
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA full monolayer of superoxide: oxygen activation on the unmodified Ca3Ru2O7(001) surface†
Daniel
Halwidl
 a,
Wernfried
Mayr-Schmölzer
ab,
Martin
Setvin
a,
David
Fobes
c,
Jin
Peng
c,
Zhiqiang
Mao
c,
Michael
Schmid
a,
Florian
Mittendorfer
ab,
Josef
Redinger
ab and
Ulrike
Diebold
*a
a,
Wernfried
Mayr-Schmölzer
ab,
Martin
Setvin
a,
David
Fobes
c,
Jin
Peng
c,
Zhiqiang
Mao
c,
Michael
Schmid
a,
Florian
Mittendorfer
ab,
Josef
Redinger
ab and
Ulrike
Diebold
*a
aInstitute of Applied Physics, TU Wien, Wiedner Hauptstrasse 8-10/134, 1040 Vienna, Austria. E-mail: diebold@iap.tuwien.ac.at
bCenter for Computational Materials Science, TU Wien, Wiedner Hauptstrasse 8-10/134, 1040 Vienna, Austria
cDepartment of Physics and Engineering Physics, Tulane University, 2001 Percival Stern Hall, New Orleans, LA 70118, USA
First published on 5th March 2018
Abstract
Activating the O2 molecule is at the heart of a variety of technological applications, most prominently in energy conversion schemes including solid oxide fuel cells, electrolysis, and catalysis. Perovskite oxides, both traditionally-used and novel formulations, are the prime candidates in established and emerging energy devices. This work shows that the as-cleaved and unmodified CaO-terminated (001) surface of Ca3Ru2O7, a Ruddlesden–Popper perovskite, supports a full monolayer of superoxide ions, O2−, when exposed to molecular O2. The electrons for activating the molecule are transferred from the subsurface RuO2 layer. Theoretical calculations using both, density functional theory (DFT) and more accurate methods (RPA), predict the adsorption of O2− with Eads = 0.72 eV and provide a thorough analysis of the charge transfer. Non-contact atomic force microscopy (nc-AFM) and scanning tunnelling microscopy (STM) are used to resolve single molecules and confirm the predicted adsorption structures. Local contact potential difference (LCPD) and X-ray photoelectron spectroscopy (XPS) measurements on the full monolayer of O2− confirm the negative charge state of the molecules. The present study reports the rare case of an oxide surface without dopants, defects, or low-coordinated sites readily activating molecular O2.
Introduction
Oxygen plays a pivotal role in solid oxide fuel cells, electrolysis and catalysis, where complex ternary oxides are intensively investigated materials.1 Of particular interest are perovskite-type oxides with the basic formula ABO3 (where A stands for an alkali, alkaline earth, or rare earth metal, while B refers to a transition metal) and variations of this structure, such as double perovskites, A′A′′B2O5+δ or the Ruddlesden–Popper series An+1BnO3n+1.1 Molecular O2 in its triplet ground state is an inert species and requires activation to undergo catalytic reactions or dissociation into atomic oxygen.2 The O2 activation is a crucial step in the oxygen reduction reaction3 (ORR) that is potentially a major rate limiting factor for applications.4 Therefore a microscopic understanding of the oxygen surface chemistry on perovskite-type oxides is of great importance.5In general, the activation of O2 is achieved by either exciting the neutral molecule to one of its two singlet states6 or by charging the molecule thus forming a superoxide (O2−) or a peroxide (O22−) ion. If available, the electrons for charging the O2 can be transferred directly from the oxide surface, but a substantially unperturbed oxide (i.e. stoichiometric, step- and defect-free) rarely provides electrons for a spontaneous formation of charged oxygen species. Often the oxide has to be activated by photoinduced electron transfer, surface intermolecular electron transfer,7 or the introduction of dopants, defects, or low-coordinated sites.8
Superoxo and peroxo species have been predicted and/or observed on a few catalytically relevant binary oxides. In experiments on anatase TiO2(101) it was recently experimentally shown that molecular oxygen accepts charge from subsurface Nb+ dopants, resulting in a mixed adsorption of neutral O2 and superoxide ions9 at low temperature (5 K). Charged oxygen species have been predicted for reduced CeO2(111),10 (110) and (100) surfaces,11 and their formation has been experimentally confirmed on polycrystalline CeO2.12,13 Calculations for pure ZrO2 show only a weak physisorption of O2 while yttria stabilized zirconia should facilitate dissociation via a superoxo species.14 Similarly, O2 is predicted to adsorb weakly on pure ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0)15 but to form a superoxo species when the ZnO is Al-doped.16 Charged oxygen species on SnO2 play a crucial role in chemical sensing, although the correct interpretation of experimental results has been questioned.17
0)15 but to form a superoxo species when the ZnO is Al-doped.16 Charged oxygen species on SnO2 play a crucial role in chemical sensing, although the correct interpretation of experimental results has been questioned.17
The binary alkaline-earth oxide (001) surfaces are closely related to AO-terminated perovskite surfaces. Molecular oxygen does not adsorb on the unperturbed CaO(001)18 and MgO(001)19 surface. Only after introducing defects by ultraviolet or gamma irradiation, thermal activation, or doping with transition metal ions the formation of charged oxygen species was observed.18,20 Ultrathin MgO films supported on a metal (Ag or Mo) allow for an electron transfer from the substrate to activate adsorbed oxygen, explaining the catalytic properties of such systems.19,21,22
Turning to ternary oxides, direct experimental evidence on microscopic reaction pathways is sparse. One of the most studied perovskite oxides is SrTiO3.5 Calculations have shown that on the technologically relevant SrO-terminated (001) surface O2 is activated to O2− only if surface oxygen vacancies are present.23 In contrast, the defect free LaO-terminated La2NiO4(001) surface is theoretically predicted to readily chemisorb oxygen either as superoxo or peroxo species.24
This paper combines theory and experiment to investigate on the first step in oxygen activation on a ternary oxide, namely electron transfer to adsorbed O2. Ca3Ru2O7, the prototypical perovskite material considered here, is a Ruddlesden–Popper perovskite (alternating ABO3 and AO layers) and cleaves easily between adjacent CaO layers. It is shown that the pristine CaO-terminated (001) surface facilitates the activation of O2 to a superoxide ion without a need for prior surface treatment, or the presence of steps, oxygen-vacancies or dopants. Surface spectroscopy in combination with atomically-resolved Scanning Probe Microscopy and DFT calculations confirm the charge state of the as-formed oxygen and provide detailed models for the adsorption geometry of isolated molecules and the dense (2 × 1) O2− overlayer formed at higher coverage.
Methods
DFT calculations
The surface was modeled using one double-layer thick slabs of Ca3Ru2O7, separated by 15 Å of vacuum. The calculations were performed with the Vienna Ab initio Simulation Package25 (VASP). The van-der-Waals contributions were described by the optB86b26,27 functional which has been employed successfully in previous studies of water adsorption on ruthenate surfaces.28,29 The low-coverage case was described using a (3 × 3) unit cell with one O2 molecule adsorbed on it, yielding a coverage of 1/9, while the high-coverage model cell consisted of a (2 × 1) surface unit cell with two added O2 molecules. For the large (3 × 3) unit cell a 2 × 2 × 1 Gamma-centered k-point grid was sufficient to achieve electronic convergence while for the smaller (2 × 1) model a 3 × 6 × 1 Gamma centered mesh was used. The energy cutoff was set to 400 eV. The hybrid calculations were performed using the range-separated HSE06 (ref. 30) functional which implements the Fock exchange interaction in the PAW formalism.25 For the advanced many-electron calculations a modern, low-scaling algorithm was used to obtain both the ACFDT-RPA total energies31,32 and the G0W0 densities of states.33 Due to the computational cost involved, the k-point grid was reduced to a 2 × 4 × 1 mesh for the (2 × 1) model cell for both the hybrid functional (HSE06) and the many-electron (G0W0/RPA) calculations. The O 1s core level binding energies were calculated in the initial state approximation.Experimental setup
XPS and STM measurements were carried out in an ultrahigh vacuum (UHV) system consisting of a preparation chamber and an STM chamber with base pressures of 2 × 10−11 and 6 × 10−12 mbar, respectively. A low-temperature STM (commercial Omicron LT-STM) was operated at 78 K in constant-current mode using an electrochemically etched W-tip. XPS was performed in the preparation chamber using non-monochromatized Mg Kα X-rays and a SPECS PHOIBOS 100 electron analyser at normal emission with a pass energy of 15 eV. O2 was dosed in the preparation chamber while keeping the sample at 110 K.Combined STM/nc-AFM measurements were performed at 4.8 K in a UHV system consisting of a preparation chamber and an analysis chamber with base pressures below 2 × 10−11 mbar, equipped with a commercial Omicron q-Plus LT head. Tuning-fork-based AFM sensors with a separate wire for the tunnelling current were used34 (k = 1900 N m−1, f0 = 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 Hz, Q ≈ 30000). A custom-design cryogenic differential preamplifier was used for measuring the cantilever deflection.35 Electrochemically etched W-tips were glued to the tuning fork and cleaned in situ by field emission and self-sputtering in 10−6 mbar Ar.36 O2 was either dosed directly into the cryostat in the analysis chamber at 5.5 K or in the preparation chamber while keeping the sample at 123 K.
500 Hz, Q ≈ 30000). A custom-design cryogenic differential preamplifier was used for measuring the cantilever deflection.35 Electrochemically etched W-tips were glued to the tuning fork and cleaned in situ by field emission and self-sputtering in 10−6 mbar Ar.36 O2 was either dosed directly into the cryostat in the analysis chamber at 5.5 K or in the preparation chamber while keeping the sample at 123 K.
For all STM measurements the bias voltage was applied to the sample; positive or negative bias voltages result in STM images of the unoccupied or occupied states, respectively. All STM images shown were corrected for distortions as described elsewhere.37
Sample preparation
High-quality calcium ruthenate single crystals were grown by the floating zone technique using a mirror-focused furnace.38 The composition of the samples was determined via inductively coupled plasma mass spectroscopy (ICP-MS) using laser ablation (LA) for direct analysis of the solid samples. Before insertion into the UHV the samples were fixed on stainless-steel sample plates with conducting silver epoxy glue (EPO-TEK H21D, Epoxy Technology Inc.), and a metal stud was glued on top with another epoxy adhesive (EPO-TEK H77, Epoxy Technology Inc.). The crystals were cleaved in UHV at 110 K by removing the metal stub with a wobble stick.Results
The pristine surface
As a sample, Ca3Ru2O7 was chosen. This is the n = 2 member of the Ruddlesden–Popper series Can+1RunO3n+1, which consists of n perovskite-like CaRuO3 layers separated by adjacent CaO layers along the [001] direction (orthorhombic unit cell: a = 5.365 Å, b = 5.562 Å, c = 19.525 Å). The RuO6 octahedra are alternately tilted with respect to the c axis, and alternately rotated in the ab plane,39 where the a, b and c axes correspond to the [100], [010] and [001] directions, respectively, see Fig. 1a, b. The preferred cleaving plane is between two CaO layers, with a cleaving energy of 3.62 eV (DFT value) per unit cell. This low cleaving energy is consistent with experiments, where only the CaO termination is consistently observed upon cleaving single crystals in UHV. These CaO terraces are typically at least a few hundred nm in size.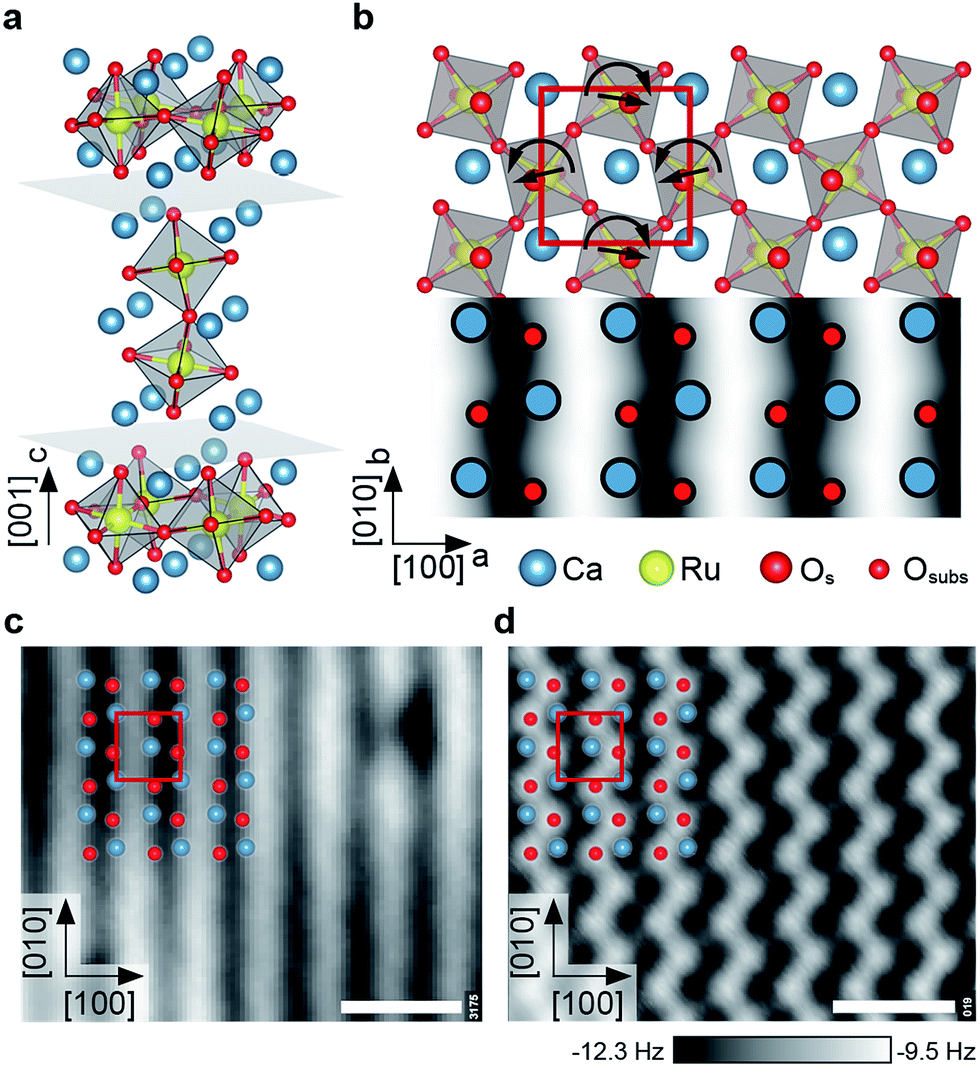 | ||
| Fig. 1 The calcium ruthenate structure. All scale bars correspond to 1 nm. (a) Unit cell of the n = 2 member of the Can+1RunO3n+1 Ruddlesden Popper series. Cleavage planes are marked in gray. (b) Top view and STM simulation (Tersoff–Hamann approximation40) of the CaO-terminated Ca3Ru2O7(001) surface. The RuO6 octahedra are alternately tilted with respect to the c axis (most pronounced in the ac plane), and rotated in the ab plane as indicated by the straight and curved arrows, respectively. The red box marks the orthorhombic unit cell (a = 5.365 Å, b = 5.562 Å). (c) STM image of the cleaved surface. The dark (bright) substrate lines in [010] direction correspond to areas where the apical oxygen atoms of the RuO6 octahedra are tilted towards (away from) each other, compare to (b). The point defects are attributed to spurious impurities (see ESI†). STM parameters: Vsample = +0.8 V, Itunnel = 0.1 nA, Tsample = 78 K; image rotated and cropped; fast scan direction is 68° clockwise from horizontal. (d) Constant-height nc-AFM image, different area than in (c). Shown is the frequency shift, Δf. The apical oxygen atoms are imaged as bright dots (less negative Δf, more repulsive), arranged in zig-zag lines in [010] direction. AFM parameters: A = 100 pm, Vsample = +0.25 V, Tsample = 4.8 K; fast scan direction is 50° clockwise from horizontal. | ||
The tilt and rotation of the octahedra leads to a distorted surface oxygen sublattice that was found essential in affecting the ordering of a hydroxyl overlayer on this surface.29 STM shows alternating bright and dark lines along the [010] direction, see Fig. 1c. According to a STM simulation (Tersoff–Hamann approximation)40 the dark (bright) substrate lines correspond to areas where the apical oxygen atoms of the RuO6 octahedra are tilted towards (away from) each other (Fig. 1b). All non-contact atomic force microscopy (nc-AFM) images in this work show the frequency shift, Δf, and are recorded in constant-height mode, where darker color means higher attractive force. Pioneering works on oxide surfaces show that the AFM contrast is governed by the electric charge of the tip apex.41,42 ‘Positively terminated’ tips interact attractively with anions, and repulsively with cations. ‘Negative tips’ show the opposite behaviour. The contrast in Fig. 1d corresponds to the negative termination; simultaneously measured STM and AFM images show that the apical oxygen atoms interact repulsively with the tip and thus are imaged as bright dots arranged in zig-zag lines in [010] direction.
The observed point defects are attributed to impurities (Ti, Sr, Mg, Ba) in the material (see Table S1†) rather than intrinsic defects that would stem from the cleaving process. The calculated creation energies for O and Ca vacancies, 3.9 and 5.3 eV, respectively, are significantly higher than the cleaving energy per broken Ca–O bond of 0.91 eV. Also, the appearance in STM of O vacancies created by electron irradiation is known from previous work.29
Low coverage of O2−
The theoretical description of complex oxides is challenging and the currently-available approximations deliver only incomplete descriptions. Here results from DFT calculations including van-der-Waals interactions (vdW-DFT) are discussed first, for a more detailed analysis using ‘beyond-DFT’ methods, see section ‘Electronic Properties’. The vdW-DFT calculations predict adsorption of O2 as a charged molecule with an adsorption energy of 1.4 eV. The molecule adsorbs as a superoxo species at a Ca–Ca bridge, labelled B1 in Fig. 2a. The octahedral tilt results in two non-equivalent bridge sites, the one chosen by the molecule (B1, equivalent to B2) lies on the bright substrate lines in STM images (Fig. 1b). Due to the charge transfer, the intramolecular O–O bond length is increased to a value of 1.35 Å. The molecule is rotated by 34° with respect to the bright substrate lines, and tilted by 29° with respect to the surface plane, see Fig. 2b. One O of the molecule is located almost at the center of the Ca–Ca bridge (O–Ca1 = 2.37 Å, O–Ca2 = 2.46 Å), the other O sits far away from Ca1 (O–Ca1 = 3.69 Å), but maintains a similar distance to Ca2 (O–Ca2 = 2.34 Å) as the first O due to the tilt of the molecule. The molecule slightly distorts the substrate as the tilt of the two nearest RuO6 octahedra is increased to 15° compared to 13° in the pristine surface. In addition, the volume of these octahedra decreases by 1.64% and 0.26%, respectively, due to a shortening of the Ru–Osurf bond. Adsorption at B3 or B4, i.e., at positions where STM shows a dark line, is disfavoured by 0.4 eV due to the shorter distances to the surface oxygen atoms and the resulting repulsion between Osurf2− and (O2−)ads. Dissociative adsorption results in an energy penalty close to 1 eV.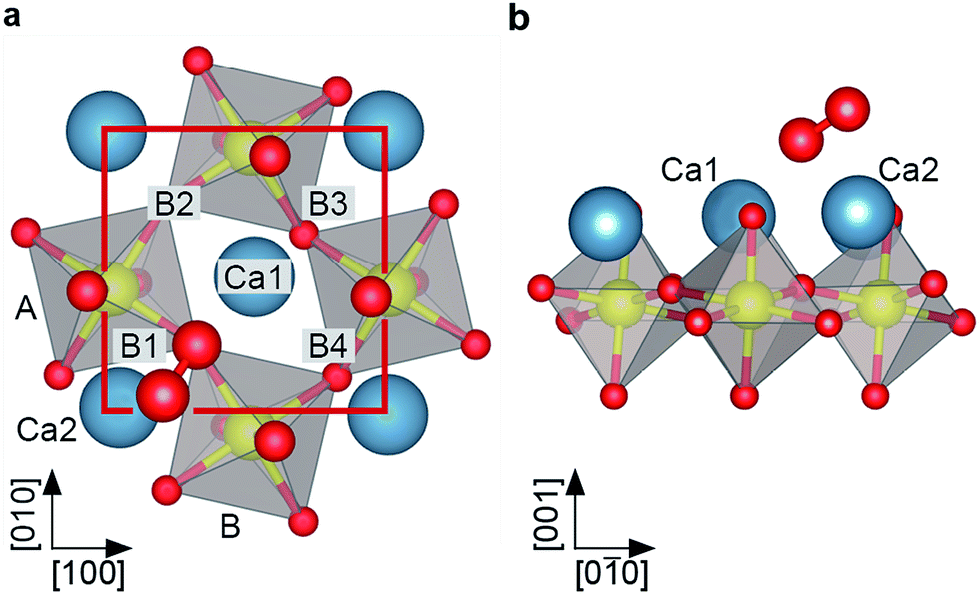 | ||
| Fig. 2 DFT model of O2− adsorbed on Ca3Ru2O7. (a) Top view and (b) side view of the vdW-DFT model for O2− with Eads = 1.4 eV. The molecule adsorbs as a tilted superoxo species close to a Ca–Ca bridge (here B1). | ||
When the Ca3Ru2O7(001) surface is exposed to a low dose of O2 at 5 K, AFM images taken at 0 V sample bias voltage show dark spots of varying contrast and size, see Fig. 3a. When applying a slightly negative or positive sample bias voltage, tunnelling current starts to flow and a few adsorbates interact with the tip, see Fig. 3b. In Fig. 3c two larger, dark spots change their position and contrast in the consecutive image in Fig. 3d and an additional feature appears in the vicinity of the two spots. This suggests that the varying contrast and size of the dark spots at zero bias voltage (Fig. 3a) can be attributed to molecules adsorbed at surface defects or in non-equilibrium positions, or molecules adsorbed too close to each other to be resolved.
 | ||
| Fig. 3 Adsorbed O2− in constant-height nc-AFM. Molecular oxygen was dosed at 5.5 K. All panels show the same area (common AFM parameters: A = 40 pm, Tsample = 4.8 K, scale bars 2 nm) but varying bias voltages Vsample and tip-sample distances Δz. (a) Vsample = 0 V. (b) Vsample = −120 mV, Δz = +25 pm. (c) Vsample = +150 mV, Δz = 0 pm. (d) Vsample = +220 mV, Δz = +28 pm. Open triangles in (c, d) mark the same position. The full triangle marks a new feature, suggesting that the darker spots contain more than one molecule. | ||
It is well-known that the tip's electric field and/or the tunnelling current can influence adsorbates.43,44 The calculated O–O stretch mode for the adsorbed O2− lies at 139 meV (1121 cm−1); exciting this mode could be the reason for the increase in mobility observed in Fig. 3c, d. In comparison, STM images of a similarly-prepared sample taken at 78 K show only streaks across surface defects, see Fig. 4. Occasionally the streaks continue along the [010] direction after the tip first scanned across a defect, indicating that a molecule was being moved along the bright substrate line. This shows that the molecules first adsorb at surface defects and the difficulty of imaging weakly-bound adsorbates with STM, see also Fig. S1.†
 | ||
| Fig. 4 STM image after dosing 0.025 L O2 at 110 K. The molecules appear as streaks across point defects. STM parameters: Vsample = +1.0 V, Itunnel = 0.1 nA, Tsample = 78 K; fast scan direction is 58° clockwise from horizontal. The scale bar corresponds to 3 nm. | ||
High coverage of O2−
At a higher coverage of one molecule per primitive unit cell, DFT calculations predict adsorption in a zig-zag arrangement, where all O2− are spaced one lattice constant apart in [010] direction and adjacent rows are shifted by half a lattice constant in [010] direction, see Fig. 5. With the O2− in one row adsorbed exclusively at B1 and in the adjacent one adsorbed exclusively at B2, they form a (2 × 1) overlayer. Although the O2− is still adsorbed as an intact superoxide ion with bond length 1.35 Å and a vibrational stretching frequency of 1121 cm−1, the vdW-DFT adsorption energy is reduced to 1.19 eV per O2−. This decrease of approximately 0.2 eV is mainly attributed to the electrostatic repulsion, as the minimal distance between the O2− molecules is decreased from 16 Å to 6 Å. The O2− are located on the bright substrate lines, between a Ca and an apical O atom. In contrast to the low-coverage case the adsorbate is only slightly rotated azimuthally to the [010] direction; tilted by just 4° with respect to the surface plane. The tilting of the two RuO6 closest to the adsorbate is 13.4°, slightly more than for the pristine surface (13.0°). A parallel arrangement, where adsorbed O2− in adjacent rows are not shifted by half a lattice constant in [010] direction is less favored by 164 meV per O2−. Adsorption in B3 or B4, i.e. the dark substrate line, is disfavored by 448 meV, similar to the low-coverage case.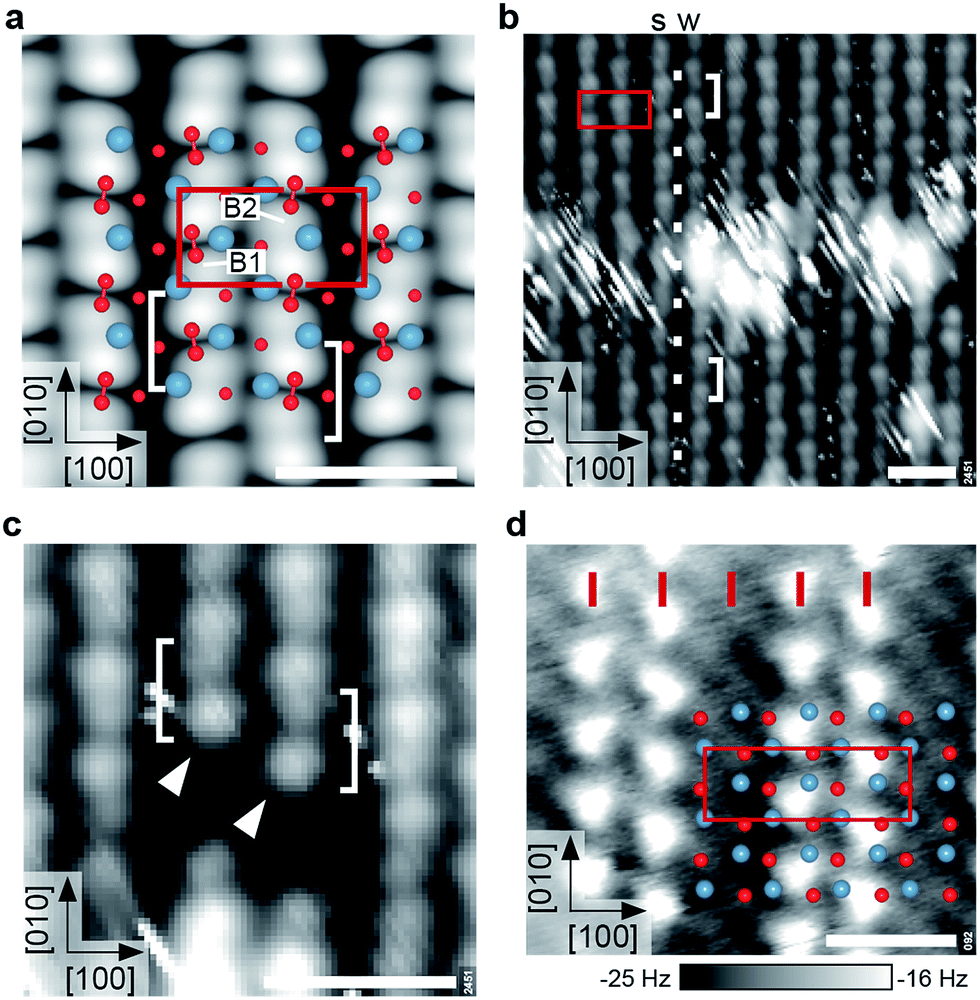 | ||
| Fig. 5 Ordered superoxide overlayers. All scale bars correspond to 1 nm. (a) DFT model and STM simulation (Vsample = +0.8 V) of the (2 × 1) overlayer with one molecule per primitive unit cell. Bridge sites B1 and B2 are labeled as in Fig. 2. Note that the O2− appear as bright elongated spots with a dark node in the center (brackets). (b) Experimental STM image of the (2 × 1) overlayer. The bright, streaky area at the center is a domain boundary as indicated by the registry of the white dots. The O2− rows show a weak (‘w’) or strong (‘s’) degree of undulation. (STM parameters: Vsample = +1.0 V, Itunnel = 0.05 nA, Tsample = 78 K; fast scan direction is 59° clockwise from horizontal.) (c) Detailed STM image (same parameters) of O2− adsorbed next to a surface defect. Both rows begin with a bright spot (arrows) followed by the distinct node and a slightly weaker spot, in agreement with (a). (d) AFM image of a partially covered sample, showing a (3 × 1) overlayer. Red ticks mark lattice-constant intervals along [100]. The O2− and surface O are imaged as intense and faint, bright spots, respectively (corresponding to repulsion). AFM parameters: A = 500 pm, Vsample = 0 V, Tsample = 4.8 K. | ||
The STM simulation of the (2 × 1) overlayer shows bright, elongated spots corresponding to a superposition of two lobes of the neighboring O2−, separated by a dark node at the center of the molecular bond, rather than between two molecules (see Fig. 5a). The electronic contrast leads to an apparently alternating spacing of the rows in STM although the O2− molecules are equally spaced in [100] direction. Here only a STM simulation is shown as the simulation of atomically-resolved nc-AFM is more involved, since the tip has to be taken into account explicitly.
When going from low coverage towards saturation, the molecules become less mobile and STM images less streaky, until the (2 × 1) overlayer forms, see Fig. S1.† The experimental saturation coverage of close to one monolayer (ML, where 1 ML is defined as one adsorbate per primitive unit cell) is shown in Fig. 5b. The O2− covered part of the surface appears as rows of pairs of a bright and a slightly weaker spot on a black background, running in [010] direction. The distance between the pairs along the row is one lattice constant, and every other gap between the bright and slightly weaker spots is more pronounced. Neighbouring rows are shifted by half a lattice constant in [010] direction, and appear subtly different as the spots in one row strongly (weakly) undulate with respect to the [100] direction, marked by s (w) in Fig. 5b. The O2− rows have alternatingly narrow and wide gaps in [100] direction. The bright, streaky areas are domain boundaries: the spacing of the rows labelled s and w is narrow in the upper half of the image, but wide in the lower half. The row that undulates strongly in the upper half of the image undulates weakly in the lower half and vice versa. This is because the molecules are shifted half a lattice constant in [010] direction across the domain boundary, where the row with all O2− adsorbed in B1 sites becomes a row with all O2− adsorbed in B2 sites and vice versa (compare to the white dots that mark lattice-constant-intervals along [010]). The streaky appearance of the domain boundaries indicates weakly-bound molecules that cannot be imaged with STM. This results in an apparent coverage of only 0.9 ML when counting all O2− on the rows.
Fig. 5c shows two O2− rows (marked by arrows) that start next to an unknown, dark surface defect. Both rows begin with a bright spot followed by the more pronounced gap and a slightly weaker spot. This agrees well with the STM simulation showing a distinct node in the center of the molecules, and a weaker depression between two molecules. To confirm that the O2− are adsorbed in either B1 or B2 sites, i.e., on the bright substrate lines, a part of the overlayer was removed by scanning with higher bias voltage (+2.0 V), see Fig. S2.†
While, as pointed out above, STM disturbs molecules at low coverage too much for reliable imaging, this is possible with AFM. Fig. 5d shows an AFM image of a sample partially covered with O2−. It should be noted that after the tip approach with non-zero sample bias voltage the tip was moved to a different region with 0 V bias voltage to exclude a possible charging or manipulation of the adsorbate upon the initial scan. In AFM the molecules are imaged as bright spots (less negative Δf, more repulsive). The surface oxygen atoms of the uncovered sample between the O2− rows are imaged as faint, bright spots. The overlay shows the surface atoms of the pristine surface aligned to the uncovered part of the sample, and confirms that the O2− molecules adsorb close to a Ca–Ca bridge. In contrast to STM images the rows are equally spaced by one lattice constant in [100] direction. Thus the alternating row spacing in STM images (Fig. 5b) is mainly due to a difference in electronic contrast.
Electronic properties
The Ca3Ru2O7 substrate shows a first-order transition from a metallic to a non-metallic phase at 48 K.45 This is reflected in the theoretical description: while the standard vdW-DFT calculations yield the metallic ground state due to the common underestimation of the electronic band gap, more involved approaches such as hybrid functional (HSE06) calculations predict the formation of a gap of 1.27 eV. A single iteration of many-electron (G0W0) corrections33 does not qualitatively change this finding.A detailed analysis of the electronic properties of the (2 × 1) overlayer shows that the O2 molecule does not adsorb as a neutral molecule. The density of states show an additional, occupied π* state in the minority channel, leading to the formation of a charged O2− species, regardless of the level of theory, see Fig. 6. An analysis of the differential charge shows that the electron is transferred into the π* orbital oriented towards the nearest Ca, filling this orbital, see Fig. 7a, b. This is also reflected in the magnetic properties: the neutral gas phase O2 molecule has two half-filled π* orbitals in the triplet state resulting in a magnetic moment of 2 μB. Upon adsorption, one π* orbital becomes filled, leaving the second orthogonal π* state singly occupied. This orbital carries the remaining magnetic moment of about 1 μB (see Fig. 7c). The electron is transferred from the RuO states at the Fermi level in case of the metallic substrate phase described by vdW-DFT, and from the Ru of the closest RuO6 octahedron in the insulating substrate phase described by the HSE06 functional. As a consequence of the more localized electron transfer in the insulating phase the RuO6 octahedra in the surface layer closest to the O2− molecule contract by 5.2% while the remaining two expand by 3.0%, see Fig. S3a.† In vdW-DFT approximation, only a slight contraction (3%) of the octahedra close to the molecule takes place (Fig. S3b†), where the bond between the central Ru and the apical O atom is shortened. Compared to the metallic phase, only minor structural changes occur in the HSE06 calculations: the O2− molecules rotate by 4.5° clockwise and 4.3° counterclockwise, and the tilting angle is increased to a value of 12° with respect to the surface plane. Although the O2− adsorption geometries are almost identical, the adsorption energy decreases from 1.19 eV to 0.58 eV and the HOMO–LUMO gap of the O2− increases from 0.7 eV to 3.1 eV when comparing the HSE06 to the vdW-DFT result. The HSE06 functional certainly improves the description of the electronic structure of the molecular adsorbate, but does not capture the metallic character of the substrate and tends to underestimate the screening properties of metals and small-gap semiconductors.46 However, the HSE06 adsorption energy of 0.58 eV agrees well with the experimental estimate of 0.6 eV (see below).
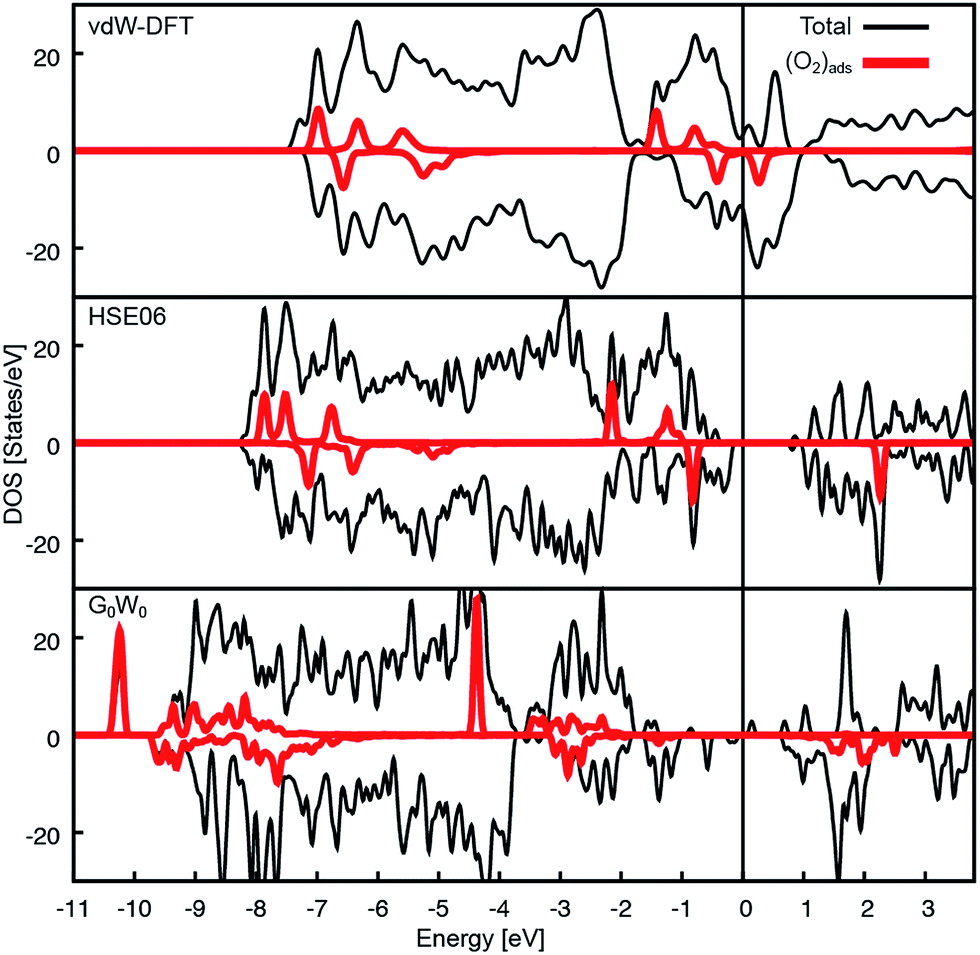 | ||
| Fig. 6 Density of states for the (2 × 1) overlayer. Total and O2 DOS for the different theoretical approaches. In all cases charge is transferred into the split π* states of the O2 in the spin down channel. The HOMO–LUMO gap is 0.7, 3.1 and approximately 1.7 eV on the DFT, HSE06 (insulating phase) and G0W0 level, respectively. | ||
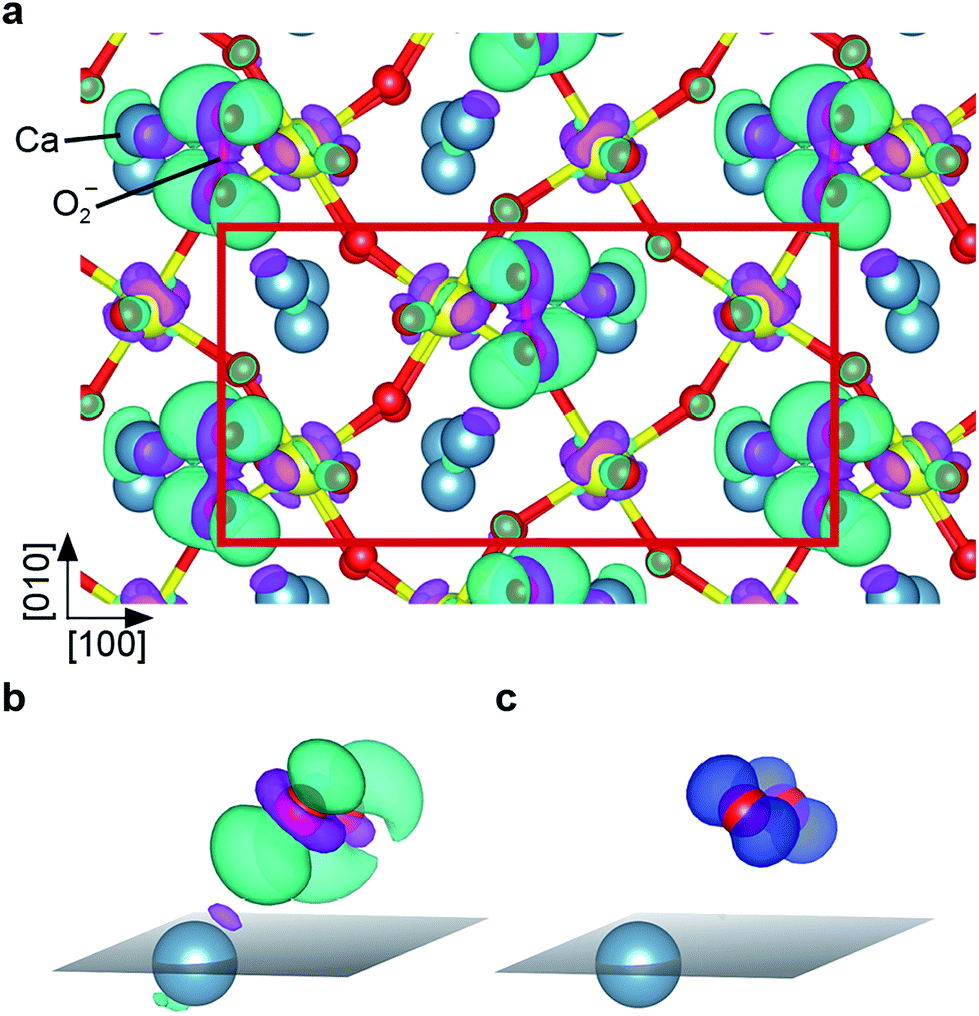 | ||
| Fig. 7 (a) Charge density difference plot of the (2 × 1) overlayer, top view: charge is transferred to and from the turquoise and violet areas, respectively. (b) Detailed view (surface plane marked in grey) of the charge difference of the Ca and O2− labelled in (a): the additional electron is transferred into a π* state rotated towards the nearest Ca atom. (c) Magnetization density plot: the blue colored orbitals carry the magnetic moment of the superoxo species. These orbitals are perpendicular to the orbital that the additional electron is transferred to, compare to (b) (same perspective). | ||
To assess why the adsorption energy according to vdW-DFT is too high compared to the experimental value, and whether the strong decrease in adsorption energy in the HSE06 calculation is due to the insulating substrate phase or the increased HOMO–LUMO gap, a state-of-the-art many-electron-approach in the random phase approximation47 (RPA) was used. The RPA calculations predict an O2− adsorption energy of 0.72 eV and the corresponding G0W0 calculations result in the metallic substrate phase with a HOMO–LUMO gap of the O2− of approximately 1.7 eV (see Fig. 6c), which is significantly larger compared to the vdW-DFT calculation. The increase in the adsorption energy of 0.14 eV compared to HSE06 is attributed to additional non-local contributions such as vdW interactions that are not considered in HSE06.
The comparison of different theoretical approaches (see Table 1) indicates several contributions to the adsorption energy: comparing the PBE to the PBE+U results shows that the overestimation of the polarizability of the surface on the GGA level contributes about 0.2 eV, a similar increase as obtained by including vdW corrections. A third contribution stems from the underestimation of the HOMO–LUMO gap: since the charge state of the molecule changes, a correct description of the electron affinity, i.e., the cost to add an additional electron to the O2 molecule, plays a key role for the energy balance. Comparing the PBE and the HSE06 functional, advanced quantum-chemical (CCSD(T)) calculations48,49 show an electron affinity difference of gas phase O2 of 0.15 eV, which is close to the predicted difference in adsorption energy when comparing the DFT+U and the HSE06 results.
| Computational method | Adsorption energy [eV] |
|---|---|
| PBE | 0.93 |
| PBE+U (U − J = 4 eV) | 0.68 |
| vdW-DF | 1.19 |
| HSE06 | 0.59 |
| RPA | 0.72 |
Spectroscopies on the (2 × 1) overlayer
To gain experimental information on the charge state of the O2 overlayer, point Kelvin probe spectroscopy51 was performed. For a quantitative analysis it was assumed that the Cu(110) sample used for tip preparation has a work function (Φ) of 4.5 eV.52 The measured local contact potential difference (LCPD) of the Cu sample of +0.4 V gives a tip Φ of 4.1 eV, according to VLCPD = (Φsample − Φtip)/|e|, with e the charge of the electron. This tip was then used to measure the LCPD on a sample where part of the (2 × 1) overlayer was removed (similar to one shown in Fig. S2†) allowing to measure on the (2 × 1) overlayer and the pristine surface in close succession, thus excluding a tip change between the measurements. LCPD values of −1.45 V and +0.2 V were found for the pristine surface and the (2 × 1) overlayer, respectively, see Fig. 8a. Combining the known tip Φ and the measured LCPDs gives a Φ of 2.7 and 4.3 eV for the pristine surface and the (2 × 1) overlayer, respectively. Such a work function increase is due to a negatively charged adsorbate overlayer.53 DFT calculations give a Φ of 3.1 eV and 5.4 eV for the pristine surface and the (2 × 1) overlayer, respectively.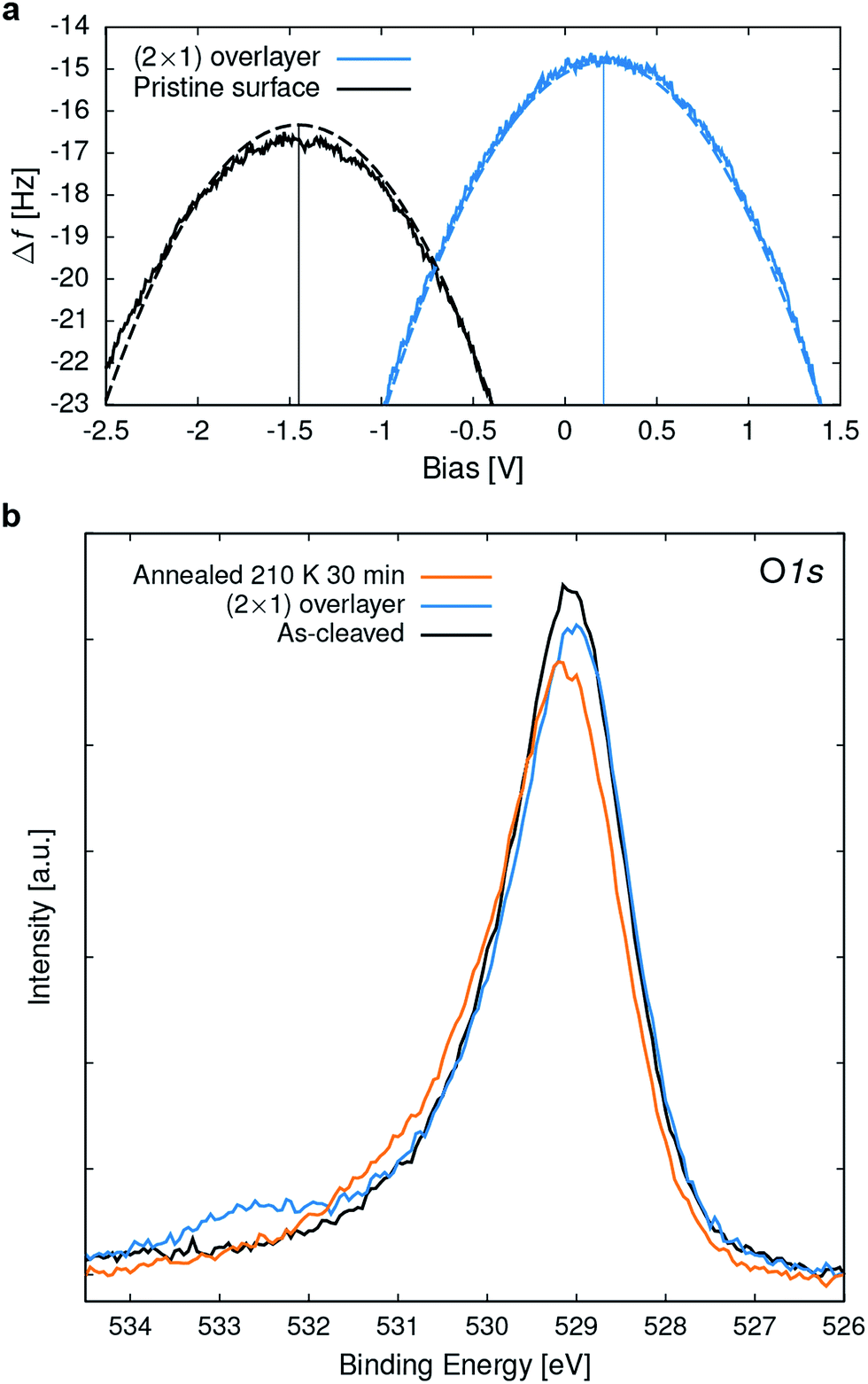 | ||
| Fig. 8 Surface spectroscopies. (a) Point Kelvin probe spectroscopy of the pristine surface and the (2 × 1) O2− overlayer at 78 K. Shown are the measured (full line) and the numerically fitted (dashed lines) Kelvin parabolas, reflecting a change in work function (see main text for analysis details). (b) O 1s XPS (Mg Kα, Tsample = 110 Κ) spectra of the as-cleaved surface, the (2 × 1) O2− overlayer and after annealing at 210 K for 30 min. | ||
In X-ray photoelectron spectroscopy (XPS) the pristine surface exhibits an asymmetric O 1s peak at 529.1 eV binding energy (BE), broadened towards the high BE side, see Fig. 8b. According to vdW-DFT calculations the O 1s core level for the O atoms in the rock-salt like CaO planes is shifted to 0.7 eV higher BE compared to the RuO2 planes, which serve as DFT reference. On a structurally related Sr2RuO4 sample,28 two distinct O 1s peaks were observed and attributed to the apical and equatorial O atoms. The observation of only one asymmetric peak for Ca3Ru2O7 is attributed to the limited resolution of the experimental setup, the 3:4 ratio of O atoms in the CaO and RuO2 planes, and potentially the different electronic structure of these materials. Exposing the sample to 2.5 L O2 at 110 K results in a (2 × 1) overlayer and an O 1s peak at 532.6 eV BE; i.e. 3.5 eV above the peak of the pristine surface. In addition, the lattice oxygen peak shifts towards lower BE due to an upward band bending of 0.1 eV. The O2− peak lies approximately half way between reported BEs for physisorbed, neutral O2 of 536 to 538 eV (ref. 9 and 54) and the double negatively charged bulk O at 529.1 eV. Thus this XPS result is also consistent with a superoxo species. The calculation for the adsorbed O2− gives an initial-state shift of 1.3 eV (which would result in 530.4 eV BE), which is significantly lower than the experimental peak at 532.6 eV. Calculations including the final state approximation have been attempted but were ultimately not considered, as they resulted in an unphysical over-screening of the core-hole: the unoccupied O2 π* orbital calculated just above the Fermi level is pulled below it and localizes an additional electron at the molecule, which directly leads to an underestimation of the binding energy.55 While using a hybrid functional approach (such as HSE06) partially cures this problem by shifting the unoccupied O2 π* orbital away from the Fermi level, it also leads to an insulating substrate and therefore was not considered for the evaluation of the core level shifts.
For the superoxo species a double peak with a 1:3 ratio is expected due to the coupling of the O1s hole spin to the O2p valence states resulting in a single or triplet final state configuration. Similarly, adsorbed neutral O2 leads to a double peak with a 2:4 ratio (doublet and quadruplet) and a splitting of roughly 1 eV.9,54 The observation of only one broad peak in Fig. 8b is attributed to the relatively small splitting of 0.25 eV (estimated from DFT calculations) and the limited experimental resolution.
When annealing the O2− saturated sample to room temperature, the molecule desorbs, the band bending vanishes, and subsequent STM images show the pristine surface. The superoxo peak in the O 1s XPS starts to disappear at approximately 200 K, see Fig. 8b. The slight increase at higher binding energy is probably due to hydroxyls that accumulated during this experiment.29 A rough estimate for the adsorption energy based on the desorption temperature is 0.6 eV, which is in good agreement with the calculated adsorption energy of 0.72 eV (RPA).
Discussion
These results provide clear evidence of the adsorption geometry and electronic structure of O2− on a well-defined perovskite-type surface and it is interesting to draw comparisons to studies on metals and binary metal oxides.The spectroscopic results and DFT calculations conclusively show that molecular oxygen readily adsorbs as O2− on the as-cleaved Ca3Ru2O7(001) surface. The calculated bond length of 1.35 Å and the internal stretching frequency of 1121 cm−1 agrees well to the values found for O2− on late transition metals.56 The source of electrons for charging the O2 are the Ru d-states in the RuO2 subsurface layer below the CaO-terminated surface. According to the calculations, in the metallic phase the electron is transferred from the valence band, while in the insulating phase it is transferred mainly from a neighboring RuO6 octahedron, which is also reflected in a stronger contraction of that octahedron (see Fig. S3a†). This charge transfer is crucial for the adsorption: on CaO(001) chemisorption is only observed after introducing Mo impurities that donate electrons to the oxide,18 while the RuO2 layers in Ca3Ru2O7 can be seen as intrinsic electron donors. Consequently a full monolayer of adsorbed O2− can form. The transfer of a second electron, i.e., the formation of a peroxo species, is hindered by a too weak hybridization of the states of the substrate and the molecule. This is supported by the density of states that shows that the remaining unoccupied π* state is located substantially above EF (Fig. 6).
In an earlier study it was found that O2 adsorbs as a neutral species on defect-free SrTiO3(001),23 but as an activated species on La2NiO4(001).24 This observation was correlated to the charge transferred from the A-site ions:24 while Sr2+ does not deviate from its formal ionic charge and no charge transfer is observed, the La charge changes from +3 to +2 for both adsorption modes (peroxo and superoxo), suggesting a covalent bond character between La and the charged molecule. In the present work no major charge transfer from the Ca ions is found: while the Ca ions are polarized, the Ca Bader charge stays +2, similarly to Sr2+ on SrTiO3(001),23 as the charge is transferred from the subsurface Ru to the ionically bonded O2− at the surface. Preliminary DFT calculations (not shown) show a similar charge transfer mechanism on the related, SrO-terminated Sr3Ru2O7(001) surface, suggesting that the difference is due to the energetic levels of the B-site ions.
Ultrathin oxide films that are only a few layers (L) thick are often considered as model systems for metal oxide surface chemistry. Calculations for ultrathin MgO films supported on a metal (Mo and Ag) concluded that charge can be transferred from the metal support through the oxide film to the adsorbed oxygen. A comparison of O2− adsorbed on MgO(2L)/Ag (Φ = 3.1 eV)57 and MgO(3L)/Mo (Φ = 2.05 eV)57 showed that the adsorption energy19 is smaller for the system with the higher work function. This suggests that the higher the work function the harder it is to transfer an electron to an adsorbate. The work function and O2− adsorption energy on Ca3Ru2O7(001) of 2.7 and 0.72 eV, respectively, are similar to the values for the MgO(2L)/Ag system, 3.1 and 0.64 eV.
The formation of a full, adsorbed overlayer of O2−, and the availability of atomically-resolved nc-AFM allows to directly inspect the configurations of the adsorbate, thus providing a tight connection to the theoretical calculations. At low coverage, single O2− could be imaged by AFM, but the application of a relatively low sample bias voltage (∼100 mV) already resulted in interactions between the scanning tip and the molecules (Fig. 3 and 4). The interactions occurred for positive as well as negative sample bias voltages, and the exact threshold varied between 100 and 150 mV depending on the tip-sample distance and the particular molecule. At higher sample bias voltages of roughly −1 and +2 V the O2− is desorbed by the tip and the pristine surface is revealed (see Fig. S2†). The desorption at negative sample bias voltage, where the electrons tunnel from the molecule to the tip, might be facilitated by discharging the O2− as shown on anatase TiO2(101).9
At increasing coverages the O2− form an ordered (2 × 1) overlayer up to an apparent (in STM) saturation coverage of close to 1 ML. In contrast to isolated O2− the molecules in the overlayer can be imaged by STM without perturbation by the tip, except for the overlayer domain boundaries (Fig. 5b). Within the ordered overlayer the molecules are not moved about by the STM tip, probably because they are locked in by adjacent molecules, while at the boundaries tip-induced movement of molecules is possible. This agrees with the observation that after desorbing the O2− from a specific area by scanning at higher bias voltage, molecules from the surrounding area diffuse back to the area again during subsequent STM images taken at the usual, lower sample bias voltage (see Movie S1†).
Applications utilizing the ORR usually operate at much higher temperatures than the experimental temperatures of the present work. Therefore it is interesting how the interaction with O2 would change at realistic working conditions. As the tilting/rotation of the RuO6 octahedra strongly influences the adsorption energy of O2− in the different adsorption sites (B1/B2 and B3/B4, see Fig. 2) it is appropriate to consider what happens to the RuO6 as the temperature is raised. The temperature dependence of the Ca3Ru2O7 structure was studied by Yoshida et al.58 using neutron scattering up to 292 K. Up to this temperature their data suggests that the RuO6 tilting angle slightly decreases but the structure does not undergo any dramatic changes. The orthorhombic structure of Ca2RuO4 and Ca3Ru2O7 is caused by RuO6 octahedral rotation and tilt. For Ca2RuO4, when both rotation and tilt disappear at elevated temperatures (>357 K), its structure becomes tetragonal.59 If Ca3Ru2O7 follows a similar trend (this has not been studied), adsorption of O2 as superoxo species would still be expected as it was calculated for tetragonal Sr3Ru2O7 (not published). However, due to the rather low adsorption energy of approximately 0.6 eV, the oxygen partial pressure would have to be substantially increased to achieve any O2− coverage at elevated temperatures: for example, if a simple Langmuir adsorption model is assumed, the O2 partial pressure necessary for a coverage of 0.5 ML at 900 K is 1 bar. Regarding a possible change of the surface termination at elevated temperatures it can only be speculated that a RuO2-terminated surface would reconstruct. This is based on the fact that the CaO termination is by far the lowest-energy configuration, and that SrTiO3 (the only perovskite, where a significant number of surface studies are available) is known to form a variety of complex reconstructions with a structure that is sensitively dependent on the composition.60 However, in the present work no different termination than CaO was observed and studies of different perovskite-type materials after high temperature (1000 °C) treatment under oxidizing conditions suggest that the surfaces are AO-terminated.61
Conclusion
The as-cleaved and unmodified Ca3Ru2O7(001) surface activates molecular oxygen to O2− upon adsorption, with the charge stemming from the subsurface RuO2 layer. While the adsorption site and configurations are reflected well at the level of vdW-DFT, more accurate theoretical approaches are necessary for a correct modelling of the charge redistribution and realistic binding energies. The advantages of a combined STM and nc-AFM approach in case of adsorbates sensitive to electric field or tunnelling current are demonstrated; as single superoxo species are strongly influenced by the STM tip. When increasing the coverage, the O2− forms a full, (2 × 1) ordered monolayer that can be resolved by STM and AFM. The negative charge state of the molecules is evident in Kelvin probe and X-ray photoelectron spectroscopy. No dopants, defects, or low-coordinated sites are necessary to facilitate the activation of molecular O2 on this oxide surface.Author contributions
D. H. performed the STM and XPS experiments and data analysis. D. H. and M. Se. performed the AFM measurements. W. M. S., F. M., and J. R. performed the DFT calculations. D. F., J. P., and Z. M. grew the sample. U. D. directed and supervised the project. D. H., W. M. S., F. M., M. Sc., and U. D. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Austrian Science Fund (FWF project F45), the ERC Advanced Grant “OxideSurfaces”, and the Vienna Scientific Cluster (VSC). Structural drawings were prepared using the program VESTA.62 The work at Tulane is supported by the U.S. Department of Energy under EPSCoR Grant No. DE-SC0012432 with additional support from the Louisiana Board of Regents.References
- J. A. Kilner and M. Burriel, Materials for intermediate-temperature solid-oxide fuel cells, Annu. Rev. Mater. Res., 2014, 44, 365–393 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. Alnashef, Superoxide ion: generation and chemical implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- G. M. Rupp, A. K. Opitz, A. Nenning, A. Limbeck and J. Fleig, Real-time impedance monitoring of oxygen reduction during surface modification of thin film cathodes, Nat. Mater., 2017, 16, 640–645 CrossRef CAS PubMed.
- R. Merkle and J. Maier, How is oxygen incorporated into oxides? A comprehensive kinetic study of a simple solid-state reaction with SrTiO3 as a model material, Angew. Chem., Int. Ed., 2008, 47, 3874–3894 CrossRef CAS PubMed.
- C. Schweitzer and R. Schmidt, Physical mechanisms of generation and deactivation of singlet oxygen., Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed.
- M. Anpo, M. Che, B. Fubini and E. Garrone, Generation of superoxide ions at oxide surfaces, Top. Catal., 1999, 8, 189–198 CrossRef CAS.
- M. Setvin, X. Hao, B. Daniel, J. Pavelec, Z. Novotny, G. S. Parkinson, M. Schmid, G. Kresse, C. Franchini and U. Diebold, Charge trapping at the step edges of TiO2 anatase (101), Angew. Chem., Int. Ed., 2014, 53, 4714–4716 CrossRef CAS PubMed.
- M. Setvin, J. Hulva, G. S. Parkinson, M. Schmid and U. Diebold, Electron transfer between anatase TiO2 and an O2 molecule directly observed by atomic force microscopy, Proc. Natl. Acad. Sci., 2017, 114, E2556–E2562 CrossRef CAS PubMed.
- Y. Zhao, B. T. Teng, X. D. Wen, Y. Zhao, Q. P. Chen, L. H. Zhao and M. F. Luo, Superoxide and peroxide species on CeO2(111), and their oxidation roles, J. Phys. Chem. C, 2012, 116, 15986–15991 CAS.
- M. Nolan, Healing of oxygen vacancies on reduced surfaces of gold-doped ceria, J. Chem. Phys., 2009, 130, 144702 CrossRef PubMed.
- Y. M. Choi, H. Abernathy, H. T. Chen, M. C. Lin and M. Liu, Characterization of O2–CeO2 interactions using in situ Raman spectroscopy and first-principle calculations, ChemPhysChem, 2006, 7, 1957–1963 CrossRef CAS PubMed.
- V. V. Pushkarev, V. I. Kovalchuk and J. L. D'Itri, Probing defect sites on the CeO2 surface with dioxygen, J. Phys. Chem. B, 2004, 108, 5341–5348 CrossRef CAS.
- X. Xia, R. J. Oldman and C. R. A. Catlow, Oxygen adsorption and dissociation on yttria stabilized zirconia surfaces, J. Mater. Chem., 2012, 22, 8594 RSC.
- Y. Yan, M. M. Al-Jassim and S.-H. Wei, Oxygen-vacancy mediated adsorption and reactions of molecular oxygen on the ZnO(100) surface, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 161307 CrossRef.
- D. Ma, Z. Wang, H. Cui, J. Zeng, C. He and Z. Lu, First-principles study of O2 adsorption on Al-doped ZnO(100) surface, Sens. Actuators, B, 2016, 224, 372–380 CrossRef CAS.
- A. Gurlo, Interplay between O2 and SnO2: Oxygen ionosorption and spectroscopic evidence for adsorbed oxygen, ChemPhysChem, 2006, 7, 2041–2052 CrossRef CAS PubMed.
- Y. Cui, X. Shao, M. Baldofski, J. Sauer, N. Nilius and H. J. Freund, Adsorption, activation, and dissociation of oxygen on doped oxides, Angew. Chem., Int. Ed., 2013, 52, 11385–11387 CrossRef CAS PubMed.
- P. Frondelius, H. Häkkinen and K. Honkala, Adsorption and activation of O2 at Au chains on MgO/Mo thin films., Phys. Chem. Chem. Phys., 2010, 12, 1483–1492 RSC.
- M. Che and A. J. Tench, Characterization and reactivity of molecular oxygen species on oxide surfaces, Adv. Catal., 1983, 32, 1–148 CAS.
- A. Hellman, S. Klacar and H. Grӧnbeck, Low temperature CO oxidation over supported ultrathin MgO films, J. Am. Chem. Soc., 2009, 131, 16636–16637 CrossRef CAS PubMed.
- A. Gonchar, T. Risse, H. J. Freund, L. Giordano, C. Di Valentin and G. Pacchioni, Activation of oxygen on MgO: O2− radical ion formation on thin, metal-supported MgO(001) films, Angew. Chem., Int. Ed., 2011, 50, 2635–2638 CrossRef CAS PubMed.
- A. Staykov, H. Téllez, T. Akbay, J. Druce, T. Ishihara and J. Kilner, Oxygen activation and dissociation on transition metal free perovskite surfaces, Chem. Mater., 2015, 27, 8273–8281 CrossRef CAS.
- T. Akbay, A. Staykov, J. Druce, H. Téllez, T. Ishihara and J. A. Kilner, The interaction of molecular oxygen on LaO terminated surfaces of La2NiO4, J. Mater. Chem. A, 2016, 4, 13113–13124 CAS.
- G. Kresse, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Van der Waals density functionals applied to solids, Phys. Rev. B: Condens. Matter Mater. Phys.B, 2011, 83, 195131 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, Chemical accuracy for the van der Waals density functional., J. Phys.: Condens. Matter, 2010, 22, 22201 CrossRef PubMed.
- D. Halwidl, B. Stöger, W. Mayr-Schmölzer, J. Pavelec, D. Fobes, J. Peng, Z. Mao, G. S. Parkinson, M. Schmid, F. Mittendorfer, J. Redinger and U. Diebold, Adsorption of water at the SrO surface of ruthenates, Nat. Mater., 2015, 15, 450–455 CrossRef PubMed.
- D. Halwidl, W. Mayr-Schmölzer, D. Fobes, J. Peng, Z. Mao, M. Schmid, F. Mittendorfer, J. Redinger and U. Diebold, Ordered hydroxyls on Ca3Ru2O7(001), Nat. Commun., 2017, 8, 23 CrossRef PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- M. Kaltak, J. Klimeš and G. Kresse, Cubic scaling algorithm for the random phase approximation: Self-interstitials and vacancies in Si, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 54115 CrossRef.
- M. Kaltak, J. Klimeš and G. Kresse, Low scaling algorithms for the random phase approximation: imaginary time and Laplace transformations, J. Chem. Theory Comput., 2014, 10, 2498–2507 CrossRef CAS PubMed.
- P. Liu, M. Kaltak, J. Klimeš and G. Kresse, Cubic scaling GW: towards fast quasiparticle calculations, Phys. Rev. B, 2016, 94, 165109 CrossRef.
- F. J. Giessibl, Sensor for Noncontact Profiling of a Surface, US Pat. 2012/0131704 A1, 2012.
- F. Huber and F. J. Giessibl, Low noise current preamplifier for qPlus sensor deflection signal detection in atomic force microscopy at room and low temperatures, Rev. Sci. Instrum., 2017, 88, 73702 CrossRef PubMed.
- M. Setvín, J. Javorský, D. Turčinková, I. Matolínová, P. Sobotík, P. Kocán and I. Ošt’ádal, Ultrasharp tungsten tips - characterization and nondestructive cleaning, Ultramicroscopy, 2012, 113, 152–157 CrossRef.
- J. I. J. Choi, W. Mayr-Schmölzer, F. Mittendorfer, J. Redinger, U. Diebold and M. Schmid, The growth of ultra-thin zirconia films on Pd3Zr(0001), J. Phys.: Condens. Matter, 2014, 26, 225003 CrossRef CAS PubMed.
- W. Bao, Z. Q. Mao, Z. Qu and J. W. Lynn, Spin valve effect and magnetoresistivity in single crystalline Ca3Ru2O7, Phys. Rev. Lett., 2008, 100, 247203 CrossRef PubMed.
- G. Cao, K. Abboud, S. McCall, J. E. Crow and R. P. Guertin, Spin-charge coupling for dilute La-doped Ca3Ru2O7, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 998–1003 CrossRef CAS.
- J. Tersoff and D. R. Hamann, Theory of the scanning tunneling microscope, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805–813 CrossRef CAS.
- A. Yurtsever, D. Fernández-Torre, C. González, P. Jelínek, P. Pou, Y. Sugimoto, M. Abe, R. Pérez and S. Morita, Understanding image contrast formation in TiO2 with force spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 1–9 CrossRef.
- R. Bechstein, Ć. Gonźalez, J. Schütte, P. Jeĺinek, R. Ṕerez and A. Kühnle, ‘All-inclusive’ imaging of the rutile TiO2(110) surface using NC-AFM, Nanotechnology, 2009, 20, 505703 CrossRef PubMed.
- H. J. Lee and W. Ho, Single-bond formation and characterization with a scanning tunneling microscope, Science, 1999, 286, 1719–1722 CrossRef CAS PubMed.
- Y. Kim, K. Motobayashi, T. Frederiksen, H. Ueba and M. Kawai, Action spectroscopy for single-molecule reactions - Experiments and theory, Prog. Surf. Sci., 2015, 90, 85–143 CrossRef CAS.
- G. Cao, S. McCall, J. E. Crow and R. P. Guertin, Observation of a metallic antiferromagnetic phase and metal to nonmetal transition in Ca3Ru2O7, Phys. Rev. Lett., 1997, 78, 1751–1754 CrossRef CAS.
- J. Paier, M. Marsman and G. Kresse, Dielectric properties and excitons for extended systems from hybrid functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 1–4 CrossRef.
- L. Schimka, J. Harl, A. Stroppa, A. Grüneis, M. Marsman, F. Mittendorfer and G. Kresse, Accurate surface and adsorption energies from many-body perturbation theory, Nat. Mater., 2010, 9, 741–744 CrossRef CAS PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Atomic and molecular electron affinities: photoelectron experiments and theoretical computations, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed.
- National Institute of Standard and Technology, NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database No. 101., 2006 Search PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- S. Sadewasser and T. Glatzel, Kelvin Probe Force Microscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012 Search PubMed.
- P. O. Gartland, S. Berge and B. J. Slagsvold, Photoelectric work function of a copper single crystal for the (100), (110), (111), and (112) faces, Phys. Rev. Lett., 1972, 28, 738–739 CrossRef CAS.
- L. Gross, F. Mohn, P. Liljeroth, J. Repp, F. J. Giessibl and G. Meyer, Measuring the charge state of an adatom with noncontact atomic force microscopy., Science, 2009, 324, 1428–1431 CrossRef CAS PubMed.
- C. Puglia, A. Nilsson, B. Hernnäs, O. Karis, P. Bennich and N. Mårtensson, Physisorbed, chemisorbed and dissociated O2 on Pt(111) studied by different core level spectroscopy methods, Surf. Sci., 1995, 342, 119–133 CrossRef CAS.
- H. Li, J. I. J. Choi, W. Mayr-Schmölzer, C. Weilach, C. Rameshan, F. Mittendorfer, J. Redinger, M. Schmid and G. Rupprechter, Growth of an ultrathin zirconia film on Pt3Zr examined by high-resolution X-ray photoelectron spectroscopy, temperature-programmed desorption, scanning tunneling microscopy, and density functional theory, J. Phys. Chem. C, 2015, 119, 2462–2470 CAS.
- M. M. Montemore, M. A. van Spronsen, R. J. Madix and C. M. Friend, O2 activation by metal surfaces: implications for bonding and reactivity on heterogeneous catalysts, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.7b00217.
- L. Giordano, F. Cinquini and G. Pacchioni, Tuning the surface metal work function by deposition of ultrathin oxide films: Density functional calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 1–6 CrossRef.
- Y. Yoshida, S.-I. Ikeda, H. Matsuhata, N. Shirakawa, C. Lee and S. Katano, Crystal and magnetic structure of Ca3Ru2O7, Phys. Rev. B, 2005, 72, 54412 CrossRef.
- C. Alexander, G. Cao, V. Dobrosavljevic, S. McCall, J. Crow, E. Lochner and R. Guertin, Destruction of the Mott insulating ground state of Ca2RuO4 by a structural transition, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, R8422–R8425 CrossRef CAS.
- Z. Wang, A. Loon, A. Subramanian, S. Gerhold, E. McDermott, J. A. Enterkin, M. Hieckel, B. C. Russell, R. J. Green, A. Moewes, J. Guo, P. Blaha, M. R. Castell, U. Diebold and L. D. Marks, Transition from reconstruction toward thin film on the (110) surface of strontium titanate, Nano Lett., 2016, 16, 2407–2412 CrossRef CAS PubMed.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Surface termination and subsurface restructuring of perovskite-based solid oxide electrode materials, Energy Environ. Sci., 2014, 7, 3593–3599 CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: An STM movie in avi format is available (Movie S1). See DOI: 10.1039/c8ta00265g |
| This journal is © The Royal Society of Chemistry 2018 |