Bruker NMR spectrometer (Ultrashield 400 MHz). The polymers were dissolved in deuterated chloroform (CDCl3) for analysis.
Size exclusion chromatography
Size exclusion chromatography (SEC) was conducted with DMF as the mobile phase containing 5 mM NH4BF4 additive at 70 °C at a flow rate of 1.00 mL min−1. The SEC system was equipped with a solvent pump (Viscotek VE 1121), a degasser (Viscotek VE 7510), two Styragel columns (MGHHR-M E0057 and MGHHR-M E0058) and a refractive index detector (Viscotek VE 3580). DMF was used as the mobile phase at a flow rate of 1 mL min−1. The apparatus was calibrated prior to analysis by using linear poly(methyl methacrylate) polymers with narrow molecular weight distributions as standards. Then, 100 μL of polymer aliquots in DMF (40 mg mL−1) was injected to the instrument to determine the average molecular weight (Mn) and index of polydispersity (ĐM).
Determination of the polymers’ lower critical solution temperature (LCST)
The polymers (5 mg) were dissolved in 1 mL of phosphate-buffered saline (PBS) pH 7.4 and gradually heated by immersing the solution in water bath of predetermined temperature and the LCST value was determined as the onset of optical turbidity for each sample by a Agilent Cary series UV-vis spectrophotometer at 550 nm.
Determination of the polymers’ critical micelle concentration (CMC)
The critical micelle concentration of the polymers was studied using the standard pyrene protocol.22 Briefly, 20 μL aliquots of pyrene in acetone (6.5 × 10−5 M) were added to series of glass vials and the acetone was allowed to evaporate in open air. Subsequently, 1 mL of various concentrations of block copolymer samples in water (from 0.002 to 10 mg mL−1) was added to each glass vial and left to equilibrate overnight under vigorous stirring. Fluorescent intensity of pyrene was measured using a spectrofluorometer (SpectraMax multi-mode microplate reader, molecular device) where the excitation spectra were scanned from 280 to 350 nm and emission wavelength was fixed at 370 nm.
Formulation of drug loaded micelles
The thin-film hydration method was used to load the drug molecules in the polymeric micelles. Polymer P1 (5 mg mL−1) and PTX/Sq-Gem (500 μg mL−1 each) were dissolved in acetone (2 mL) and transferred in a 10 mL round bottom flask, followed by slow vacuum drying until a thin film was formed on the walls of the flask. Subsequently, hydration with 1 mL of PBS (pH 7.4) under vigorous stirring at room temperature for 5 min took place in order to form the micelles. The as prepared micelles were further sonicated for 5 min before passing them through a 0.2 μm filter in order to isolate them as a nearly monodispersed suspension. The suspension was further centrifuged (Sigma 3–16 KL) at 13300 rpm for 10 min at room temperature to confirm the complete absence of pellet formation, indicative of virtually quantitative loading efficiency under this protocol.
HPLC analysis for drug loading and release determination
An HPLC assay for the quantification of both PTX and Sq-Gem was developed using an Agilent Technologies 1200 Series HPLC system. The chromatographic separation was achieved using a Phenomenex Synergi™ 4 μm Polar-RP 80 Å, LC Column 250 × 4.6 mm. Methanol and water (85:15) was introduced to the column as mobile phase under isocratic conditions. The mobile phase was pumped through the column at a flow rate of 1 mL min−1. The UV detector was set at 250 nm and the injection volume was 20 μL. The PTX and Gem-Sq standard solutions used for quantification were prepared by suitably diluting a 100 μg mL−1 working standard of each drug in a mixture of 80:20 methanol:water. The data was acquired and analysed using ChemStation for LC, software by Agilent Technologies, UK.
Dynamic light scattering (DLS)
Particle size measurements of the polymeric micelles were recorded using a Zetasizer Nano ZS, Malvern Instruments Ltd, UK. The scattered laser beam (632.8 nm) was measured at an angle of 175°. Samples were prepared using polymer (5 mg mL−1) and PTX/Sq-Gem (500 μg mL−1 each) dispersed in 1 mL of PBS pH 7.4 (0.01 M). All samples were measured in a quartz cuvette at 25 °C and the data were recorded by Malvern Zetasizer software 7.11.
Transmission electron microscopy (TEM)
The morphology and size of the polymeric micelles was determined by TEM on a FEI/PHILIPS CM120 BioTwin. A carbon coated copper grid was soaked by a drop of distilled water and after a few seconds it was removed by filter paper. Then a drop of the micellar suspension was placed on the grid and left to dry in open air. Before imaging, a drop of a staining solution (1% uranyl acetate reagent) was applied to enhance the contrast of the sample and a few seconds later it was also gently absorbed by filter paper. After drying, the grid was transferred to the sample holder ready to be introduced to the electron beam. The recorded imaged were analysed by ImageJ to construct a frequency histogram and to determine the average diameter of the micelles.
Determination of in vitro drug release
The drug release profile from the micelles was evaluated using a Slide-A-Lyzer dialysis cassette (MWCO 7 kDa, Spectrum laboratories). Drug loaded micelles (polymer 5 mg mL−1, and PTX/Sq-Gem 500 μg mL−1 each) dispersed in 5 mL of PBS pH 7.4 (0.01 M) were transferred in the dialysis cassette by using a syringe, which was then immersed in 200 mL PBS pH 7.4 (0.01 M) containing 1% Tween 20. The drug release experiments were performed under mild stirring below and above LCST, 37 °C and 40 °C respectively, in order to compare the drug release profiles at different temperatures. At predetermined time intervals, aliquots (0.5 mL) were withdrawn from the cassette, which were replenished with fresh PBS pH 7.4 (0.01 M). The collected samples were centrifuged (Thermo Scientific Heraeus Fresco 17) at 13300 rpm for 10 min and the drug(s) released were collected as pellets and analyzed by HPLC to measure the drug concentration released at different time intervals.
In vitro cytotoxicity assay
The cytotoxicity studies were performed with the use of the MTT assay. MiaPaca-2 cells were seeded in a 96-well plate at a density of 1 × 104 cells per well. Cells were incubated in Dulbecco's modified eagle's medium-high glucose and supplemented with 10% fetal bovine serum, 1% penicillin–Streptomycin and 1% L-glutamine. Incubation was performed either at 37 °C (below LCST) or 40 °C (above LCST) in humidified atmosphere with 5% CO2 for 24 h before the assay. Then, the medium was replaced with 200 μL medium containing either empty block copolymer micelles or loaded with 0.0001–10 μM of each drug. The free drugs were dissolved in DMSO and then diluted in the medium; briefly, concentrated stock solutions of Sq-Gem and PTX were prepared (1 mg/200 μL of DMSO). 3 μL of stock solution was diluted in 1997 μL of medium to achieve 10 μM for each drug. Further dilutions were carried out to obtain less concentrated samples. After incubation for 72 h, the medium was replaced by 100 μL of fresh medium and 25 μL of MTT stock solution (5 mg mL−1 in PBS) and incubated for an additional 4 h. Subsequently, the medium was removed and the formazan crystals were dissolved in 200 μL of DMSO. The plates were shaken for 2 min at room temperature before measuring the optical density (OD) at 570 nm on a SpectraMax® M2/M2e Multimode Microplate Reader, with SoftMax® Pro Software.
Calculation of the combination index
The combination index (CI) was investigated in order to measure the combinatorial therapeutic effect resulting from the co-delivery of PTX and Sq-Gem. CI > 1 implies antagonistic behavior, CI = 1 corresponds to additive behavior and CI < 1 represents synergistic behavior. The CI was calculated based on the IC50 values obtained from the MTT assay by using the formula (eqn (1)):| | 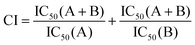 | (1) |
where IC50 (A) and IC50 (B) are the IC50 values obtained from each drug separately. IC50 (A + B) is the IC50 value of both drugs in combination.
Thermo-dependent cellular uptake of fluorescent polymer
The influence of the temperature on the cellular uptake of thermo-responsive nanoparticles below and above their thermal transition temperatures was studied using fluorescence microscopy. MiaPaCa-2 cells were seeded in 6-well plates at a concentration of 1 × 105 cells per well and incubated at 37 °C and 5% CO2 to allow the cells to attach and reach confluency. The media in each well was replaced with 2 mL of media containing 2 mg of fluorescent polymer and incubated at 37 °C (below LCST) or 40 °C (above LCST) for 30 min. The media was removed and the cells were rinsed with Dulbecco's phosphate buffered saline (DPBS) once, at room temperature. The washed cells were then fixed using 4% paraformaldehyde phosphate buffer solution for 20 min and rinsed twice with DPBS. The cells were observed in 6-well plates using EVOS® FL Imaging microscope.
Flow cytometry analysis
MiaPaCa-2 cells were plated in 6-well plate at a density of 1 × 105 cells per well. After the cells reached confluency, each well was treated with 2 mg of poly(DiEGMA-co-OEGMA300)-b-(EHMA-co-FOMA) dissolved in 2 mL of culture medium. After 30 min, the cells were washed with cold PBS (4 °C) several times to stop the intake. 1 mL of cold PBS (4 °C) was added, the cells were scraped and transferred into Nalgene® centrifuge tubes to be analyzed by MACSQuant Analyzer 10 Flow Cytometer.
Clonogenic cell survival assay
2 × 105 MiaPaCa-2 cells were seeded in 25 cm2 T-flask (using 5 mL of culture medium). Once the cells reached confluency, each flask was treated with different concentrations of drugs in combinations. One flask was remained untreated as a control. After 24 hours, the treated and untreated flasks were trypsinized, counted and subsequently 250 cells from each flask were seeded in the 6 well plate. After 14 days, the colonies were washed with PBS and then fixed using methanol and acetic acid in ratio 3:1. The colonies were stained with 0.5% crystal violet solution (diluted with methanol) for 5 min. The stained plates were rinsed in the tray full of distilled water and left in the fume hood overnight to dry. Colonies appeared as clusters of violet stained cells which could be visualized with the naked eye. The number of air-dried colonies for the average of three colony counts for each plate was recorded. A cluster of 50 or more cells was counted as one colony.
The plating efficiency (PE) was calculated (eqn (2)) by dividing the number of colonies counted by the number of cells plated and then multiplying by 100:
| | 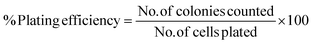 | (2) |
PE was determined to investigate the percentage of the single cells seeded in the plates that formed a colony. PE of the control was considered as 100%. By determining the PE, the survival fraction of the single cells seeded in the plates was also calculated (
eqn (3)) by dividing the PE of the treated cells by the PE of the control and then multiplying by 100:
| | 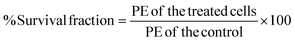 | (3) |
Results and discussion
Polymer synthesis and design rationale
Our proposed system (shown in Scheme 1) comprises the synthesis of a thermoresponsive block copolymer with a hydrophobic block serving as the core and is made of poly(2-ethylhexyl methacrylate) (p(EHMA)). This polymer synthon was chosen by preliminary experiments which were performed with lauryl-methacrylate and n-butyl methacrylate as potential hydrophobic building blocks; p(EHMA) was shortlisted as it exhibited the lowest critical micelle concentration and optimum colloidal stability compared to the other two candidates (data not shown). The thermoresponsive corona was based on the copolymer of di(ethylene glycol)methyl ether methacrylate (DiEGMA) and oligo(ethylene glycol)methyl ether methacrylate (OEGMA300);23–25 these co-monomers were chosen as they resemble the structure of poly(ethylene glycol) which is biocompatible, and protein repellent which allows for prolonged circulation of the carriers in the bloodstream with minimum opsonisation by the mononuclear phagocyte system.26 In addition, this monomer system may be polymerized by controlled polymerization reactions, allowing for precise control on the final polymer structure and it is also possible to tune the LCST of the polymer by adjusting the DiEGMA:OEGMA300 monomer feed ratio.
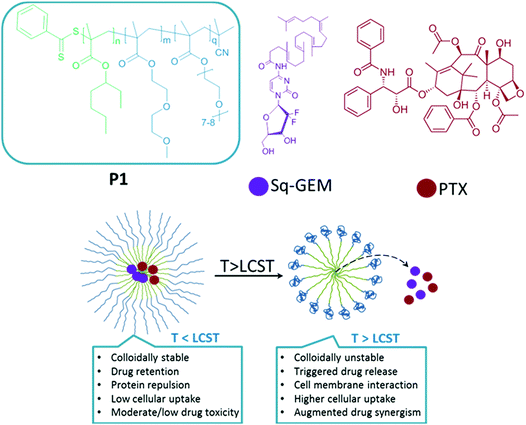 |
| | Scheme 1 The chemical structures of poly(2-ethylhexyl methacrylate)-b-poly[di(ethylene glycol)-oligo(ethylene glycol)methyl ether methacrylate] (P1), squalene-gemcitabine (Sq-GEM), paclitaxel (PTX) and key features of our proposed formulation below and above the lower critical solution of the polymer micelles. | |
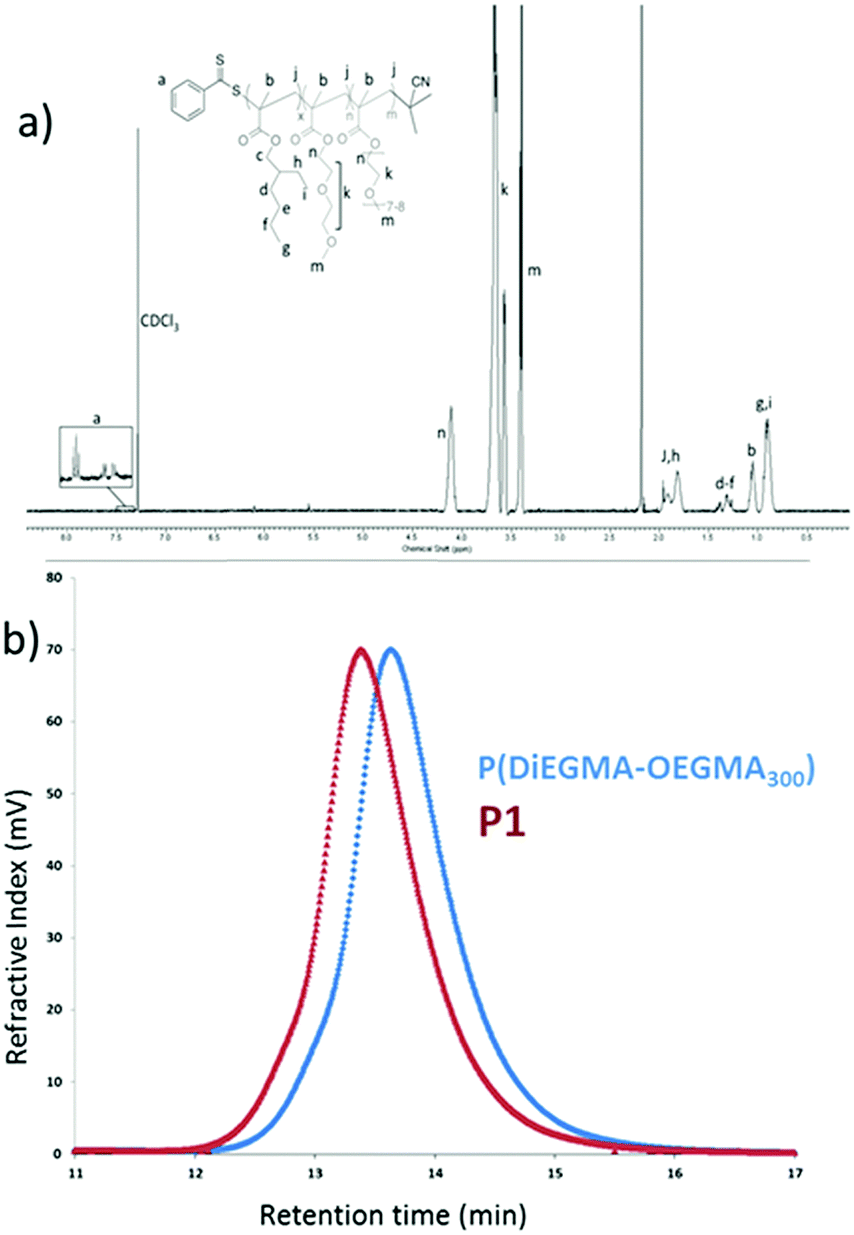
The block copolymer was synthesized by RAFT polymerization of the thermoresponsive block at DiEGMA:OEGMA300 ratio of 60:40 in order to achieve an optimum LCST onset of 40 °C, slightly above physiological temperature (Fig. S1, ESI†). The corona block was then used as a macro-RAFT transfer agent to grow the p(EHMA) block. The final polymer structure was confirmed by 1H NMR where all the proton peaks could be assigned successfully (Fig. 1a). From the GPC data it was possible to confirm that the obtained products had a Mn of 31600 Da and 39500 Da for p(DiEGMA–OEGMA300) and final P1, respectively; the degree of polymerization (DP) was close to the initial feed for both blocks (DPGPC for p(EHMA) and p(DiEGMA–OEGMA) was 43 and 116, respectively); also the final polymer had a narrow polydispersity index of 1.3 ĐM. Only a very minor tailing of the GPC trace was observed which was also persistent in the final P1 chromatogram, but this was negligible given the relatively low ĐM value. The critical micelle concentration of the polymer was determined in water by the pyrene assay and was found to be ca. 5 mg L−1 (Fig. S2, ESI†), which is in accord with similar studies27 and also verify the colloidal stability of the polymer in solution.
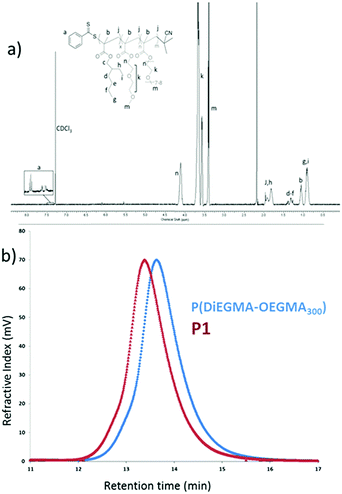 |
| | Fig. 1 (a) 1H NMR spectrum of P1 and (b) the GPC trace of the thermoresponsive block and P1. | |
Drug loading and triggered release studies
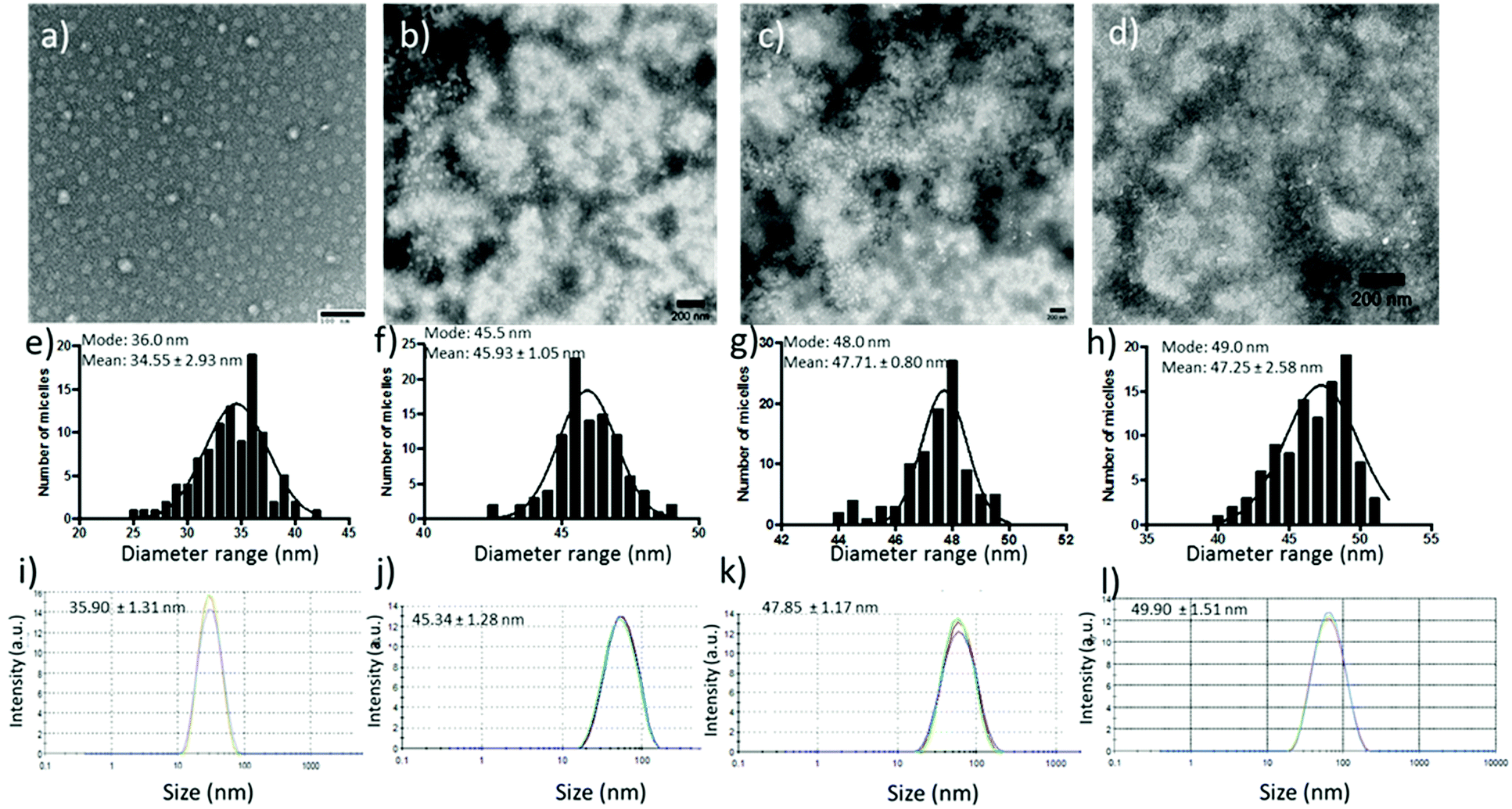
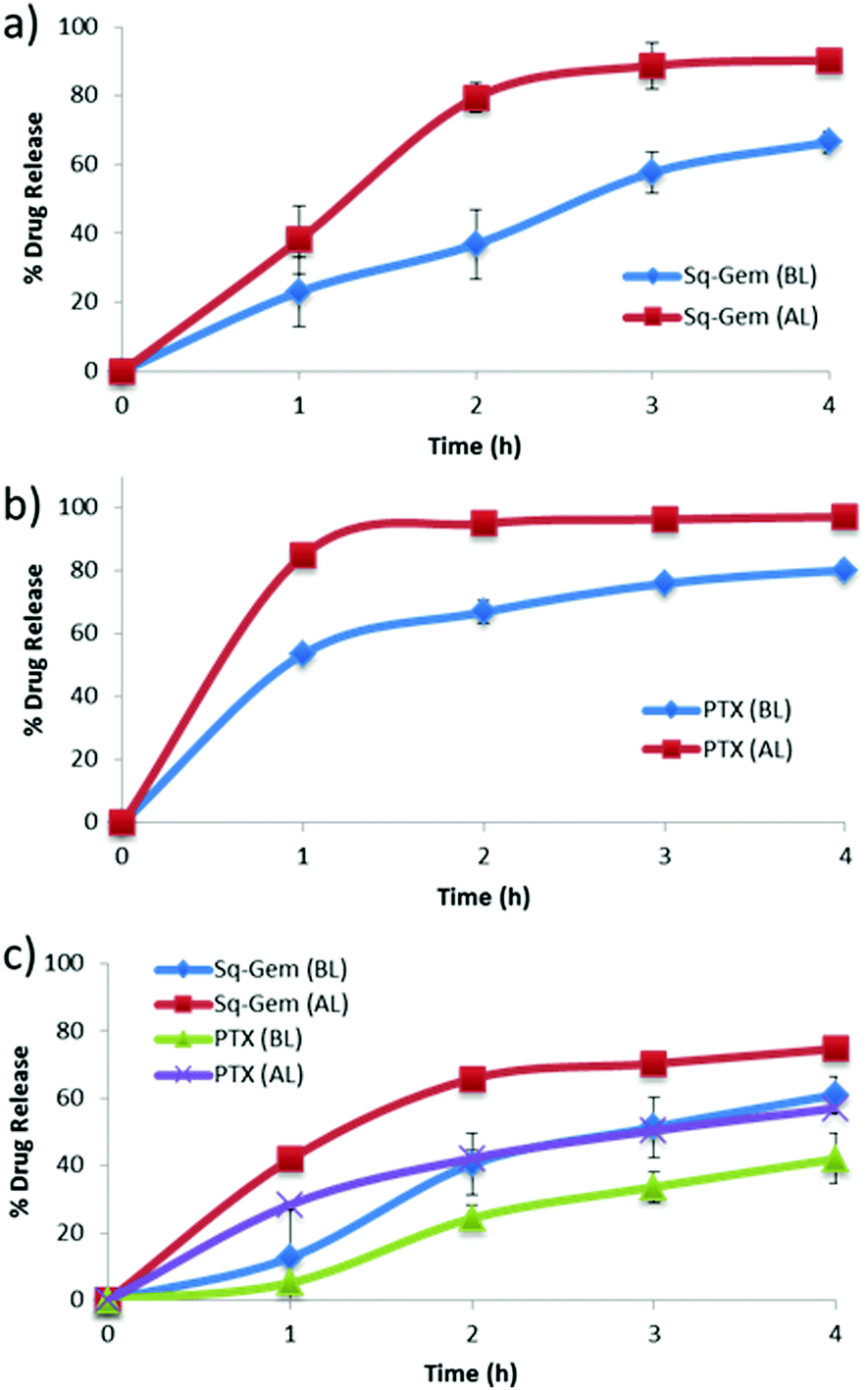
Paclitaxel and squalene-gemcitabine were chosen as a combinational drug ensemble as they exert synergistic activity and are currently indicated in clinical practice for patients with locally advanced pancreatic cancer (LAPC). PTX is already highly lipophilic and could be incorporated in the hydrophobic compartment of the polymer micelles, whereas gemcitabine in its parent form is highly water soluble and hence a synthetic more lipophilic28 derivative was chosen; squalene-gemcitabine was synthesized by the amide coupling of squalenic acid with gemcitabine at the 4-amino position that results in the formation of an amphiphilic drug with its own self-assembly properties: Sq-gem forms nanoparticles of ca. 130 nm in solution. Therefore, it was hypothesized that the squalene moiety could interact with the p(EHMA) segment of the micelles so that Sq-GEM could be co-compartmentalized with PTX in the micellar core. The polymer could self-assemble in PBS pH 7.4 (0.01 M) into relatively narrow dispersed spherical micelles with an average diameter of 34.55 nm as measured by TEM (Fig. 2a and e). The size and the morphology of the drug loaded micelles was also studied by TEM (Fig. 2b–d and f–h); it was found that the loaded micelles had larger average diameter compared to the empty ones (45.93 nm, 47.71 nm, and 47.25 nm, for Sq-GEM, PTX, and Sq-GEM/PTX co-loaded micelles, respectively) which was expected due to the loading of the molecular cargo. The results were further confirmed by DLS measurements which were in almost perfect agreement with the TEM results (Fig. 2i–l). Intriguingly, it was observed that the doubly loaded micelles had only a small increase in their diameter compared to the singly loaded ones, however a careful inspection of the TEM histograms showed that the population of the micelles in the frequency bands between 40 and 45 nm slightly increased. Moreover, it is likely that Sq-GEM further enhanced the colloidal stability of the system owing to its own amphiphilic properties which could in turn enhance the drug loading capacity of the system without significant increase in the overall diameter of the formulation. Indeed, it was possible to load PTX, Sq-GEM, or both, in the micelles at a drug loading efficiency of 10% per mass of polymer (i.e. 0.5 mg of each drug per 5 mg of polymer) without any measurable loss of drug during preparation. Remarkably, it was possible to retain an average size below 50 nm in the micelles co-loaded with both drugs. Also, from the DLS size distribution histogram, we could not trace the presence of larger particles implying the complete absence of free Sq-GEM particles (Fig. 2j–l). This means that it was possible to achieve nearly quantitative encapsulation efficiency with our drug loading protocol. We next studied the drug release profile of the two drugs separately loaded or co-loaded, below and above the polymers’ LCST (Fig. 3). In the case of Sq-GEM (Fig. 3a), below the LCST the release rate followed a zero-order rate, reaching ca. 37% and 67% within 2 and 4 hours, respectively. Above the LCST, the release rapidly reached nearly 80% which plateaued at ca. 90% in 4 hours. For PTX (Fig. 3b), it was found that 50% of the drug was released in one hour followed by a slower release rate which eventually resulted in ca. 80% release in 4 hours, below the LCST. Above LCST, the PTX release was significantly more pronounced, reaching ca. 85% in 1 hour before plateauing to >90% release within 4 hours. In the case of the sq-GEM and PTX co-loaded micelles (Fig. 3c), it was observed that PTX exhibited a more linear release profile both below and above the LCST and the release reached ca. 57% and 40% above and below the LCST, respectively. The temperature effect was more pronounced in the release pattern of sq-GEM where it was found that ca. 65% was released within 2 hours above the LCST (vs. 42% below the LCST) and reached >70% in 4 hours above the LCST compared to ca 57% below the LCST. Overall, it can be concluded that the effect of the micelle disruption due to the thermal collapsing of the p(DiEGMA–OEGMA300) corona had a more pronounced impact on the formulations of singly loaded drugs and to lesser extent to the PTX/Sq-GEM co-loaded micelles; it may be assumed that in the co-loaded sample, hydrophobic intermolecular interactions between PTX, Sq-GEM and the p(EHMA) polymer segments may be stronger and hence somewhat stabilize the overall formulation against the thermal collapsing of the micellar corona. The possible interaction of PTX with Sq-GEM can be further supported by the higher drug loading efficiency that could be achieved (i.e. 20%) compared to the singly loaded formulations (10%); it should be noted that Sq-GEM is colloidally stable due to its own amphiphilic character and hence it could possibly contribute to the further stabilization of the formulation.
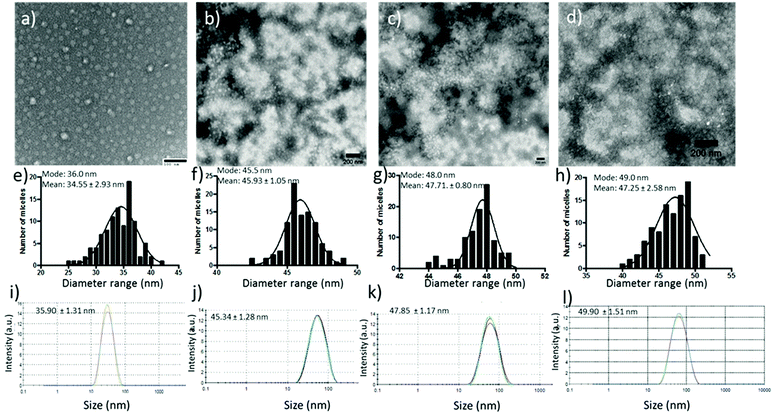 |
| | Fig. 2 TEM images of blank, Sq-GEM loaded, PTX loaded and Sq-GEM/PTX co-loaded micelles in (a–d) with their frequency histograms in (e–h), and their DLS measurements in (i–l). | |
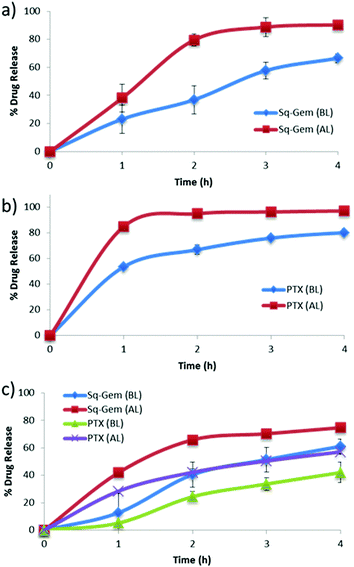 |
| | Fig. 3 Drug release of (a) Sq-GEM, (b) PTX, and (c) Sq-GEM and PTX, from P1 micelles below and above the LCST. | |
In vitro cytotoxicity studies
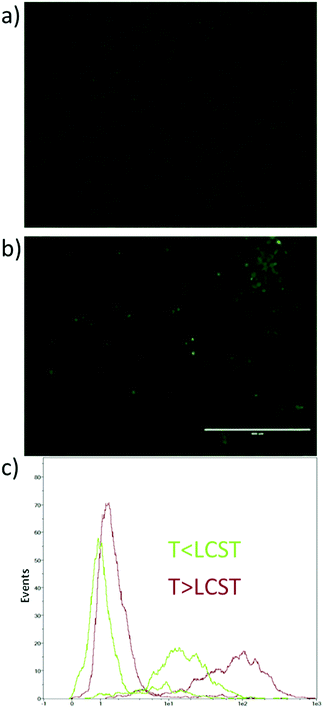
The cytotoxicity of the formulations was tested against model pancreatic cancer cell line MiaPaCa-2, using the MTT assay. Initially, we tested the possible toxicity of temperature increase (to 40 °C), DMSO (which was used as a co-solvent to dissolve free drugs), and the polymer without drugs loaded above and below its LCST; it was found that these parameters did not induce any detectable cytotoxicity (Fig. S3, ESI†). Having established that the experimental conditions were not toxic to the cells, we determined the half maximal inhibitory concentration (IC50) of the drugs, the formulations and their combinations under various conditions. First, Sq-GEM was found to have an IC50 of 1122 nM which is significantly higher than the parent drug (Table 1).21,29 Also, the obtained IC50 value is considerably higher than the IC50 of Sq-GEM in its nano-assembled form (i.e. 40 nM on MiaPaCa-2 cells30), which signifies the marked variances in the cytotoxicity profile of the same molecule under different formulations. Remarkably, Sq-GEM that was loaded in the micelles did not show any cytotoxicity under the conditions tested and it was not possible to extract an IC50 value (Table 1). Conversely, above the polymers’ LCST, the Sq-GEM-loaded formulation had an IC50 of 310.5 nM which is a 3.6 fold improvement compared to the non-encapsulated sample. Second, PTX was found to have an IC50 of 9.44 nM, which is in accord with previous reports.31 The micelles could reduce the cytotoxicity of the parent drug by almost 3-fold as the formulation had an IC50 of 29.22 nM which could be fully restored (and marginally improved) above the LCST (IC50 = 7.25 nM) (Table 1). Finally, the simultaneous addition of the two drugs resulted in a reduced IC50 (3.03 nM) which was significantly lower than each drug individually, indicative of the synergistic activity of the two drugs. In the micellar form, the IC50 was merely increased below the LCST (IC50 = 4.23 nM) but was significantly enhanced above the LCST (IC50 = 1.93 nM) (Table 1). The synergistic activity of the two drugs was verified by the Chou-Talalay32 combination index (CI)(eqn (1)); it was found that the CI of the non-loaded drugs was 0.32 which verifies the synergism of the two drug molecules (CI < 1 is indicative of synergistic activity). It was not possible to calculate a CI value for the micelles below the LCST as Sq-GEM loaded micelles were virtually non-toxic under the experimental conditions. Above LCST, the CI was 0.27 indicating a further enhancement of the synergistic activity of PTX and Sq-GEM in the formulated form. Overall, these results indicate that in all cases, the encapsulation of the drugs, individually or in combination, resulted in retention or significant enhancement of the cytotoxicity above the LCST and in a reduction of the cytotoxicity below the LCST. However, a careful interpretation of the data based merely on the drug release patterns cannot justify the large differences observed. Therefore, we hypothesized that the polymer itself may be actively interacting with cellular organelles (i.e. the cell membrane) in a temperature dependent manner which could further augment the drug uptake rates above the LCST. For this, we synthesized a block copolymer with a fluorescent tag that could be trackable with fluorescence spectrometry. The polymer was incubated with MiaPaCa-2 cells for 30 min below or above the LCST, and then the cells were visualized by fluorescence microscopy. Interestingly, it was found that the polymer only adsorbed onto the cellular membrane and did not permeate inside cells when incubated below LCST (at 37 °C) (Fig. 4a) as the fluorescence signal was persistently tracked at the periphery of the cells. On the other hand, when incubated with the micelles above the LCST (at 40 °C), the cells displayed stronger fluorescent signal inside the cell cytosol (Fig. 4b). These results indicated that increasing the temperature effectively increases the cell internalization of the fluorescent polymer first by eliciting a direct interaction of the lipophilic pEHMA block of the polymer with the cell membrane followed by enhanced internalization, likely driven by the thermal collapsing of the PEGMA block.
Table 1 IC50 values with their standard deviation from three experiments (S.D.) of Sq-GEM, PTX, and their combination in non-encapsulated (N.E.) and encapsulated formulations below and above the LCST
| Drug |
LCST |
IC50 (nM) |
S.D. |
| Sq-Gem |
N.E. |
1122 |
±258.59 |
| Below |
— |
— |
| Above |
310.4 |
±55.44 |
|
|
| PTX |
N.E. |
9.44 |
±0.17 |
| Below |
29.22 |
±1.75 |
| Below |
7.25 |
±0.16 |
|
|
| Sq-Gem and PTX |
N.E. |
3.03 |
±0.30 |
| Below |
4.24 |
±0.16 |
| Above |
1.93 |
±0.10 |
 |
| | Fig. 4 Fluorescence microscopy images of MiaPaCa-2 cells incubated with P1 (a) below, and (b) above the LCST of a fluorescent P1 derivative with their respective flow cytometry graphs in (c). Scale bar is 400 μm. | |
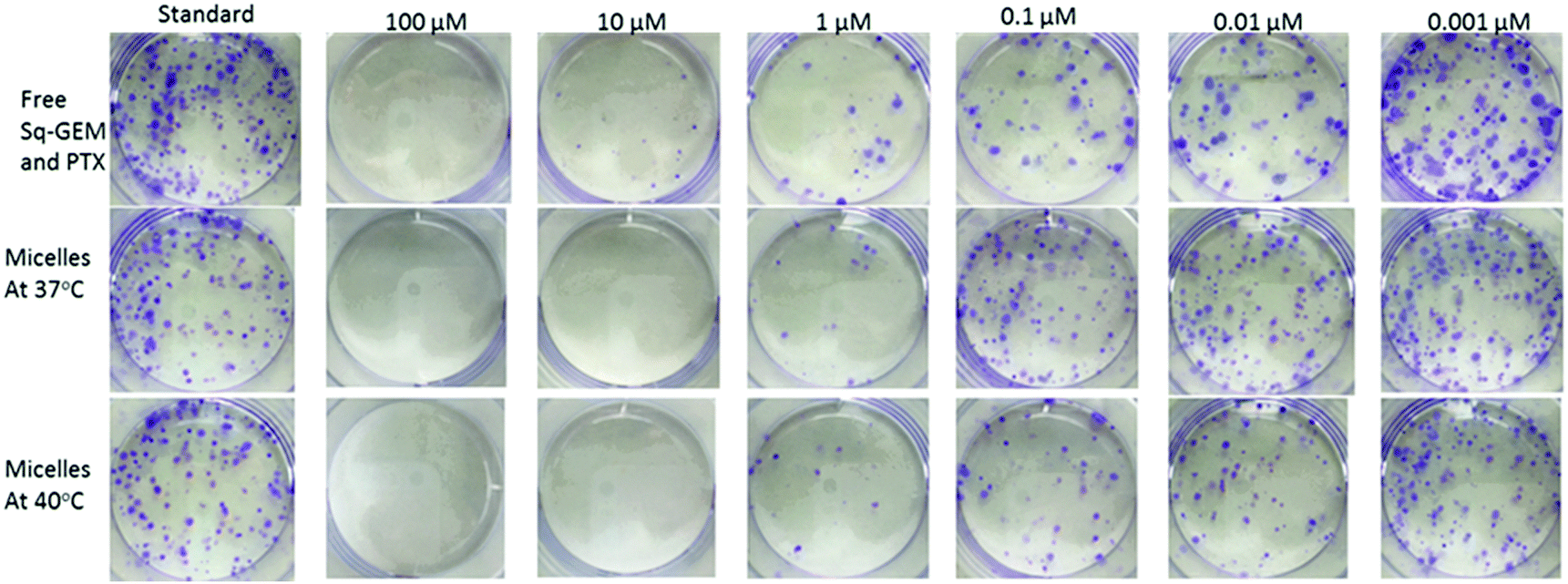
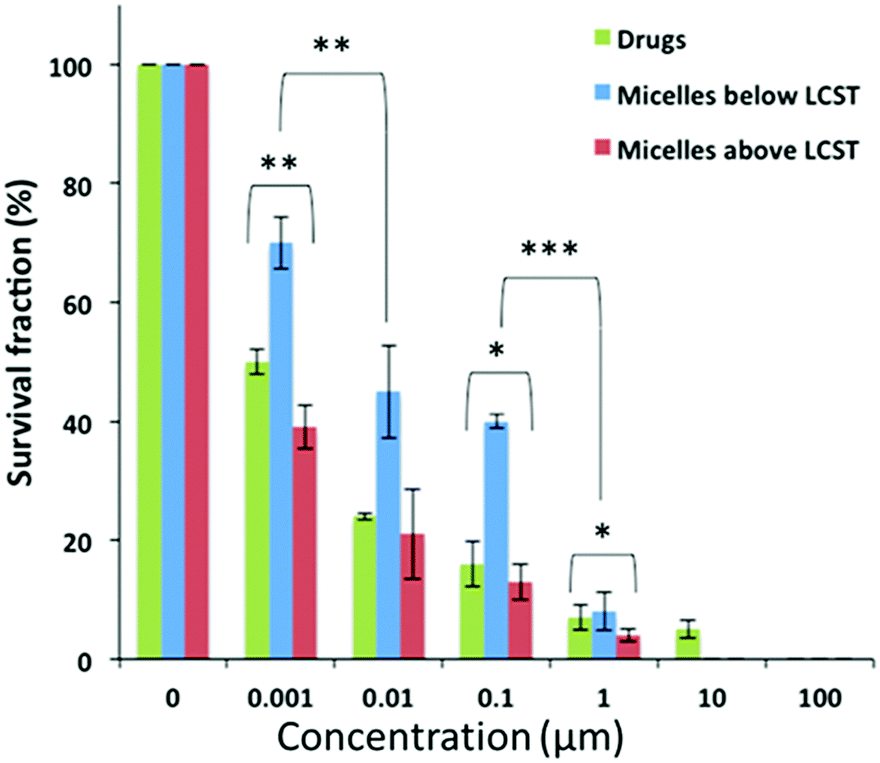
The consistency of this internalization pattern was further confirmed by flow cytometry experiments. Fig. 4c shows a standard curve divided into two curves, one with lower intensity and the other one shifting towards higher intensity. The first band was attributed to the fluorescence signal from the polymer adsorbed on the cell membrane while the second band corresponded to the signal resulting from the cell internalized polymer. The flow cytometry profile above the polymers’ LCST showed more cells with more fluorescent polymer adsorbed on the cell membrane when compared to the curve below LCST. Moreover, the fluorescent signal corresponding to internalized polymer increased significantly above the LCST as evidenced by the pronounced right-shifting of the fluorescent band. These results corroborate with our hypothesis that the polymer was initially adsorbed on the cell membrane via hydrophobic interactions primarily due to the pEHMA followed by enhanced cellular uptake driven by the collapsing of the thermoresponsive corona. In effect, this mechanism could adequately explain the enhanced cytotoxicity profiles and point to a more complex and collective cytotoxic mechanism pattern which is justified by: (1) The enhanced drug release controlled by the temperature stimulus which effectively leads to increased drug translocation across the cell membrane, (2) The direct interaction of the polymer with the cell membrane which potentially results in an increased permeability of the cell membrane above the polymers LCST which could further augment the translocation rates of both lipophilic drugs and 3. The enhanced cellular uptake of the micelles above the LCST via endocytosis mechanism.33–35 Finally, a clonogenic cell survival assay was conducted with MiaPaCa-2 cells to investigate the proliferative ability of survived cells post treatment (Fig. 5). The clonogenic cell survival assay determines the long-term cytotoxic effects of drugs by measuring their ability to propagate from a single cell to a clone and produce a viable colony.36 Initially MiaPaCa-2 cells were grown on 25 cm2 T-flask which were then treated with different concentrations of both drugs in the formulated or non-formulated form, below and above the polymers’ LCST. The survival fraction was calculated 14 days post treatment in order to probe the cytotoxic properties of the micelles in the longer term. As expected, it was found that the survival fraction decreased as the concentration of the drugs increased (Fig. 6). Interestingly, the micelles below the LCST, were found to decrease the survival fraction induced by the drugs at concentrations from 0.001 μM to 0.1 μM. However, above the LCST, the survival fraction was still more diminished and was very close to the values for the free drugs (note that for 0.001 μM Sq-GEM/PTX it was statistically improved) (Fig. 6). This longer term effect of the polymer micelles further supports our proposed mechanism of action as previously discussed, especially given the fact that the release of the drugs is controlled only for a few hours by temperature (Fig. 3); therefore, a longer term mechanism as evidenced by the cell membrane interaction studies could indeed explain the longer term effects of temperature on the cytotoxicity profiles of the drugs.
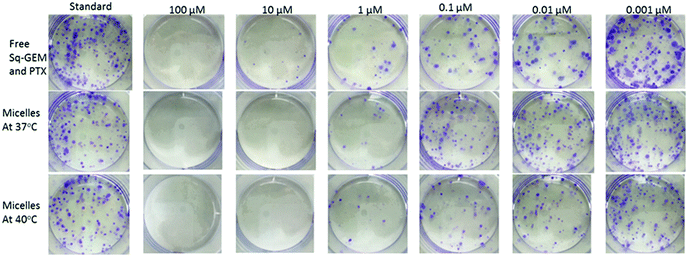 |
| | Fig. 5 Digital photographs of the clonogenic assay of MiaPaCa-2 cells performed with the combination of free Sq-GEM and PTX, or loaded in micelles below and above the LCST for 14 consecutive days in 6-well plates (the diameter of each well is approx. 34 mm). | |
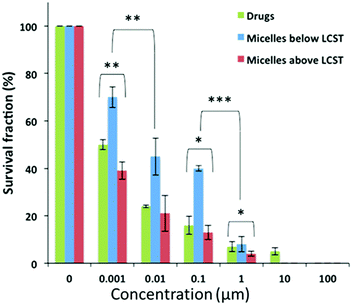 |
| | Fig. 6 The survival fraction percentile of MiaPaCa-2 cells post treatment with free Sq-GEM and PTX, and Sq-GEM/PTX loaded micelles for 14 days. *, **, and *** denote moderate (0.01 > p > 0.05), high (0.001 > p > 0.01), and very high (0.0001 > p > 0.001) statistical difference respectively, based on p values from an unpaired Student's t-test. | |
Conclusions
This study introduces a new thermoresponsive block copolymer that self assembles in sub-50 nm micelles and can be co-loaded with two potent anticancer drugs, namely, PTX, and Sq-GEM. We demonstrated that the polymer micelles could modulate the release profiles of two drugs simultaneously and elicit synergistic cytotoxicity against a model pancreatic cell line. More importantly, it was experimentally demonstrated that the polymer actively interacted with the cell membrane which in turn thermally modulated and augmented the synergistic activity of the drugs in the longer term. Although, there are many studies in thermoresponsive block copolymer micelles, the potential role of the actual carrier, that is the polymer, is almost systematically neglected and hence we anticipate that our study will be insightful for the design of polymer based nanomedicinal formulations for cancer, especially for formulations aimed for drug combinations where the therapeutic window is far more challenging to exploit. In addition, the proposed system could be suitable for deep tissue triggered drug release via high intensity focused ultrasound probes37,38 or other types of remotely activated39,40 therapeutic modalities.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the Engineering and Physical Sciences Research Council (G. P., EPSRC EP/M014649/1) and University College London (G. P., Excellence Fellowship) for financial support. We also thank Ms Mina Emamzadeh for training and helping with the in vitro studies.
Notes and references
- M. Malvezzi, P. Bertuccio, F. Levi, C. La Vecchia and E. Negri, Ann. Oncol., 2014, 25, 1650–1656 CrossRef CAS PubMed.
- D. Li, K. Xie, R. Wolff and J. L. Abbruzzese, Lancet, 2004, 363, 1049–1057 CrossRef CAS.
- H. A. Burris, 3rd, M. J. Moore, J. Andersen, M. R. Green, M. L. Rothenberg, M. R. Modiano, M. C. Cripps, R. K. Portenoy, A. M. Storniolo, P. Tarassoff, R. Nelson, F. A. Dorr, C. D. Stephens and D. D. Von Hoff, J. Clin. Oncol., 1997, 15, 2403–2413 CrossRef PubMed.
- E. Mini, S. Nobili, B. Caciagli, I. Landini and T. Mazzei, Ann. Oncol., 2006, 17, v7–v12 CrossRef PubMed.
- L. A. Shipley, T. J. Brown, J. D. Cornpropst, M. Hamilton, W. D. Daniels and H. W. Culp, Drug Metab. Dispos., 1992, 20, 849–855 CAS.
-
https://www.fda.gov/ohrms/dockets/ac/06/briefing/2006-4254b_11_04_KP%20GemcitabineFDAlabel42005.pdf
.
- T. Conroy, F. Desseigne, M. Ychou, O. Bouché, R. Guimbaud, Y. Bécouarn, A. Adenis, J. L. Raoul, S. Gourgou-Bourgade, C. de la Fouchardière, J. Bennouna, J.-B. Bachet, F. Khemissa-Akouz, D. Péré-Vergé, C. Delbaldo, E. Assenat, B. Chauffert, P. Michel, C. Montoto-Grillot and M. Ducreux, N. Engl. J. Med., 2011, 364, 1817–1825 CrossRef CAS PubMed.
- D. D. Von Hoff, T. Ervin, F. P. Arena, E. G. Chiorean, J. Infante, M. Moore, T. Seay, S. A. Tjulandin, W. W. Ma, M. N. Saleh, M. Harris, M. Reni, S. Dowden, D. Laheru, N. Bahary, R. K. Ramanathan, J. Tabernero, M. Hidalgo, D. Goldstein, E. Van Cutsem, X. Wei, J. Iglesias and M. F. Renschler, N. Engl. J. Med., 2013, 369, 1691–1703 CrossRef CAS PubMed.
- K. K. Frese, A. Neesse, N. Cook, T. E. Bapiro, M. P. Lolkema, D. I. Jodrell and D. A. Tuveson, Cancer Discovery, 2012, 2, 260–269 CrossRef CAS PubMed.
- R. Alvarez, M. Musteanu, E. Garcia-Garcia, P. P. Lopez-Casas, D. Megias, C. Guerra, M. Munoz, Y. Quijano, A. Cubillo, J. Rodriguez-Pascual, C. Plaza, E. de Vicente, S. Prados, S. Tabernero, M. Barbacid, F. Lopez-Rios and M. Hidalgo, Br. J. Cancer, 2013, 109, 926–933 CrossRef CAS PubMed.
- A. K. Singla, A. Garg and D. Aggarwal, Int. J. Pharm., 2002, 235, 179–192 CrossRef CAS PubMed.
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC.
- C. d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- X. Guo, D. Li, G. Yang, C. Shi, Z. Tang, J. Wang and S. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 8549–8559 CAS.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- T. Hosoki, Am. J. Roentgenol., 1983, 140, 959–965 CrossRef CAS PubMed.
- A. Sofuni, H. Iijima, F. Moriyasu, D. Nakayama, M. Shimizu, K. Nakamura, F. Itokawa and T. Itoi, J. Gastroenterol., 2005, 40, 518–525 CrossRef PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed.
- B. Al-Lazikani, U. Banerji and P. Workman, Nat. Biotechnol., 2012, 30, 679–692 CrossRef CAS PubMed.
- P. Couvreur, B. Stella, L. H. Reddy, H. Hillaireau, C. Dubernet, D. Desmaële, S. Lepêtre-Mouelhi, F. Rocco, N. Dereuddre-Bosquet, P. Clayette, V. Rosilio, V. Marsaud, J. M. Renoir and L. Cattel, Nano Lett., 2006, 6, 2544–2548 CrossRef CAS PubMed.
- M.-C. Jones and J.-C. Leroux, Eur. J. Pharm. Biopharm., 1999, 48, 101–111 CrossRef CAS PubMed.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- G. Pasparakis and C. Alexander, Angew. Chem., 2008, 120, 4925–4928 CrossRef.
- J. F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- X. Zhao, Z. Poon, A. C. Engler, D. K. Bonner and P. T. Hammond, Biomacromolecules, 2012, 13, 1315–1322 CrossRef CAS PubMed.
- D. Sobot, S. Mura, S. O. Yesylevskyy, L. Dalbin, F. Cayre, G. Bort, J. Mougin, D. Desmaële, S. Lepetre-Mouelhi, G. Pieters, B. Andreiuk, A. S. Klymchenko, J. L. Paul, C. Ramseyer and P. Couvreur,
, 2017, 8, 15678 CAS.
- S. Réjiba, L. H. Reddy, C. Bigand, C. Parmentier, P. Couvreur and A. Hajri, Nanotechnol. Biol. Med., 2011, 7, 841–849 CrossRef PubMed.
- J. Caron, A. Maksimenko, J. Mougin, P. Couvreur and D. Desmaele, Polym. Chem., 2014, 5, 1662–1673 RSC.
- Y. Lin, F. M. Richards, B. F. Krippendorff, J. L. Bramhall, J. A. Harrington, T. E. Bapiro, A. Robertson, D. Zheleva and D. I. Jodrell, Br. J. Cancer, 2012, 107, 1692–1701 CrossRef CAS PubMed.
- T. C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed.
- S. R. Abulateefeh, S. G. Spain, K. J. Thurecht, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Biomater. Sci., 2013, 1, 434–442 RSC.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Mol. Pharmaceutics, 2010, 7, 926–935 CrossRef CAS PubMed.
- Y. Hiruta, M. Shimamura, M. Matsuura, Y. Maekawa, T. Funatsu, Y. Suzuki, E. Ayano, T. Okano and H. Kanazawa, ACS Macro Lett., 2014, 3, 281–285 CrossRef CAS.
- P. R. Roper and B. Drewinko, Cancer Res., 1976, 36, 2182–2188 CAS.
- A. Z. Zardad, Y. Choonara, L. du Toit, P. Kumar, M. Mabrouk, P. Kondiah and V. Pillay, Polymers, 2016, 8, 359 CrossRef.
- N. Rapoport, A. M. Kennedy, J. E. Shea, C. L. Scaife and K.-H. Nam, Mol. Pharmaceutics, 2010, 7, 22–31 CrossRef CAS PubMed.
- Y. Wang, G. Wei, X. Zhang, F. Xu, X. Xiong and S. Zhou, Adv. Mater., 2017, 29, 1605357 CrossRef PubMed.
- Y. Guo, K. Jiang, Z. Shen, G. Zheng, L. Fan, R. Zhao and J. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 43508–43519 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb02899g |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a