
Macrophage responses to the physical burden of cell-sized particles†

Abstract
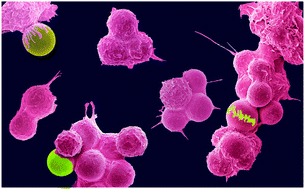
The role of a biophysical signal on cell response has excited tremendous interest recently. Herein, we exploited an “intake method” together with uniform autofluorescent cell-sized particles to investigate macrophage responses against different particle burdens. Our work not only revealed an insatiable macrophage uptake of cell-sized microparticles (MPs) but also widened the theoretical size and volume range for particle entry. MPs outperform NPs in the utmost volume of intracellular particles, indicating a converse size-associated event than originally anticipated. Such a superior volume burden (2.64-fold of the cell volume) for MPs is highly correlated to less membrane loss (19-fold below that for NPs) and more evident deformation of the cell membrane/nucleus. In addition, the cells with a high burden of MPs exhibit moderate migration activity, which is in line with mild cytokine release. These results highlight the indispensable role of physical burden on the regulation of macrophage functions, providing important views for desired biological outcomes and minimal side effects.
- This article is part of the themed collection: 2017 Journal of Materials Chemistry B HOT Papers
 Please wait while we load your content...
Please wait while we load your content...

