
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of ammonolysis and of sol–gel titanium oxide nitride coating on carbon fibres for use in flexible supercapacitors†
Junxian
Zhang
ab,
Andrew L.
Hector
 *a,
Samantha
Soulé
a,
Qinghua
Zhang
*b and
Xin
Zhao
b
*a,
Samantha
Soulé
a,
Qinghua
Zhang
*b and
Xin
Zhao
b
aChemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: A.L.Hector@soton.ac.uk
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science & Engineering, Donghua University, Shanghai 201620, China. E-mail: qhzhang@dhu.edu.cn
First published on 12th March 2018
Abstract
Smooth titanium oxide nitride coatings have been deposited on carbon fibres using a non-oxide sol–gel method based on self-condensation of titanium alkylamide species. Both the coating and a nitridation of the fibres resulting from the coating process increase the capacitance of the fibres measured in aqueous acid conditions, with significant redox capacitance contributions. When flexible symmetric capacitors were made from the coated fibres and an acid gel electrolyte they provided competitive energy and power densities. Their capacitance was retained very effectively on bending up to 180°.
Introduction
Electrical charge storage in batteries and capacitors typically involves devices that are rigid in shape and cannot be incorporated into flexible objects without accommodating a bulky component. Fibre-based supercapacitors with flexible gel electrolytes are a promising way to integrate charge storage into items such as clothing without compromising on design.1,2 Carbon fibres have the advantages that they are relatively inexpensive, can maintain electronic conductivity over long distances and can be woven directly into flexible sheets.In order to increase the charge storage capacity of flexible supercapacitors there are two key options, to increase the surface area of the carbon in order to maximise double layer capacitance or to incorporate redox functionalities. Polymers such as polyaniline can be incorporated onto fibre surfaces to provide this functionality.3 Various composites of redox-active metal oxides with carbon particles, tubes, fibres, sheets or 3-dimensional structures have also been tested in flexible supercapacitors.4 Oxides used include TiO2,5,6 V2O5,7 WO3,8 Mn2O3,9 MnO2,10–13 RuO2,14 Co3O4,15 NiO16 and CuO,17 most of which have relatively low electronic conductivities.
Metal nitrides including TiN,18–28 VN,29–39 TiVN,40 NbN,41,42 Nb4N5,43,44 CrN,45 Mn3N2,46 MoN,47 Mo2N,48–52 Fe2N,53,54 RuN55 and WN56 and GaN57 are increasingly studied for supercapacitor applications, with higher conductivities than the respective oxides a key driver. In VN the capacity has been found to increase over initial cycles with surface oxide formation and high capacity has been attributed to this surface oxide.29 In TiN this is more complex, with some authors noting a decrease in capacitance18 but others noting “surface activation”.20 Electrochemical oxidation prior to assembly of devices can enhance capacitance significantly.23 The performance of the native oxide surface on TiN has been linked to its nitrogen content, with a nitrogen-rich material achieving more capacity during cycling due to nitrogen doping of the surface oxide that enhances its conductivity.21 Titanium oxynitride nanostructures have also shown good supercapacitor performance.58
Non-oxide sol–gel methods can be used to produce a range of morphologies including metal nitride powders, films and porous structures.59–63 We recently showed that sols based on Ti(NMe2)4 and nPrNH2 could be used to coat nanocrystalline LiFePO4 powders providing an effective conductive surface when the powders were incorporated into lithium battery cathodes.64 Herein we use an adaptation of that process to coat carbon fibres with the aim of exploiting a redox functionality in the coatings to provide increased capacitance.
Experimental
Tetrakisdimethylamido titanium (Ti(NMe2)4; Epichem) was distilled in vacuo before use. n-Propylamine (Aldrich) was distilled from barium oxide. THF and hexane were purchased from Fisher and distilled from sodium/benzophenone ketyl ether. Carbon fibres were purchased from Zhongfu-Shenying Carbon Fiber Co., Ltd, and were immersed in concentrated sulfuric acid (AR, Pinghu Chemical Reagent Co., Ltd) at room temperature for 2 h, then washed with deionized water several times and dried in the oven for 2 h (designated CF). Anhydrous ammonia (BOC) was dried by passing through a column of molecular sieves before use in ammonolysis reactions, and samples were loaded into furnace tubes that allowed air-free handling of samples prior to heating. Some carbon fibres were annealed under ammonia at 1000 °C for 6 h before use (designated CF–NH3).Sol preparation and fibre coating were carried out under nitrogen using Schlenk or glove box methods. Ti(NMe2)4 was dissolved in hexane (or THF) and nPrNH2 was added, the quantities of each component are shown in Table 1. The solution gradually changed colour from yellow to red-orange. The hexane sols underwent some initial precipitation but this re-dissolved, as reported previously.62 After stirring at room temperature overnight the sols were used to coat carbon fibres by dipping into the sol and allowing the solvent to evaporate in the glove box. After coating with the titanium amide-based gel, the carbon fibres were heated under a flow of NH3 at 2.5 °C min−1 to 800 °C and maintained for 6 h before cooling. The prepared samples with different sol concentrations were designated as CF–TiN-x (x = 1, 2, 3, 4, 5 depending on concentration, Table 1).
| x = 1 | x = 2 | x = 3 | x = 4 | x = 5 | ||
|---|---|---|---|---|---|---|
| Ti(NMe2)4 | Volume/cm3 | 0.43 | 0.64 | 0.85 | 1.27 | 1.49 |
| Moles/mmol | 1.80 | 2.69 | 3.59 | 5.39 | 6.27 | |
| n PrNH2 | Volume/cm3 | 0.30 | 0.44 | 0.59 | 0.89 | 1.04 |
| Moles/mmol | 3.59 | 5.39 | 7.18 | 10.78 | 12.57 | |
| Hexane or THF | Volume/cm3 | 7.50 | 7.50 | 7.50 | 7.50 | 7.50 |
X-ray diffraction (XRD) patterns were obtained from fibres in transmission mode using a Rigaku Smartlab (0.7 mm silica capillaries) or from powders using a Bruker D2 Phaser, both with Cu-Kα radiation. Phase matching used Rigaku PDXL2 and Rietveld refinements used the WPPF routine in PDXL2 with structure models from ICSD.65 Scanning electron microscopy (SEM) used a Philips XL30-ESEM or a Jeol JSM-6500F. Energy dispersive X-ray analysis was carried out on the XL30 with a Thermofisher Ultradry detector and Noran System 7 processing. Thermogravimetric analysis (TGA) was carried out with a Netzsch TG209 F1 Libra, with a flowing O2/Ar mixture (30 cm3 min−1/20 cm3 min−1) and a heating rate of 5 °C min−1. X-ray photoelectron spectroscopy (XPS) used a ThermoFisher Theta Probe spectrometer with focused, monochromatic Al Kα radiation (analysis chamber pressure <3 × 10−9 Pa; hν = 1486.6 eV; spot size 400 μm; constant pass energy of 40 eV). Short acquisition time spectra were recorded at the beginning and at the end of each experiment to check that the samples did not suffer from degradation and/or charging during the measurements. The Casa XPS software package was used for data analysis. The binding energy scale was calibrated from the graphitic carbon at 284.6 eV. Core peaks were analysed using a nonlinear Shirley-type background.66 Peak positions and areas were optimized by a weighted least-squares fitting method using 70% Gaussian and 30% Lorentzian line shapes. Quantification was performed based on Scofield's relative sensitivity factors.67 For each sample, several XPS analyses were performed at different positions to ensure the results were statistically reliable.
Cyclic voltammetry (CV), galvanostatic charge–discharge curves, electrochemical impedance spectroscopy and cycling stability measurements were conducted using a Biologics SP150 potentiostat. The electrochemical measurements were carried out in a three electrode electrochemical cell containing 1.0 mol dm−3 H2SO4 aqueous solution as the electrolyte, a platinum gauze counter electrode and Hg/HgSO4 (0.1 mol dm−3 K2SO4) as the reference electrode at room temperature.
The gel electrolyte fibre supercapacitors were fabricated by sandwiching two CF–NH3 or CF–TiN-1 fibre electrodes with a polypropylene non-woven fabric (Shanghai Heqi Glassware Co., Ltd.) separator. Each electrode and the separator were dipped twice into a PVA/H2SO4 gel electrolyte. The gel electrolyte was prepared by a modification of a literature68 method: 3 g polyvinyl alcohol (PVA, molecular weight 1750 ± 50, Sinopharm Chemical Reagent Co., Ltd) and 4.5 g concentrated H2SO4 were mixed with 30 cm3 deionized water. The mixture was heated at 85 °C for 4 h and then allowed to cool to room temperature before use.
Results and discussion
Carbon fibres were coated with titanium oxynitride using a sol–gel approach that we have previously applied to make a number of titanium nitride morphologies.60,62–64 A similar approach was employed by Yin et al. to coat graphite fibres with Ti(C,N) to increase their oxidation resistance.69 Our method relies on the self-condensation of primary amide ligands that result from transamination of [Ti(NMe2)4] using n-propylamine. Initially sols were produced in THF, but when these were used in attempt to coat carbon fibres distinct blocks of material, rather than a coating, were formed (ESI, Fig. S1†). Switching the solvent to hexane, sols formed that were effective in coating the carbon fibres, presumably due to better wetting of the hydrophobic carbon fibre surfaces by hexane compared with THF. Fig. 1 shows these coatings to be relatively smooth and continuous after firing in ammonia. Coatings were made with various sol concentrations (see Experimental) and the samples are then labelled as CF–TiN-x where x = 1–5. | ||
| Fig. 1 SEM image and Ti, C and N EDX maps of carbon fibres coated with Ti(O,N) (CF–TiN-4) and fired under ammonia at 800 °C. | ||
In order to deconvolute any changes in the fibre composition or behaviour due to the coating process from the effects of the coating itself, comparisons were made between the carbon fibres, carbon fibres fired in ammonia and Ti(O,N) coated carbon fibres fired in ammonia. The XRD patterns of the uncoated fibres, including those fired in ammonia, contained only broad features that can be attributed to the graphitic content of the carbon (Fig. 2). The coated fibres had TiN features superimposed onto the carbon pattern, but also always contained TiO2 in the rutile and anatase forms. Hence coatings are referred to as Ti(O,N). The ammonia used in firing was carefully dried, so gels produced by removing the solvent from the sols in vacuo were also fired under the same conditions. The crystalline oxide phases were not observed in their XRD patterns. Hence, the oxide observed in the coatings is believed to derive from reactions of the oligomers within the sol with oxygen-containing groups on the carbon fibre surfaces. Sulfuric acid treatment of carbon fibres can be used to improve bonding to polymers in composite material production, and results in multiple oxygen-containing groups on the fibre surfaces.70 Rietveld fits to the XRD data showed TiN to be the major phase with smaller amounts of anatase and rutile (ESI, Fig. S2 and Table S1†). The TiN lattice parameter of 4.18–4.19 Å is typical of those reported in the literature.65
 | ||
| Fig. 2 XRD patterns of carbon fibres (CF), carbon fibres heated in ammonia at 800 °C (CF–NH3) and carbon fibres coated with TiN (CF–TiN-1). | ||
TGA showed a single mass loss step in all cases, with an onset temperature around 450 °C (Fig. 3). The uncoated carbon fibres combusted completely, but a white residue was left from the coated samples that was used to calculate how much titanium was in the samples. This residue was identified by XRD as rutile-type TiO2 (ESI, Fig. S3†), confirming that the TiN component had also been completely oxidised. Combining the Rietveld derived phase fractions (Table S1†) and the carbon contents from the TGA (Fig. 3) the overall compositions of the fibres can be calculated (Table 2).
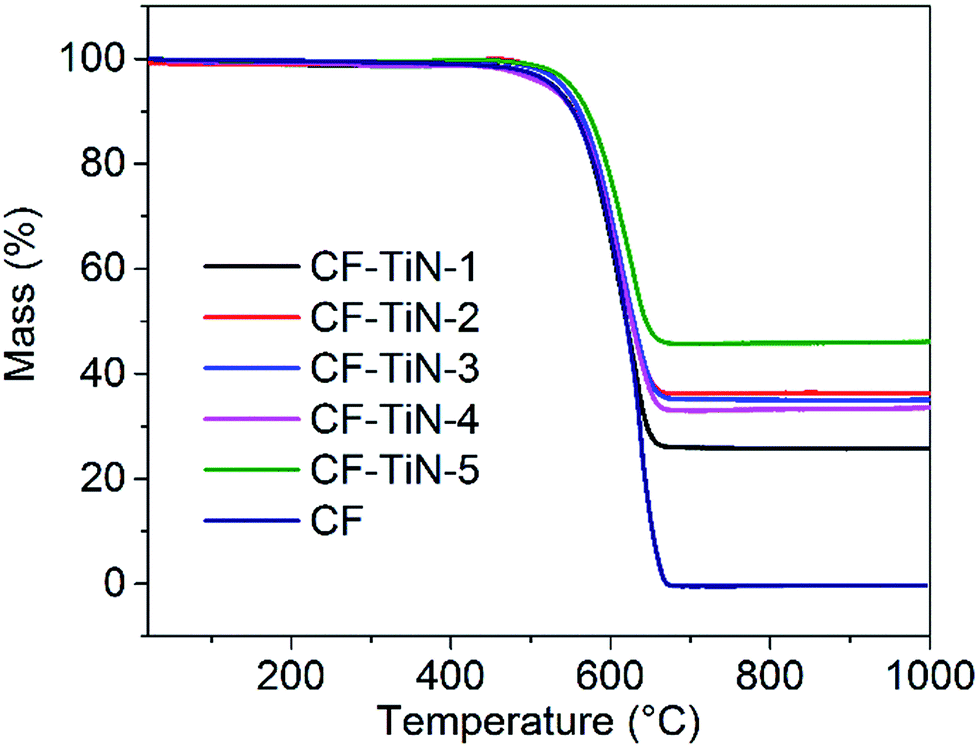 | ||
Fig. 3 TGA profiles of Ti(O,N)-coated carbon fibres heated under a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 O2/Ar mixture (30 cm3 min−1/20 cm3 min−1) at 5 °C min−1. Residual masses were 0.04% (CF), 26.1% (CF–TiN-1), 37.1% (CF–TiN-2), 35.3% (CF–TiN-3), 33.3% (CF–TiN-4) and 47.1% (CF–TiN-5). :2 O2/Ar mixture (30 cm3 min−1/20 cm3 min−1) at 5 °C min−1. Residual masses were 0.04% (CF), 26.1% (CF–TiN-1), 37.1% (CF–TiN-2), 35.3% (CF–TiN-3), 33.3% (CF–TiN-4) and 47.1% (CF–TiN-5). | ||
| C/wt% | TiN (osbornite)/wt% | TiO2 (rutile)/wt% | TiO2 (anatase)/wt% | |
|---|---|---|---|---|
| CF–TiN-1 | 78.3 | 14.8 | 4.0 | 2.9 |
| CF–TiN-2 | 70.4 | 24.9 | 2.6 | 2.1 |
| CF–TiN-3 | 70.2 | 18.4 | 4.8 | 6.6 |
| CF–TiN-4 | 72.6 | 19.6 | 5.2 | 2.6 |
| CF–TiN-5 | 63.0 | 29.3 | 6.7 | 1.0 |
The XPS analysis of the pristine carbon fibres evidences nitrogen (400.7 eV) in low concentration at the surface (2.5 at%) that can correspond to both pyrrolic N (H–N–(C)2) and tertiary N (N–(C)3–) (Fig. 4). After ammonia treatment the number of pyrrolic and tertiary N groups increases at the surface and a further environment at 398.1 eV is observed on the N 1s core peak (ESI, Table S2†). That new environment is associated with pyridinic N (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C).71,72 After coating, the XPS analysis shows a Ti 2p signal with two different environments (Fig. 5). The broad Ti 2p3/2–1/2 doublet at 457.5–463.0 eV is assigned to titanium oxynitride TiOxNy in agreement with the additional N 1s component located at 396.2 eV.23 The doublet at higher binding energy (459.1–464.8 eV) is characteristic of titanium dioxide. The Ti 2p3/2 peak of TiN occurs at 455 eV and is not observed, showing that the TiN component of the coatings contains a surface oxide layer. C 1s and O 1s spectra are available (ESI, Fig. S4†).
N–C).71,72 After coating, the XPS analysis shows a Ti 2p signal with two different environments (Fig. 5). The broad Ti 2p3/2–1/2 doublet at 457.5–463.0 eV is assigned to titanium oxynitride TiOxNy in agreement with the additional N 1s component located at 396.2 eV.23 The doublet at higher binding energy (459.1–464.8 eV) is characteristic of titanium dioxide. The Ti 2p3/2 peak of TiN occurs at 455 eV and is not observed, showing that the TiN component of the coatings contains a surface oxide layer. C 1s and O 1s spectra are available (ESI, Fig. S4†).
 | ||
| Fig. 4 N 1s XPS spectra of carbon fibre (CF), ammonia treated carbon fibre (CF–NH3), Ti(O,N)-coated carbon fibre (CF–TiN-1) and post-cycling TiN-coated carbon fibre samples. | ||
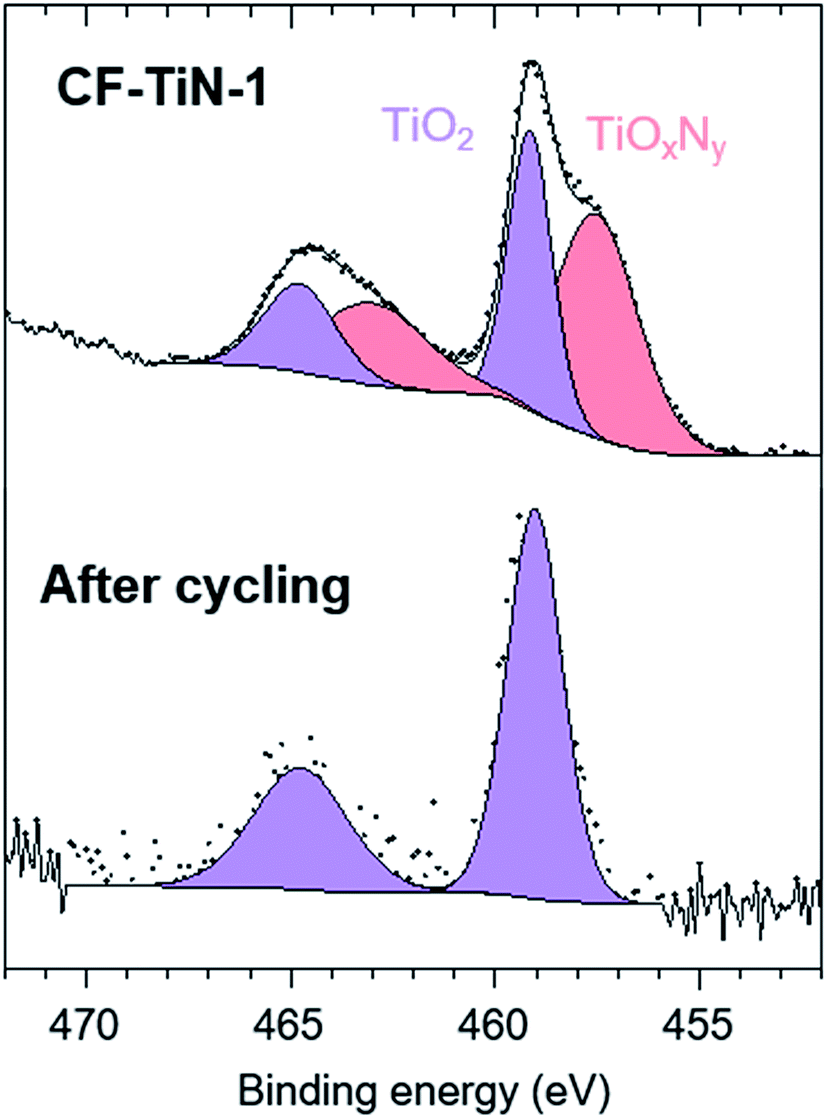 | ||
| Fig. 5 Ti 2p XPS spectra of TiN-coated carbon fibre (CF–TiN-1) and post-cycling Ti(O,N)-coated carbon fibre samples. | ||
Cyclic voltammograms of the carbon fibres were recorded in dilute sulfuric acid. The potential window was defined by recording a series of scans and limiting the potential range to avoid water oxidation or reduction, hence a window of −0.8 to +0.2 V vs. Hg/HgSO4 was chosen. The CVs of the untreated fibres showed box-shaped features typical of double layer capacitance, Fig. 6. Ammonolysis of the fibres before cell assembly resulted in a large increase in the currents observed in the CVs, retaining the same basic shape. The Ti(O,N)-coated fibre CVs showed redox features at −0.56 V (oxidation) and −0.75 V (reduction) superimposed onto the nitride fibre shape. These provided a 54% increase in the capacitance at a slow scan rate of 1 mV s−1 but much smaller increases at 10 or 300 mV s−1 (Table 3; CVs at further scan rates are provided in ESI, Fig. S4† and the method used to calculate capacitance is shown in ESI, Note 1†). The extra capacitance was found to be particularly prominent below ∼50 mV s−1 scan rate, below which the capacitance ramped up steeply (ESI, Fig. S6†). This slower charge storage performance is typical of a redox process and likely indicates intercalation of protons into a surface oxynitride component of the Ti(O,N) coating. The redox features observed for TiO2 coatings in similar conditions are expected to be at ∼0 V vs. Hg/HgSO4,70 and with TiN the features are so broad that they tend not to be observed.73
 | ||
| Fig. 6 Cyclic voltammograms (1.0 mol dm−3 H2SO4 aqueous solution, Hg/HgSO4 (0.1 mol dm−3 K2SO4), scan rates as labelled) of carbon fibres (CF), ammonia treated carbon fibres (CF–NH3) and Ti(O,N)-coated carbon fibres (CF–TiN-1). | ||
| CF/F g−1 | CF–NH3/F g−1 | CF–TiN-1/F g−1 | |
|---|---|---|---|
| 1 mV s−1 | 3.3 | 19.4 | 29.9 |
| 10 mV s−1 | 4.7 | 26.9 | 30.3 |
| 300 mV s−1 | 2.6 | 11.7 | 13.2 |
| 0.1 A g−1 | 4.6 | 22.8 | 38.3 |
| 0.5 A g−1 | 3.0 | 16.5 | 20.0 |
| 1.0 A g−1 | 2.0 | 12.0 | 16.0 |
Similar results were obtained under galvanostatic cycling conditions. Redox features associated with the Ti(O,N) coating were only observed at low potential at slower charge/discharge rates (Fig. 7), and the remainder of the profile closely resembled that of the ammonia-treated fibres (ESI, Fig. S7†). The ammonia treatment led to a significant increase in capacitance and the Ti(O,N) coating increased the capacitance further at lower currents (Table 2). Comparison of the capacitances achieved with different thicknesses of Ti(O,N) coating (ESI, Fig. S7†) showed that a thicker coating led to only small further increases, and that is why most of the work focused on the samples with the thinnest coatings. The capacitance was stable over 600 galvanostatic cycles, and low resistances were measured for these cells (Fig. 7).
 | ||
| Fig. 7 Galvanostatic potential–time plots (1.0 mol dm−3 H2SO4 aqueous solution, Hg/HgSO4 (0.1 mol dm−3 K2SO4), scan rates as labelled) for Ti(O,N)-coated carbon fibres (CF–TiN-1; top) and variations in the capacitance and IR drop (measured as the voltage drop between the last charging and the first discharging data point) of the same fibres with cycle number at 0.5 V s−1 (bottom). | ||
After cycling, the components associated with TiOxNy on both N 1s and Ti 2p core peaks disappear (Fig. 4 and 5) and the at% of TiO2 decreases suggesting some loss of the coating and the surface layer being fully oxidised. The former is unsurprising, similar results are found with TiN electrodes23 and it should be remembered that XPS is only probing around 5–10 nm of the surface. The oxide is likely the redox active component and the nitride is present mainly to retain conductivity. Note that nitrogen is present even in this thin surface layer (Fig. 4), although the N 1s core peaks displays a new component at 402.9 eV after cycling, indicating the formation of some oxidised nitrogen species. The surface oxidation could be responsible for the small increases in resistivity shown in Fig. 7, but overall the resistances remain low.
Resistance was also probed using electrochemical impedance spectroscopy. The spectra were mainly capacitive (Fig. 8), but showed small uncompensated solution resistances and charge transfer resistances. The latter were 7.6 Ω for the carbon fibres, 6.4 Ω for the ammonia treated carbon fibres and 6.1 Ω for the Ti(O,N)-coated carbon fibres.
 | ||
| Fig. 8 Electrochemical impedance spectra (1.0 mol dm−3 H2SO4 aqueous solution, 0 V vs. Hg/HgSO4 (0.1 mol dm−3 K2SO4), amplitude 10 mV, frequency range 10−2 to 105 Hz) of carbon fibres (CF), ammonia treated carbon fibres (CF–NH3) and Ti(O,N)-coated carbon fibres (CF–TiN-1). Data were fitted using ZView to a series combination of a resistor (uncompensated solution resistance), a parallel resistor/constant phase element (charge transfer resistance/interface capacitance), and a constant phase element (capacitance). | ||
Symmetric gel electrolyte fibre supercapacitors were fabricated by sandwiching two Ti(O,N)-coated carbon fibre (CF–TiN-1) electrodes with a polypropylene non-woven fabric. The electrode and separator were coated with a PVA/H2SO4 gel electrolyte prior to assembly. Similar capacitors were also assembled and tested using the ammonia treated carbon fibres (CF–NH3).
Cyclic voltammograms of the symmetric cells over a 1 V potential range and at a moderate scan rate of 10 mV s−1 showed no redox features (Fig. 9) but the Ti(O,N)-coated fibres (CF–TiN-1) still showed a larger capacitance (6.0 F g−1 based on the mass of both electrodes) than the ammonia treated fibres (CF–NH3; 3.8 F g−1). The lack of redox features can be attributed to the electrodes in this symmetric environment not being exposed to the low potentials that were needed to activate the redox chemistry in the aqueous cells with a reference electrode. Thus the higher capacitance may be due to an increased surface area in the coated fibre samples. Similar results were found under galvanostatic cycling conditions, where symmetrical charge/discharge profiles were observed (Fig. 9). The Ti(O,N)-coated fibres showed capacitances of 7.8 F g−1 at 0.1 A g−1, 6.9 F g−1 at 0.5 A g−1 and 6.6 F g−1 at 1.0 A g−1, compared with 3.4, 2.4 and 1.6 F g−1, respectively, for the ammonia-treated fibres (CF–NH3).
 | ||
| Fig. 9 Cyclic voltammograms (top) and galvanostatic potential–time plots (bottom) for symmetric supercapacitors assembled using ammonia-treated carbon fibre (CF–NH3) or Ti(O,N)-coated carbon fibre (CF–TiN-1) electrodes. The CV scan rate was 10 mV s−1 and galvanostatic current rates were as shown. | ||
Flexibility during operation is a key potential advantage of supercapacitors constructed from fibres and with gel electrolytes.1,2,74 Our symmetric supercapacitors were tested flat (0°), or bent through 90° or 180° with a 7 mm radius of curvature (geometry shown in ESI, Fig. S8†). Very little difference in behaviour was observed under CV or galvanostatic conditions (Fig. 10), showing that these devices are as flexible as they were intended to be. This good performance persisted over multiple cycles, with a small drop off in capacitance over the first ∼2000 cycles followed by quite steady capacitance over the next 6000 (Fig. 10). The retention in the capacitance over 8000 cycles was 77% at 0°, 73% at 90° and 83% at 180°. A video clip of a capacitor being flexed during galvanostatic cycling is available in the electronic ESI.† Both ammonia-treated fibres and Ti(O,N)-coated fibres showed a drop-off in the CV-measured capacitance with increasing scan rate, but this was a little more pronounced in the Ti(O,N)-coated fibres (ESI, Fig. S9†). This suggests that redox activity was playing a role in their charge storage behaviour. The CVs also showed higher resistance at fast scan rates in the supercapacitors bent through larger angles (ESI, Fig. S9†), which may indicate that the contact between the layers is being interrupted to some extent by bending.
 | ||
| Fig. 10 Cyclic voltammograms (10 mV s−1; top), galvanostatic potential–time plots (0.1 A g−1, centre) and capacitance vs. cycle number calculated from the galvanostatic data (0.5 A g−1, bottom) of symmetric supercapacitors cycled flat (0°) or bent through a 90° or 180° angle. | ||
The specific capacitances achieved in this study compare favourably with other studies into flexible supercapacitors in terms of energy and power density, as shown in Fig. 11 (the calculation methods used for Fig. 11 are presented in ESI, Note 2†). As described above, this performance can be ascribed to the increases in charge storage density that come from the combination of nitridation of the fibres and the redox activity of the Ti(O,N) surface coating. However, in the future it should be possible to take this approach further. For example, larger specific surface areas and better electron transport may be provided by 3-dimensional carbon network structures, by weaving the fibres or using foam or reticulated carbon structures. Much of the redox activity of the Ti(O,N) coatings is at low potential, so larger capacitances as well as higher voltages may be achievable in asymmetric capacitors by using such Ti(O,N)-coated carbon electrode systems in combination with an electrode that has redox activity at higher potential.
 | ||
| Fig. 11 Ragone plot comparing the volumetric energy and power densities of the solid flexible supercapacitor devices described in this study, measured under galvanostatic conditions at 0.1 A g−1 (a), 0.5 A g−1 (b) or 1 A g−1 (c) and at bending angles as indicated, with those of other flexible supercapacitors in the literature.5,6,8,30,75–78 | ||
Conclusions
A non-oxide sol–gel method has been used to produce titanium oxide nitride coatings on carbon fibres. Smooth coatings were achieved by matching the solvent hydrophobicity to that of the fibre surfaces. The oxide component in the coatings appears to be due to interaction with surface oxygen-containing functionalities on the carbon fibres. The high temperature ammonia treatment required to crystallise the metal nitride also increases the nitrogen content of the fibres themselves. Both the surface nitridation and the Ti(O,N) coatings contribute to increased capacitances in aqueous or gel acid environments. The main redox activity of the Ti(O,N) component is at low potential, but even at fast scan rates, where no redox peaks were observed in cyclic voltammetry or galvanostatic experiments, this component increased the capacitance of the electrodes. The capacitors had low resistance and were stable over hundreds of cycles. Flexible capacitors made with an acidic gel electrolyte delivered competitive energy and power densities that were unaffected by bending the devices.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
JZ thanks the China Scholarship Council for funding for a visit to the UK. SS is supported by EPSRC under EP/N035437/1 and the Smartlab diffractometer was funded under EP/K00509X/1 and EP/K009877/1. XZ is supported by the DHU Distinguished Young Professor Program and the National Natural Science Foundation of China (51403036).Notes and references
- L. Liu, Z. Niu and J. Chen, Chem. Soc. Rev., 2016, 45, 4340–4363 RSC.
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- J. Jang, J. Bae, M. Choi and S. H. Yoon, Carbon, 2005, 43, 2730–2736 CrossRef CAS.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- H. Zheng, T. Zhai, M. Yu, S. Xie, C. Liang, W. Zhao, S. C. I. Wang, Z. Zhang and X. Lu, J. Mater. Chem. C, 2013, 1, 225–229 RSC.
- X. Lu, M. Yu, G. Wang, T. Zhai, S. Xie, Y. Ling, Y. Tong and Y. Li, Adv. Mater., 2013, 25, 267–272 CrossRef CAS PubMed.
- S. D. Perera, B. Patel, N. Nijem, K. Roodenko, O. Seitz, J. P. Ferraris, Y. J. Chabal and K. J. Balkus Jr, Adv. Energy Mater., 2011, 3, 936–945 CrossRef.
- X. Xiao, T. Ding, L. Yuan, Y. Shen, Q. Zhong, X. Zhang, Y. Cao, B. Hu, T. Zhai, L. Gong, J. Chen, Y. Tong, J. Zhou and Z. Wang, Adv. Energy Mater., 2012, 2, 1328–1332 CrossRef CAS.
- R. Zhou, C. Meng, F. Zhu, Q. Li, C. Liu, S. Fan and K. Jiang, Nanotechnology, 2010, 21, 345701 CrossRef PubMed.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Kang, Electrochim. Acta, 2011, 56, 9240–9247 CrossRef CAS.
- J. Zhang, X. Zhao, Z. Huang, T. Xu and Q. Zhang, Carbon, 2016, 107, 844–851 CrossRef CAS.
- Q. J. Le, T. Wang, D. N. H. Tran, F. Dong, Y. X. Zhang and D. Losic, J. Mater. Chem. A, 2017, 5, 10856–10865 CAS.
- M. Li, M. Zhou, Z. Q. Wen and Y. X. Zhang, Journal of Energy Storage, 2017, 11, 242–248 CrossRef.
- P. Chen, H. Chen, J. Qiu and C. Zhou, Nano Res., 2010, 3, 594–603 CrossRef CAS.
- L. Yang, S. Cheng, Y. Ding, X. Zhu, Z. L. Wang and M. Liu, Nano Lett., 2012, 12, 321–325 CrossRef CAS PubMed.
- J. Li, W. Zhao, F. Huang, A. Manivannan and N. Wu, Nanoscale, 2011, 3, 5103 RSC.
- X. Zhang, W. Shi, J. Zhu, D. J. Kharistal, W. Zhao, B. S. Lalia, H. H. Hng and Q. Yan, ACS Nano, 2011, 5, 2013–2019 CrossRef CAS PubMed.
- D. Choi and P. N. Kumta, J. Electrochem. Soc., 2006, 153, A2298–A2303 CrossRef CAS.
- R. A. Janes, M. Aldissi and R. B. Kaner, Chem. Mater., 2003, 15, 4431–4435 CrossRef CAS.
- P. Yang, D. Chao, C. Zhu, X. Xia, Y. Zhang, X. Wang, P. Sun, B. K. Tay, Z. X. Shen, W. Mai and H. J. Fan, Adv. Sci., 2015, 3, 1500299 CrossRef PubMed.
- A. Achour, R. L. Porto, M.-A. Soussou, M. Islam, M. Boujtita, K. A. Aissa, L. Le Brizoual, A. Djouadi and T. Brousse, J. Power Sources, 2015, 300, 525–532 CrossRef CAS.
- D. J. Kim, J. K. Kim, J. H. Lee, H. H. Cho, Y.-S. Bae and J. H. Kim, J. Mater. Chem. A, 2016, 4, 12497–12503 CAS.
- B. M. Gray, A. L. Hector, M. Jura, J. R. Owen and J. Whittam, J. Mater. Chem. A, 2017, 5, 4550–4559 CAS.
- K. Grigoras, J. Keskinen, L. Grönberg, E. Yli-Rantala, S. Laakso, H. Välimäki, P. Kauranen, J. Ahopelto and M. Prunnila, Nano Energy, 2016, 26, 340–345 CrossRef CAS.
- P. Lu, P. Ohlckers, L. Müller, S. Leopold, M. Hoffmann, K. Grigoras, J. Ahopelto, M. Prunnila and X. Chen, Electrochem. Commun., 2016, 70, 51–55 CrossRef CAS.
- Y. Xie and R. Gao, J. Alloys Compd., 2017, 725, 1–13 CrossRef CAS.
- Y. Haldorai, D. Arreaga-Salas, C. S. Rak, Y. S. Huh, Y. K. Han and W. Voit, Electrochim. Acta, 2016, 220, 465–474 CrossRef CAS.
- E. Kao, C. Yang, R. Warren, A. Kozinda and L. Lin, Sens. Actuators, A, 2016, 240, 160–166 CrossRef CAS.
- D. Choi, G. E. Blomgren and P. N. Kumta, Adv. Mater., 2006, 18, 1178–1182 CrossRef CAS.
- X. Lu, M. Yu, T. Zhai, G. Wang, S. Xie, T. Liu, C. Liang, Y. Tong and Y. Li, Nano Lett., 2013, 13, 2628–2633 CrossRef CAS PubMed.
- R. Lucio-Porto, S. Bouhtiyya, J. F. Pierson, A. Morel, F. Capon, P. Boulet and T. Brousse, Electrochim. Acta, 2014, 141, 203–211 CrossRef CAS.
- Z. Hou, K. Guo, H. Li and T. Zhai, CrystEngComm, 2016, 18, 3040–3047 RSC.
- P. J. Hanumantha, M. K. Datta, K. Kadakia, C. Okoli, P. Patel and P. N. Kumta, Electrochim. Acta, 2016, 207, 37–47 CrossRef CAS.
- Y. Yang, K. Shen, Y. Liu, Y. Tan, X. Zhao, J. Wu, X. Niu and F. Ran, Nano-Micro Lett., 2017, 9, 1–15 CrossRef.
- Y. Wu and F. Ran, J. Power Sources, 2017, 344, 1–10 CrossRef CAS.
- B. Wang, Z. Chen, G. Lu, T. Wang and Y. Ge, Mater. Res. Bull., 2016, 76, 37–40 CrossRef CAS.
- F. Ran, Z. Wang, Y. Yang, Z. Liu, L. Kong and L. Kang, Electrochim. Acta, 2017, 258, 405–413 CrossRef CAS.
- Y. Wang, M. Jiang, Y. Yang and F. Ran, Electrochim. Acta, 2016, 222, 1914–1921 CrossRef CAS.
- Y. Liu, L. Liu, L. Kong, L. Kang and F. Ran, Electrochim. Acta, 2016, 211, 469–477 CrossRef CAS.
- A. Achour, R. Lucio-Porto, M. Chaker, A. Arman, A. Ahmadpourian, M. A. Soussou, M. Boujtita, L. Le Brizoual, M. A. Djouadi and T. Brousse, Electrochem. Commun., 2017, 77, 40–43 CrossRef CAS.
- D. Choi and P. N. Kumta, J. Am. Ceram. Soc., 2011, 94, 2371–2378 CrossRef CAS.
- P. Wang, R. Wang, J. Lang, X. Zhang, Z. Chen and X. Yan, J. Mater. Chem. A, 2016, 4, 9760–9766 CAS.
- H. Cui, G. Zhu, X. Liu, F. Liu, Y. Xie, C. Yang, T. Lin, H. Gu and F. Huang, Adv. Sci., 2015, 2, 1500126 CrossRef PubMed.
- B. Gao, X. Xiao, J. Su, X. Zhang, X. Peng, J. Fu and P. K. Chu, Appl. Surf. Sci., 2016, 383, 57–63 CrossRef CAS.
- B. Wei, H. Liang, D. Zhang, Z. Wu, Z. Qi and Z. Wang, J. Mater. Chem. A, 2017, 5, 2844–2851 CAS.
- S. I. U. Shah, A. L. Hector, X. Li and J. R. Owen, J. Mater. Chem. A, 2015, 3, 3612–3619 CAS.
- Y. Xie and F. Tian, Mater. Sci. Eng., B, 2017, 215, 64–70 CrossRef CAS.
- C. Chen, D. Zhao and X. Wang, Mater. Chem. Phys., 2006, 97, 156–161 CrossRef CAS.
- S. L. Roberson, D. Finello and R. F. Davis, J. Appl. Electrochem., 1999, 29, 75 CrossRef CAS.
- T.-C. Liu, W. G. Pell, B. E. Conway and S. L. Roberson, J. Electrochem. Soc., 1998, 145, 1882–1888 CrossRef CAS.
- S. I. U. Shah, A. L. Hector and J. R. Owen, J. Power Sources, 2014, 266, 456–463 CrossRef CAS.
- L. Chen, C. Liu and Z. Zhang, Electrochim. Acta, 2017, 245, 237–248 CrossRef CAS.
- L. Xu, L. Sun, J. Feng, L. Qi, I. Muhammad, J. Maher, X. Cheng and W. Song, RSC Adv., 2017, 7, 44619–44625 RSC.
- A. Śliwak, A. Moyseowicz and G. Gryglewicz, J. Mater. Chem. A, 2017, 5, 5680–5684 Search PubMed.
- S. Bouhtiyya, R. Lucio Porto, B. Laïk, P. Boulet, F. Capon, J. P. Pereira-Ramos, T. Brousse and J. F. Pierson, Scr. Mater., 2013, 68, 659–662 CrossRef CAS.
- A.-R. Ko, S.-B. Han, Y.-W. Lee and K.-W. Park, Phys. Chem. Chem. Phys., 2011, 13, 12705–12707 RSC.
- S. Wang, L. Zhang, C. Sun, Y. Shao, Y. Wu, J. Lv and X. Hao, Adv. Mater., 2016, 28, 3768–3776 CrossRef CAS PubMed.
- Z. Wang, Z. Li and Z. Zou, J. Power Sources, 2015, 296, 53–63 CrossRef CAS.
- A. L. Hector, Chem. Soc. Rev., 2007, 36, 1745–1753 RSC.
- A. W. Jackson and A. L. Hector, J. Mater. Chem., 2007, 17, 1016 RSC.
- A. W. Jackson, O. Shebanova, A. L. Hector and P. F. McMillan, J. Solid State Chem., 2006, 179, 1383–1393 CrossRef CAS.
- B. M. Gray, S. Hassan, A. L. Hector, A. Kalaji and B. Mazumder, Chem. Mater., 2009, 21, 4210–4215 CrossRef CAS.
- C. F. Mallinson, B. M. Gray, A. L. Hector, M. A. Mclachlan and J. R. Owen, Inorg. Chem., 2013, 9994–9999 CrossRef CAS PubMed.
- M. Zhang, N. Garcia-Araez, A. L. Hector and J. R. Owen, J. Mater. Chem. A, 2017, 5, 2251–2260 CAS.
- Inorg. Cryst. Struct. Database (ICSD, Fiz Karlsruhe) accessed via EPSRC-funded Natl. Chem. Database Serv. hosted by R. Soc. Chem.
- D. A. Shirley, Phys. Rev. B, 1972, 5, 4709–4714 CrossRef.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- G. Wang, H. Wang, X. Lu, Y. Ling, M. Yu, T. Zhai, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 2676–2682 CrossRef CAS PubMed.
- L. Yin, R. T. David, R. K. Mike, L. M. Brian and M. L. Richard, J. Am. Ceram. Soc., 1997, 80, 705–716 CrossRef.
- D. D. L. Chung, Carbon Fiber Composites, Butterworth-Heinemann, Newton MA, 2012 Search PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. Bin Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- A. Choudhury, J. H. Kim, S. Sinha Mahapatra, K. S. Yang and D. J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 2109–2118 CrossRef CAS.
- Y. Xie and D. Wang, J. Alloys Compd., 2016, 665, 323–332 CrossRef CAS.
- J. Bae, M. K. Song, Y. J. Park, J. M. Kim, M. Liu and Z. L. Wang, Angew. Chem., Int. Ed., 2011, 50, 1683–1687 CrossRef CAS PubMed.
- X. Xiao, T. Li, P. Yang, Y. Gao, H. Jin, W. Ni, W. Zhan, X. Zhang, Y. Cao, J. Zhong, L. Gong, W.-C. Yen, W. Mai, J. Chen, K. Huo, Y.-L. Chueh, Z. L. Wang and J. Zhou, ACS Nano, 2012, 6, 9200–9206 CrossRef CAS PubMed.
- X. Xiao, X. Peng, H. Jin, T. Li, C. Zhang, B. Gao, B. Hu, K. Huo and J. Zhou, Adv. Mater., 2013, 25, 5091–5097 CrossRef CAS PubMed.
- P. Yang, X. Xiao, Y. Li, Y. Ding, P. Qiang, X. Tan, W. Mai, Z. Lin, W. Wu, T. Li, H. Jin, P. Liu, J. Zhou, C. P. Wong and Z. L. Wang, ACS Nano, 2013, 2617–2626 CrossRef CAS PubMed.
- P. Xu, T. Gu, Z. Cao, B. Wei, J. Yu, F. Li, J. H. Byun, W. Lu, Q. Li and T. W. Chou, Adv. Energy Mater., 2014, 4, 1–6 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including raw data. See DOI: 10.1039/c7ta11142h |
| This journal is © The Royal Society of Chemistry 2018 |