
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon foams from emulsion-templated reduced graphene oxide polymer composites: electrodes for supercapacitor devices†
Robert T.
Woodward
 a,
Foivos
Markoulidis
b,
François
De Luca
a,
David B.
Anthony
b,
Daniel
Malko
b,
Tom O.
McDonald
c,
Milo S. P.
Shaffer
b and
Alexander
Bismarck
*ad
a,
Foivos
Markoulidis
b,
François
De Luca
a,
David B.
Anthony
b,
Daniel
Malko
b,
Tom O.
McDonald
c,
Milo S. P.
Shaffer
b and
Alexander
Bismarck
*ad
aPolymer & Composite Engineering (PaCE) Group, Department of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
bDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK
cDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
dPolymer and Composite Engineering (PaCE) Group, Institute of Materials Chemistry & Research, Faculty of Chemistry, University of Vienna, Währinger Straße 42, Vienna, Austria. E-mail: alexander.bismarck@univie.ac.at
First published on 1st January 2018
Abstract
Amphiphilic reduced graphene oxide (rGO) is an efficient emulsifier for water-in-divinylbenzene (DVB) high internal phase emulsions. The polymerisation of the continuous DVB phase of the emulsion template and removal of water results in macroporous poly(divinylbenzene) (polyDVB). Subsequent pyrolysis of the poly(DVB) macroporous polymers yields ‘all-carbon’ foams containing micropores alongside emulsion templated-macropores, resulting in hierarchical porosity. The synthesis of carbon foams, or ‘carboHIPEs’, from poly(DVB) produced by polymerisation of rGO stabilised HIPEs provides both exceptionally high surface areas (up to 1820 m2 g−1) and excellent electrical conductivities (up to 285 S m−1), competing with the highest figures reported for carboHIPEs. The use of a 2D carbon emulsifier results in the elimination of post-carbonisation treatments to remove standard inorganic particulate emulsifiers, such as silica particles. It is demonstrated that rGO containing carboHIPEs are good candidates for supercapacitor electrodes where carboHIPEs derived from more conventional polymerised silica-stabilised HIPEs perform poorly. Supercapacitor devices featured a room-temperature ionic liquid electrolyte and electrodes derived from either rGO- or silica-containing poly(DVB)HIPEs demonstrated a maximum specific capacitance of 26 F g−1, an energy density of 5.2 W h kg−1 and a power density of 280 W kg−1.
Introduction
Carbonaceous materials have great potential in a myriad of applications including heat transfer,1 water purification,2 electromagnetic interference (EMI) shielding,3 as catalyst supports4 and as electrodes in energy storage devices.5,6 Carbonaceous materials have many attractive and practical characteristics, such as low cost, low density, chemical inertness and high thermal stability. Porous carbon electrodes with high surface areas play a crucial role in energy storage devices, such as in electrochemical double layer capacitors (EDLCs) and lithium-ion batteries. EDLCs are promising candidates to address current and future storage requirements for sustainable energy, due to their rapid charge–discharge rates and excellent power densities.7,8 A wide range of carbon materials are used as EDLC electrodes, including woven or knitted fabrics, powders, coated foils and porous carbon monoliths.9 One major advantage of monolithic carbon foams is that they may be employed as binderless electrodes, in contrast to the established route of drying a slurry of powdered carbon for deposition on current collectors.10 Coatings require the use of a binder (usually at 5 or 10 wt%) which, although ensuring the integrity of the active material, typically impairs electrode porosity and electrical conductivity.11 Compared to compacted pellets featuring a binder, porous carbon monoliths have been shown to offer higher specific capacitance, lower internal resistance, better electrolyte infiltration and higher electrical conductivity (in the order of 1 S cm−1).12 Porous carbon monoliths are network structures with inherent three-dimensionality and hierarchical porosity in the form of micropores on the carbon particle surface and mesopores in-between the voids created by the carbon particles.12 This porosity, coupled with the possibility of employing binder-less electrodes, make porous carbon foam electrodes an attractive alternative to activated carbons for electrode development.13,14 Templating is one of the more attractive routes to porous carbon monoliths due to the great degree of control over carbon's textural properties. Ordinarily, a good carbon source is loaded with a templating material (or vice versa) prior to pyrolysis. If the templates survive pyrolysis, they are then chemically removed, producing templated porosity throughout the carbon. One common route to these porous carbon monoliths includes the loading of phenolic resins with a variety of templates such as zeolites, colloidal silica or block copolymers.15,16 However, activation is usually required post-pyrolysis to generate high surface areas as well as removal of hard templates using strong acids such as HF. An alternative templating route to carbon foams is the use of polymerised high internal phase emulsions (polyHIPE) as templates for the formation of macroporous carbon precursors. A HIPE is defined as an emulsion with an internal phase volume ratio >74%, the limit at which monodisperse droplets can no longer close pack without deformation. The polymerisation of a HIPE's continuous minority phase and the subsequent removal of the templating internal emulsion phase, yields an emulsion-templated polymer. A vast range of HIPE-templated porous polymers have been reported, testament to the versatility of the emulsion-templating approach.17–19 By synthesising polyHIPEs that are able to act as carbon precursors, novel monolithic carbon foams can be produced. Carbon foams derived from polyHIPEs, also called ‘carboHIPEs’, display hierarchical porosity arising from emulsion-templated macropores to micropores (<2 nm diameter) produced during activation. Emulsion-templating has a number of advantages over the use of solid inorganic templates, as strong acids are not required to remove the templating phase, post-carbonisation.20,21 In recent years, a number of studies have reported a polyHIPE route to porous ‘carboHIPEs’,22–29 however this number is relatively small when considering the huge array of work reported on both polyHIPEs and carbon foams. We have shown previously that polydivinylbenzene (polyDVB) can be used as an efficient carbon precursor in the form of a poly(DVB)HIPE derived from silica particle stabilised water-in-oil (w/o) HIPEs.30 Polymerisation of a DVB continuous phase yielded both open- and closed-cell poly(DVB)HIPEs containing emulsion-templated macropores. The use of particulate emulsifiers and the inherently cross-linked polyDVB structure allowed for the formation of carbon-foam precursors without the need for further treatments, such as sulfonation or crosslinking, prior to carbonisation. However, without further acid treatment, the silica particles used as emulsifier remained embedded in the surface of the carboHIPEs, limiting their electrical conductivity due to the presence of the insulating particles on the surface of the materials. To circumvent this problem, the use of a particulate carbon emulsifier in the form of reduced graphene oxide (rGO) is proposed. It was previously shown that rGO acts as efficient particulate emulsifier and formed closed-cell poly(styrene-co-DVB)HIPEs with a continuous layer of rGO at their surface upon polymerisation, resulting in electrically conducting polyHIPEs.31 However, as the bulk of the structure consisted of poly(styrene-co-DVB) (>99% by weight), the areas of low resistivity were limited to the percolating network of rGO at the surface of the pore walls inside polyHIPEs, resulting in relatively low conductivities (1.3 × 10−4 S m−1). In addition, poly(styrene-co-DVB)HIPEs do not survive carbonisation due to their low thermal stability and can therefore not be used as precursors for carboHIPEs without further stabilisation. Herein, we report the carbonisation of poly(DVB)HIPEs produced by polymerisation of rGO stabilised HIPEs in order to produce true ‘all-carbon’ carboHIPEs, in which decreased resistivity is no longer limited to the percolating network of rGO at the pore surface of the material. By using rGO as an emulsifier instead of silica nanoparticles, carboHIPEs with very high electrical conductivity can be produced. The applicability of these carboHIPEs was explored for use as electrodes in binder-less EDLC devices, exploiting their high surface areas and excellent electrical conductivities.Results and discussion
To produce the rGO emulsifier, a solution of graphene oxide (GO) (1 mg mL−1, 100 mL) was chemically reduced using hydrazine hydrate following a reported method.32 Both HIPE formulation and subsequent polyHIPE synthesis is described in detail in the Experimental section. After emulsification, viscous HIPEs were transferred into a sealed polypropylene centrifuge tube and polymerised overnight in a convection oven to yield, after drying, a freestanding poly(DVB)HIPE (rGO-PH1) (Fig. 1a).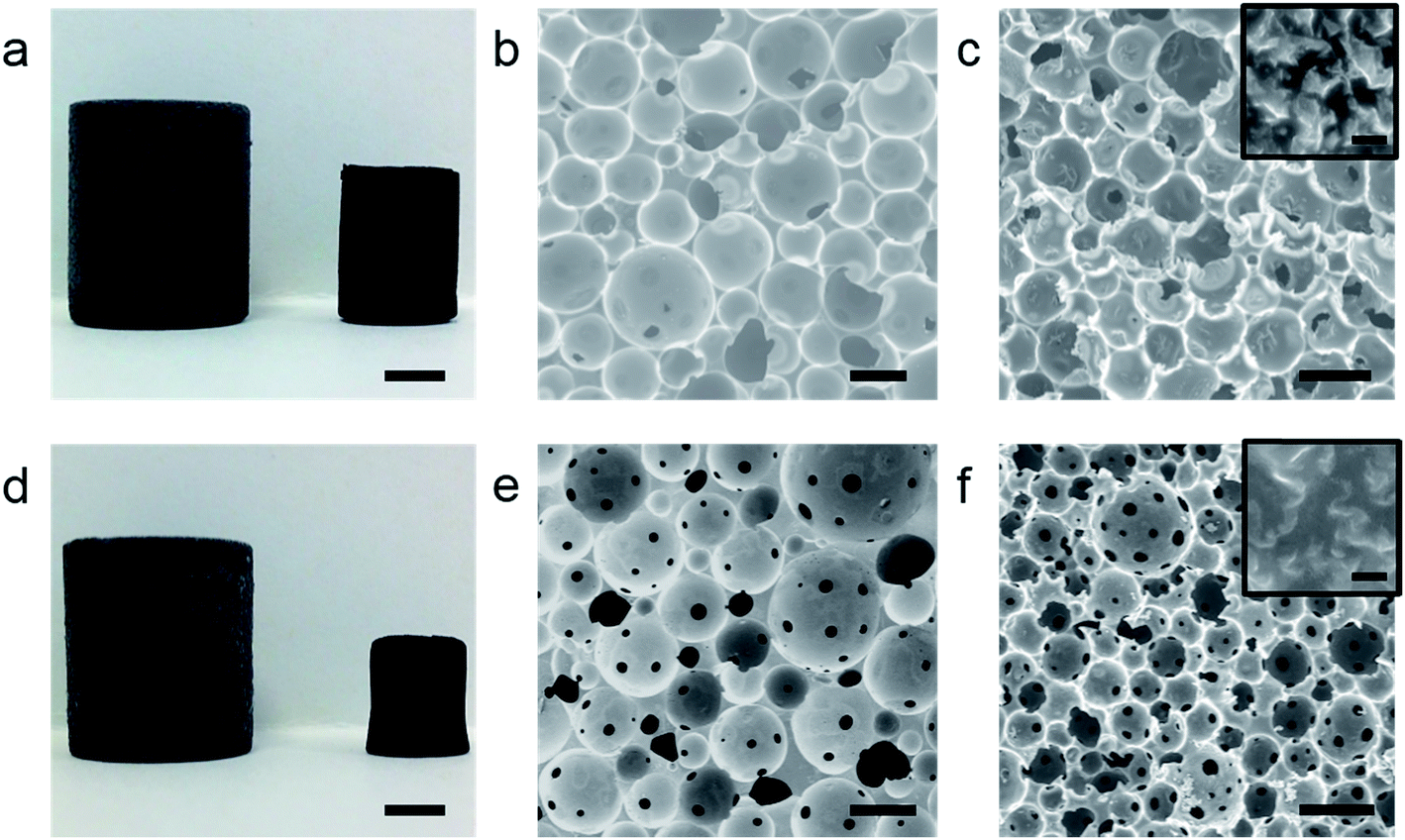 | ||
| Fig. 1 (a) Photograph of rGO-PH1 (left) and its carbonised equivalent rGO-CH1 (right), (b) SEM of rGO-PH1, (c) SEM of rGO-CH1 (inset: high magnification showing rGO platelets) (d) photograph of rGO-PH2 (left) and rGO-CH2 (right), (e) SEM of rGO-PH2, (f) SEM of rGO-CH2 (inset: high magnification showing rGO platelets). Scale bars in (a) and (d) represent 5 mm, 100 μm in all other images and 400 nm in the inset images. | ||
Scanning electron micrographs (SEM) of rGO-PH1 show a closed-cell pore structure typical of a Pickering HIPE derived polyHIPE31 with an average macropore diameter of 83 ± 29 μm (Fig. 1b). The presence of the rGO meant that the polyHIPEs were electrically conducting (2.49 × 10−3 ± 1.6 × 10−3 S m−1), in good agreement with previous findings.31 In order to prepare interconnected, or open-cell, poly(DVB)-Pickering-HIPEs a small amount of a molecular surfactant (Hypermer 2296) was added, as suggested by Ikem et al.,33 to the already formed rGO-stabilised HIPE prior to polymerisation. After addition of the surfactant the HIPE was stirred slowly for 20 s prior to polymerisation to yield interconnected poly(DVB)HIPEs (rGO-PH2) (Fig. 1d and e). The large average pore-diameter in the open-cell rGO-PH2, 94 ± 32 μm, is a good indication that the HIPE prior to polymerisation was still stabilised by particles. High-resolution FE-SEM images clearly showed rGO platelets still present at the surface of the pore walls of both rGO-containing macroporous polymers, indicating the minimal removal of rGO from the w/o interface by the molecular surfactant (Fig. 1c and f insets). However, the electrical conductivity of rGO-PH2 was 3.69 × 10−5 ± 1.10 × 10−5 S m−1, two orders of magnitude lower than its closed-cell equivalent, suggesting improved rGO dispersion caused by the surfactant and/or the removal of some rGO from the w/o interface in the HIPE prior to polymerisation, affecting the percolated rGO network covering the surfaces of the pore walls in the resulting polyHIPE.
It was hypothesised that upon carbonisation, the presence of a percolating rGO network at the surface of polyHIPEs would lead to improved electrical conductivities in subsequent carboHIPEs due their all carbon structures. The polyHIPEs were heated under N2 at a ramp rate of 2 °C min−1 to 800 °C and held there for 1 h. The resulting carboHIPEs retained the shape of the polyHIPE precursor well, albeit with significant shrinkage (Fig. 1a and d). The rGO-PH1 derived carboHIPEs (rGO-CH1) shrunk by 76 ± 4% with a carbon yield of 20 ± 1%, while the carbonised open-cell rGO-PH2 (rGO-CH2) shrank by 89 ± 1%, significantly more than the closed cell equivalent. This may be due to the gas pressure within closed-cell polyHIPEs providing some resistance against shrinkage during carbonisation or could be a result of the presence of pore throats in the open-cell structure, allowing for greater contraction of the polymer walls. In spite of the significant volume loss, the carbon yield of rGO-CH2 was 17 ± 1%, suggesting that it is not exclusively mass loss that led to the larger shrinkage of rGO-PH2. The low volume retention coupled with the larger mass retention resulted in an increased envelope density for rGO-CH2 as compared to rGO-CH1, 0.32 g cm−3 and 0.2 g cm−3, respectively, however, the increase in skeletal density upon carbonisation was similar for both rGO-PH1 and rGO-PH2 and consistent with carbonisation (Table 1).
| Sample | d p (μm) | SABET (m2 g−1) | MPV (cm3 g−1) | TPV (cm3 g−1) | P (%) | ρ s (g cm−3) | ρ e (g cm−3) |
|---|---|---|---|---|---|---|---|
| Si-PH1 (ref. 30) | 69 ± 28 | 1 | — | — | 79 | 1.11 ± 0.01 | 0.23 ± 0.01 |
| Si-PH2 (ref. 30) | 77 ± 35 | 50 | — | 0.091 | 78 | 1.09 ± 0.01 | 0.24 ± 0.01 |
| rGO-PH1 | 83 ± 29 | <1 | — | — | 80 | 1.14 ± 0.01 | 0.23 ± 0.02 |
| rGO-PH2 | 94 ± 32 | <1 | — | — | 80 | 1.09 ± 0.01 | 0.22 ± 0.01 |
| Si-CH1 | 45 ± 23 | 1399 | 0.481 | 0.594 | 91 | 1.69 ± 0.02 | 0.17 ± 0.01 |
| Si-CH2 | 49 ± 20 | 517 | 0.198 | 0.223 | 89 | 1.83 ± 0.03 | 0.22 ± 0.01 |
| rGO-CH1 | 67 ± 21 | 1820 | 0.623 | 0.786 | 88 | 1.66 ± 0.02 | 0.20 ± 0.03 |
| rGO-CH2 | 49 ± 14 | 492 | 0.169 | 0.208 | 81 | 1.58 ± 0.02 | 0.32 ± 0.02 |
SEMs showed the retention of the emulsion-templated pore structure in both cases (Fig. 1c and f); carboHIPE rGO-CH1 had a predominantly closed-cell structure and rGO-CH2 also retained the interconnected pore structure of the polyHIPE precursor. In both samples the average macropore diameter, 67 ± 21 μm and 49 ± 15 μm for rGO-CH1 and rGO-CH2, respectively, was significantly smaller than the polyHIPE precursors. The larger decrease in macropore diameter in rGO-CH2 is in good agreement with the increased shrinkage of the overall sample. Gas sorption analysis shows a steep N2 uptake at low partial pressures, indicative of a predominantly microporous structure in rGO-CH1 and rGO-CH2 (Fig. 2a and b), as confirmed by pore size distribution plots, which show a sharp peak at 8–9 Å in both cases (Fig. 2c and d). The introduction of microporosity in to the pore walls resulted in exceptionally high BET surface areas, SABET, of 1820 m2 g−1 and 492 m2 g−1 for rGO-CH1 and rGO-CH2, respectively. The SABET of rGO-CH1 was significantly higher than previously reported poly-Pickering-HIPE derived carboHIPEs30 but was in good agreement with other works that employ polyDVB as a precursor to templated carbon,34 demonstrating that lower ramp rates and optimisation of pyrolysis can improve SABET. The lower SABET of the open-cell rGO-CH2 may be due to the presence of surfactant in the polymer, reducing the stability of the materials during carbonisation by disrupting the rGO scaffold at the surface of the polymer walls, leading to reduced thermal stability. This decreased stability was reflected in the larger shrinkage and lower carbon yield measured in rGO-CH2 and led to a partial collapse of micropores within the carboHIPE. When comparing the N2 isotherms of rGO-CH1 and rGO-CH2 (Fig. 2a and b), a much lower N2 uptake was measured at low partial pressures in rGO-CH2, indicative of reduced micropore volume. This reduction was further confirmed by the micropore volume and total pore volume of all materials (Table 1).
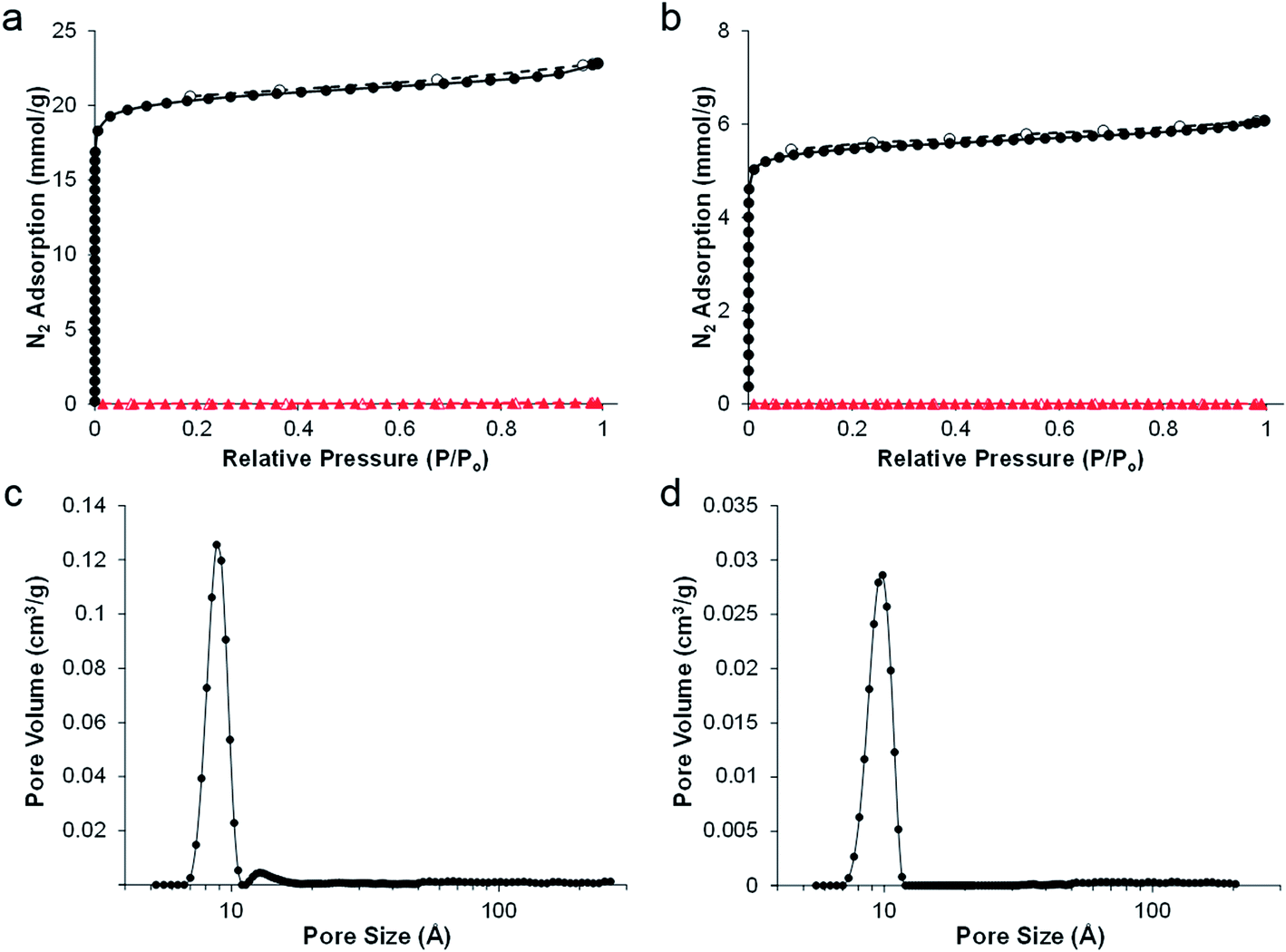 | ||
| Fig. 2 Nitrogen sorption isotherms of poly(DVB)HIPEs and carboHIPEs of (a) rGO-PH1 (red triangles) and rGO-CH1 (black circles), (b) rGO-PH2 (red triangles) and rGO-CH2 (black circles) and pore size distribution of (c) rGO-CH1 and (d) rGO-CH2. Closed shapes indicate adsorption and open shapes indicate desorption in N2 isotherms. | ||
In order to test their usefulness for energy storage applications, carboHIPEs derived from rGO-stabilised HIPEs were tested as electrodes in supercapacitor devices. For comparison, both open- and closed-cell carboHIPEs were also produced from poly(DVB)HIPEs derived from silica nanoparticle stabilised HIPEs produced adapting a previously reported method.30 The closed-cell and open-cell silica carboHIPEs are denoted as Si-CH1 and Si-CH2, respectively. The properties of the precursor silica polyHIPEs Si-PH1 and Si-PH2 were described previously30 and are summarised for comparison in Table 1. SEMs of both Si-PH1 and Si-PH2 before and after carbonisation (Fig. S1†) show also the successful retention of emulsion-templated macropores. High resolution SEM images show SiO2 particles residing in the pore wall surface of both carboHIPEs (Fig. S2†), as seen with the rGO emulsifier. N2 sorption analysis of Si-CH1 and Si-CH2 carboHIPEs showed increased adsorption at low relative pressure, indicating the presence of micropores in the sample (Fig. S3†). The introduction of microporosity during carbonisation of poly(DVB)HIPEs in the SiO2 carboHIPEs again led to huge increases in the material's SABET to 1399 and 517 m2 g−1 for Si-CH1 and Si-CH2, respectively, in good agreement with rGO equivalents. Again, carbonisation resulted in an increased skeletal density of the materials and higher porosities of the carboHIPEs in comparison to the polyHIPEs (Table 1). Raman analysis was performed on all carboHIPEs used as supercapacitor electrodes, in order to probe the degree of graphitisation within the samples (Fig. 3). All carboHIPEs showed a characteristic broad D (∼1350 cm−1) and G (∼1590 cm−1) mode, indicative of a disordered graphitic structure after carbonisation (expected as a relative low carbonisation temperature was employed), with an intensity ID/IG ratio ranging between 0.87 and 0.90 in all cases. Additionally, the subtle emergence of the 2D mode (∼2680 cm−1) in all carboHIPEs suggests some crystallinity due to graphitisation.
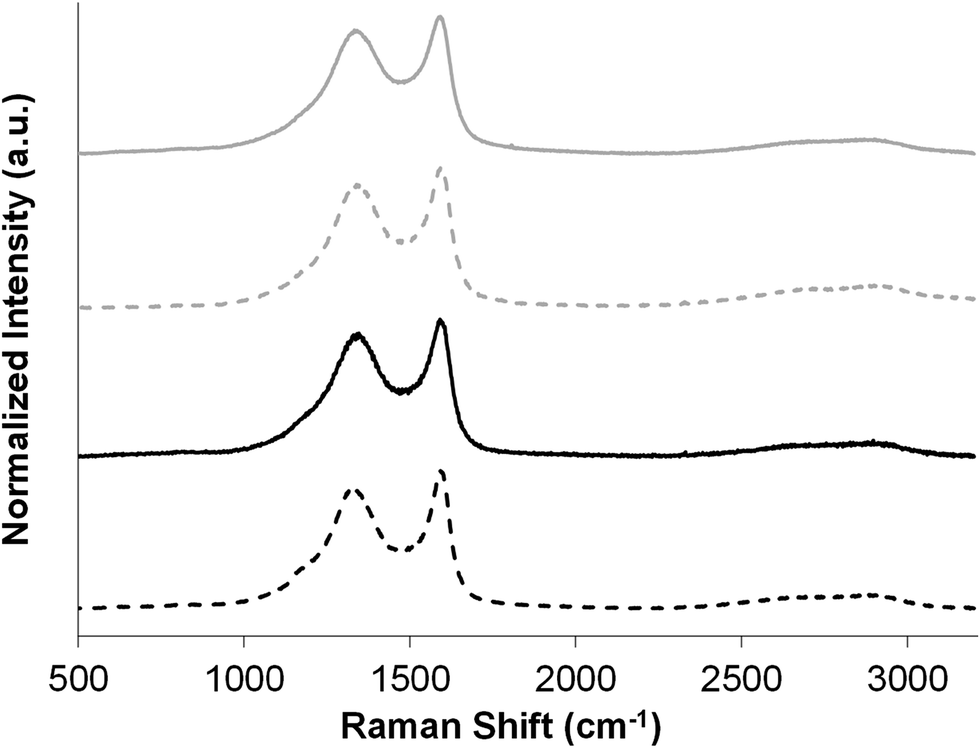 | ||
| Fig. 3 Raman spectra of both open- (solid lines) and closed-cell (dashed lines) SiO2 (grey lines) and rGO (black lines) carboHIPEs, displaying D and G bands in all cases. | ||
The electrical conductivity of each set of carboHIPEs was then investigated. The Si-CH1 and Si-CH2 carboHIPEs had excellent electrical conductivities of 78 ± 8 S m−1 for and 71 ± 14 S m−1, respectively. The electrical conductivity increased for rGO containing carboHIPEs to 277 ± 12 S m−1 and 285 ± 21 S m−1 for rGO-CH1 and rGO-CH2, respectively, around four times higher than the SiO2 equivalents and to the best of our knowledge the highest conductivity of any carboHIPE derived from a polyHIPE. The improved electrical conductivity of rGO containing carboHIPEs is attributed in part to the presence of a percolating rGO network at the pore surface of the carboHIPEs and also to the absence of insulating SiO2 particles, allowing for better contact with the probes used to measure impedance spectroscopy.
The electrochemical performance of carboHIPEs was examined using cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS). In all cases, symmetric two-electrode EDLC cells were tested within the potential window of 0 to 3 V, using the ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoro methyl sulfonyl)imide (EMI-TFSI) as electrolyte. The cyclic voltammograms of EDLCs produced using SiO2 (Si-CH1, Si-CH2) or rGO (rGO-CH1, rGO-CH2) carboHIPE composite electrodes were recorded at a scan rate of 10 mV s−1 (Fig. 4a). The rGO composite carboHIPE electrodes provided a significantly higher specific capacitance, with an associated quasi-rectangular shape. Although the specific capacitance was higher at 3 V for the closed-cell rGO-CH1 device, up to 26 F g−1, further examination of the plateau region during discharge (between 0 and 1 V) reveals that the open-cell rGO-CH2 device had a specific capacitance of around 16 F g−1. This represents a marked improvement to similar work done on SiO2 templated monolith carbon foam-based supercapacitor electrodes, with less than 13 F g−1 being measured during the discharge phase of CV at 10 mV s−1.35 Rectangular and symmetric cyclic voltammograms are both characteristics of an ideal EDLC supercapacitor,8 particularly for the rGO-CH2 device, which showed improved symmetry and serves as evidence of reversible, electric double layer-dominated processes at the electrodes. The more inclined CV plots, such as that exhibited by rGO-CH1 (Fig. 4a), suggests an undesirably high internal resistivity.36 However, the significant decrease of the maximum specific capacitance with increasing scan rate (Fig. 4b) indicates diffusion-limited electrolyte ion transport within the porous monolith structure.35,37
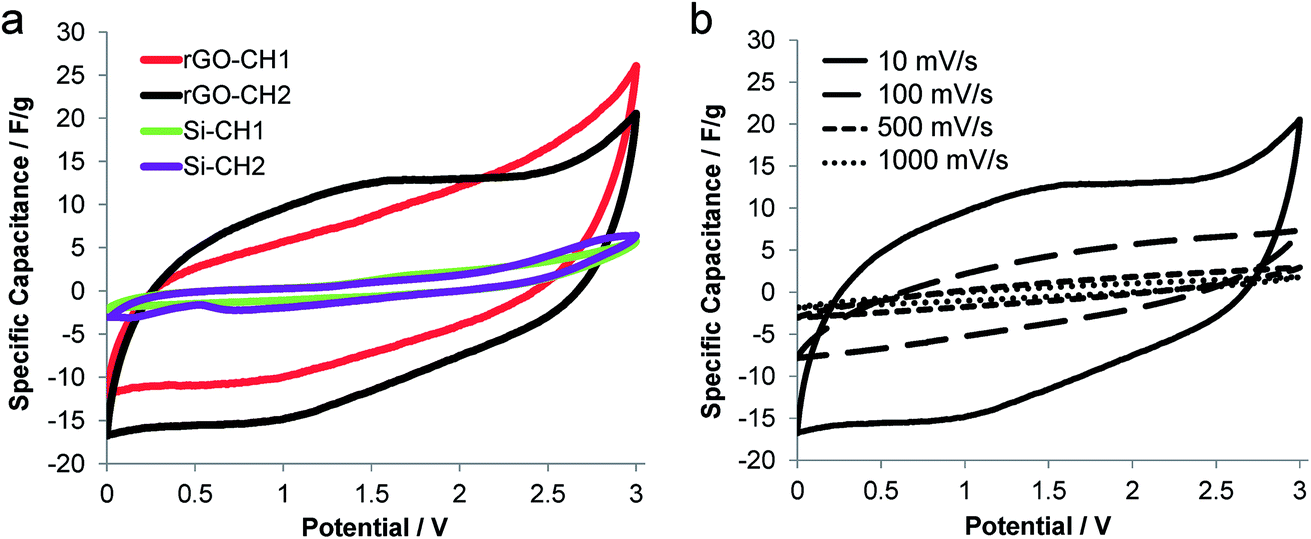 | ||
| Fig. 4 Analysis of carboHIPEs by cyclic voltammetry (a) cyclic voltammograms featuring specific electrode capacitance vs. potential of the various open and closed cell carboHIPE composite-based EDLC devices at a scan rate of 10 mV s−1. (b) Cyclic voltammograms of a rGO-PH2c-based EDLCdevice at scan rates of 10 up to 1000 mV s−1. | ||
The performance of all carboHIPE electrodes determined by GCD was evaluated within the current density range of 0.01 and 0.7 A g−1. Fig. 5a shows the discharge curves of all investigated EDLCs at the minimum current density of 0.01 A g−1. Discharging the rGO-CH2 device showed a smoother, almost linear curve below 2 V. On the other hand, the discharge curve of the less conducting rGO-CH1 containing device shows a feature at around 800 s, possibly associated to less efficient mobility of electrolyte ions within the closed-cell structure. The SiO2 carboHIPE composites, however, are discharged rapidly (<150 s) and exhibited a marked voltage drop when discharged from 3 V (as seen in Fig. 5a inset). Therefore, the rGO carboHIPE electrodes are significantly better at holding and delivering charge, over longer timescales than the SiO2 equivalents. Full charge–discharge curves (Fig. S4†) display better the large voltage drop in the transition from charge to discharge in the devices made from SiO2 carboHIPEs, leading to reduced power and suggesting an undesirably high internal resistance, whereas rGO carboHIPE devices, although not ideal symmetrical charge–discharge curves, behave more like ideal capacitors and do not exhibit any significant voltage drop prior to discharge. The Ragone plot (Fig. 5b) depicts the discharge-derived energy and power densities based on the total mass of carboHIPE per device. It is clear that the rGO carboHIPE electrodes provide the better performing device, both in terms of energy density, 5.2 W h kg−1 at minimum current density (0.01 A g−1), and also power density, 280 W kg−1 at peak current density (0.6 A g−1). The rGO-CH1 device is second-best, both in terms of energy and power densities 3.2 W h kg−1 and 209 W kg−1, respectively. The SiO2-based monoliths provide very poor electrochemical performance during GCD, consistent with the observations made after CV testing, with energy and power densities less than 0.4 W h kg−1 and 100 W kg−1, respectively. The same trend is observed in the discharge-derived specific capacitance (Fig. 5c), demonstrating the superiority of both the rGO-CH1 and rGO-CH2 electrodes, exhibiting electrode specific capacitance of 17 F g−1 at a current density of 0.01 A g−1. The lifespan of all devices were tested using cyclic GCD testing at up to at least 1000 cycles using a fixed current density of 0.6 A g−1, with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles performed on the most promising device (rGO-CH2) (Fig. 5d). The rGO-CH2 device maintained a coulombic efficiency of ∼99%, demonstrating excellent stability over 10000 cycles. All devices showed an initial conditioning over the first 200 cycles (Fig. 5d inset), after which the efficiency stabilised. The various systems stabilised at different levels, with open-cell rGO-CH2 electrodes showing the highest efficiency (99.0%), followed by the rGO-CH1 device (98.7%) and finally the two SiO2 carboHIPE EDLC devices (98.0% and 96.6%).
000 cycles performed on the most promising device (rGO-CH2) (Fig. 5d). The rGO-CH2 device maintained a coulombic efficiency of ∼99%, demonstrating excellent stability over 10000 cycles. All devices showed an initial conditioning over the first 200 cycles (Fig. 5d inset), after which the efficiency stabilised. The various systems stabilised at different levels, with open-cell rGO-CH2 electrodes showing the highest efficiency (99.0%), followed by the rGO-CH1 device (98.7%) and finally the two SiO2 carboHIPE EDLC devices (98.0% and 96.6%).
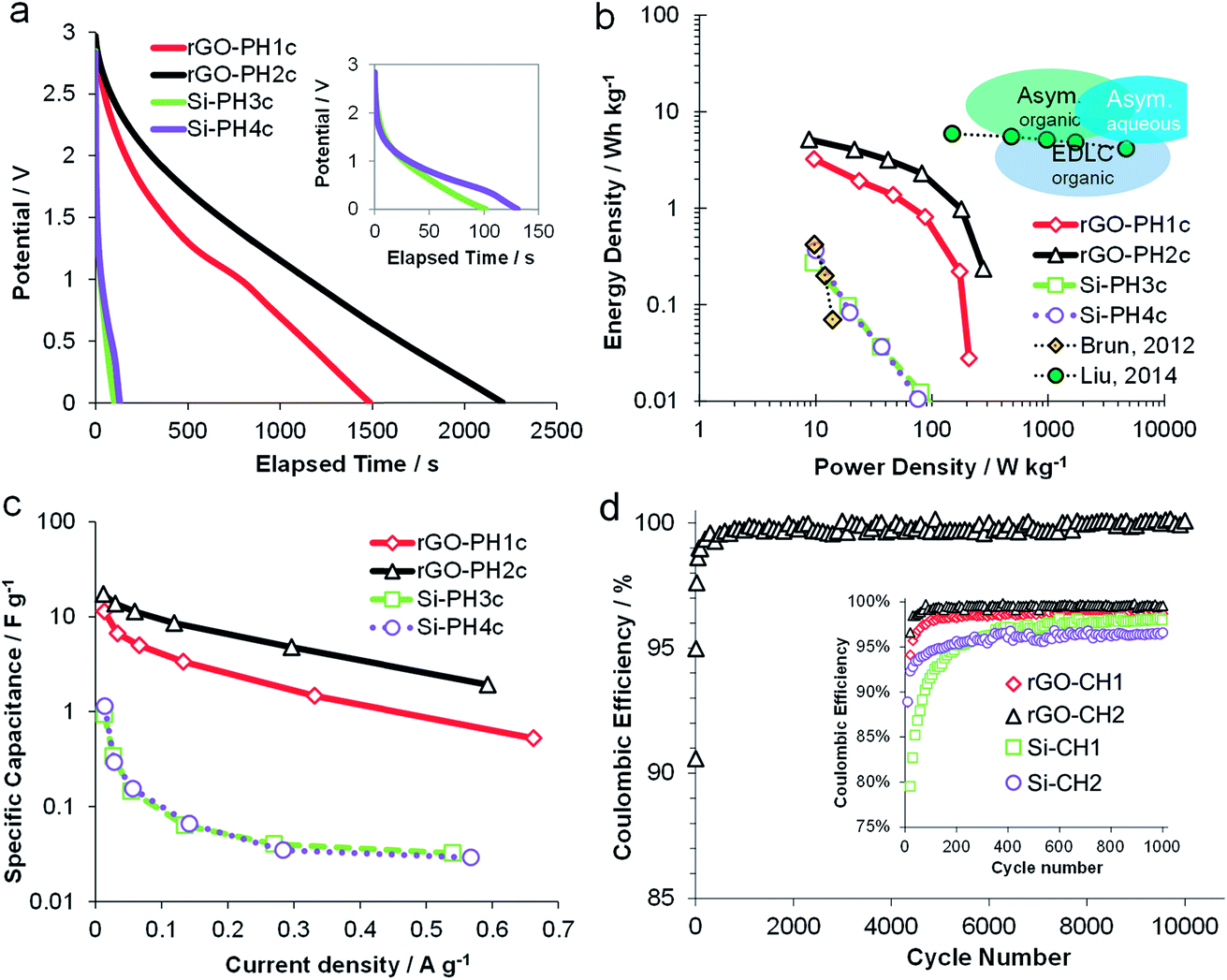 | ||
| Fig. 5 Galvanostatic charge discharge-derived plots for open and closed cell, silica and rGO-composite carboHIPEs EDLCs: (a) discharge curves at fixed current density of 0.01 A g−1, (b) Ragone plot depicting discharge-derived energy vs. power densities, containing data from two previous studies from Brun et al.35 and Liu et al.38 and with the blue bubbles demonstrating the energy characteristics of various commercially available electrochemical capacitors, (c) discharge-derived specific capacitance from 0.01 and up to 0.7 A g−1 and (d) coulombic efficiency after 10000 cycles for a rGO-CH2 cell cycles at fixed current density of 0.6 A g−1, with one data point shown every 100 cycles. The inset shows all devices up to 1000 cycles. | ||
The overall performance of the polyHIPE derived electrodes is compared to similar SiO2 HIPE-derived carbon electrodes in the Ragone plot (Fig. 5b). The SiO2–carboHIPE composite supercapacitors reported here and those reported by Brun et al. previously35 performed similarly poorly, having only an energy density of 1 W h kg−1 and a power density of 100 W kg−1. Liu et al.38 used an extra post-carbonisation KOH activation step of a nanosilica embedded carboHIPE derived from a resorcinol–formaldehyde resin, producing materials having the same energy density up to very high current densities of up to 30 A g−1. This performance is due to a number of factors, including the use of an aqueous electrolyte (6 M KOH), the high electrode BET surface area (∼1500 m2 g−1) produced by KOH activation and the grinding of samples to prepare denser carbon powders for electrodes rather than monoliths. Therefore, for monolithic binderless electrodes prepared by carbonisation of polyHIPEs, the rGO-based carboHIPEs presented here are amongst the highest performance materials reported thus far. However, as displayed by the schematic energy characteristics of various types of commercially available electrochemical electrodes (Fig. 5b), these materials still need significant improvement in order to be competitive in the electrochemical energy storage market.39
Based on the high x-axis intercept, the Nyquist plots of Si-CH1 and Si-CH2-based devices (Fig. 6b) had internal resistance or equivalent series resistance (ESR) of 6.7 and 9.6 Ω, respectively. The rGO-CH1 and rGO-CH2-based devices had an ESR of 22.7 and 13.6 Ω, respectively (Fig. 6a) meaning less internal resistance in the open-cell rGO-CH2, contributing to a better overall performance than in the closed-cell equivalent. In all cases, ESR is mainly attributed to the utilised electrolyte EMI-TFSI.40 At decreasing frequencies and within the high to medium frequency range, there is an evident semi-circle in all Nyquist plots, which is attributed jointly to the electrode's contact resistance with the carboHIPEs, as well as the electrical and charge transfer resistance of the electrolyte.41 Among the investigated carboHIPEs, the open-cell rGO-CH2 carboHIPE produced the smallest resistive semi-circle with a calculated diameter of 24.4 Ω, which can be correlated to its overall superior electrical conductivity.
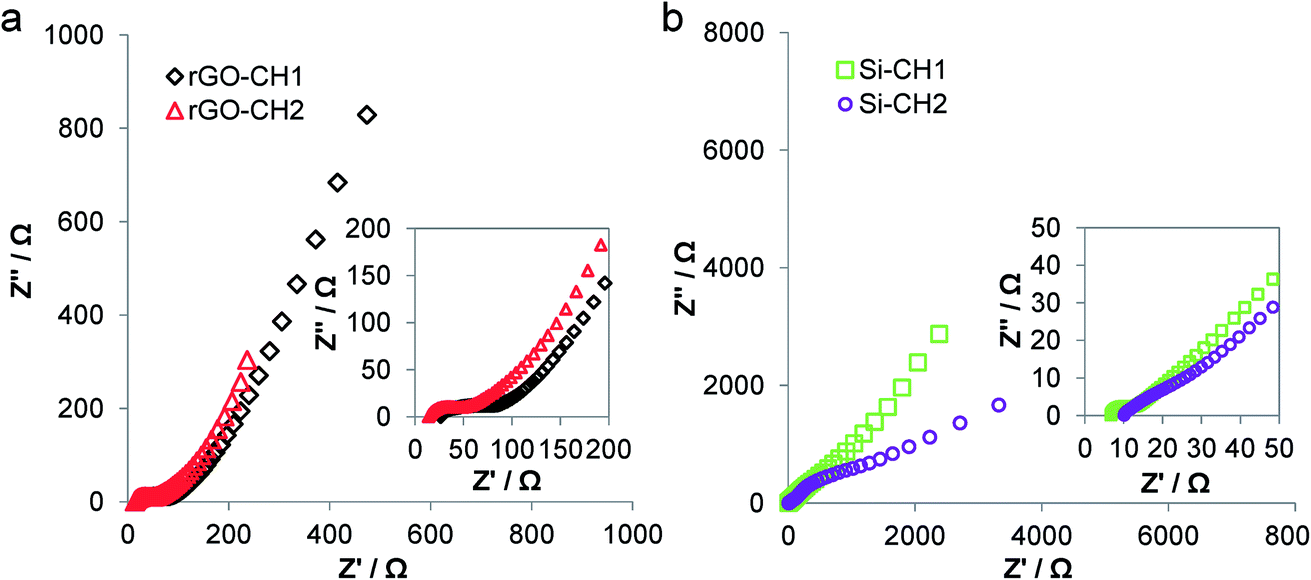 | ||
| Fig. 6 Nyquist plots derived from potentiostatic EIS of the various open and closed cell structures, (a) rGO and (b) SiO2–carboHIPE EDLC devices, measured in the frequency range of 1 MHz down to 10 mHz. | ||
Finally, the CV of rGO-CH2 was measured using a three electrode cell in order to obtain more specific electrochemical information about the best-performing carboHIPE active material (Fig. 7). The cell was equipped with a saturated calomel reference electrode, a Pt counter electrode and a KOH (6 M) aqueous electrolyte, mimicking the measurements by Liu et al.38 The CV did show deviation from ideal capacitive (i.e. non-rectangular shaped voltammogram), and deviated further from ideal behaviour with faster scan rates. This is indicative of some resistive behaviour, likely arising from the problematic diffusion of electrolyte throughout the sample, in agreement with conclusions drawn from the symmetrical devices earlier in the discussion. In addition, as monoliths are used ‘as-produced’, electrolytes will face large diffusion pathways throughout the active material, again leading to hindered ion movement. Further investigation of the effect of carboHIPE pore structure is required in order to understand this effect in more depth. Nevertheless, using the potential window described in the report by Liu et al. the capacitance was estimated to be 108 F g−1 at 1 mV s−1, with the capacitive current chosen where positive and negative sweep show similar magnitude (Fig. S5†). Although the specific capacitance remains slightly inferior to Liu et al. (172 F g−1 at 1 A g−1), when considering that carboHIPEs are used as binderless, additive-free monolithic electrode materials, this represents a significant and promising step forward in the synthesis of ‘as-produced’ electrode materials.
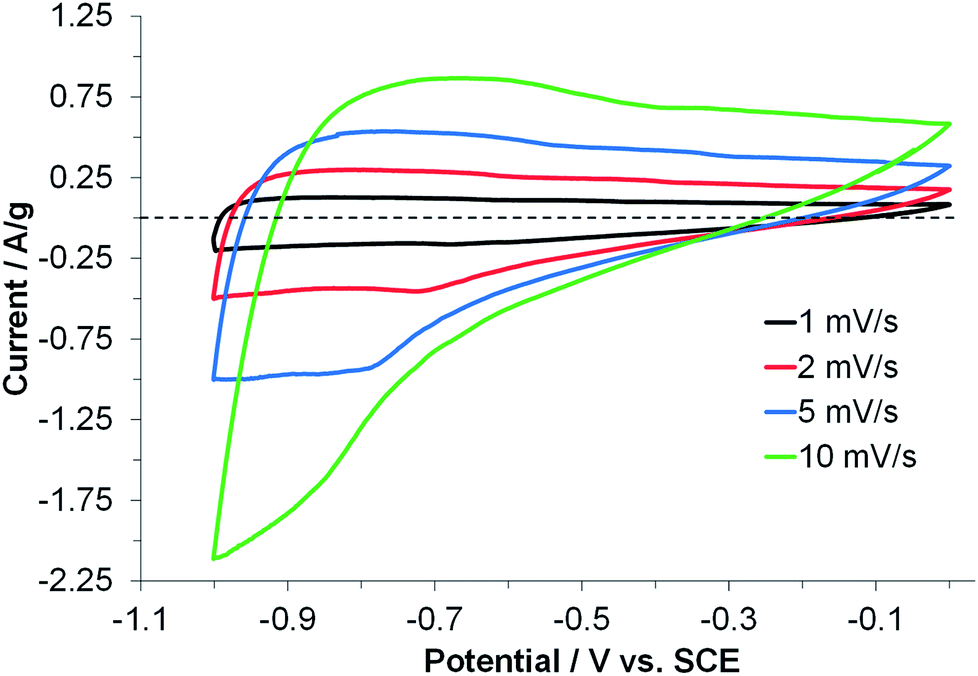 | ||
| Fig. 7 CV curves of a rGO-CH2 electrode measured at different scanning rates in a three electrode cell using a 6.0 M KOH electrolyte. The dotted line acts as a guide to the eye for a current of 0 A g−1. | ||
Conclusion
Carbon composite foams, also called carboHIPEs, were successfully produced by the carbonisation of poly(DVB)-HIPEs produced by polymerisation of the minority phase of either SiO2 nanoparticle- or rGO-stabilised water-in-DVB HIPEs. Both closed-cell and interconnected open-cell carboHIPEs were synthesised with very high surface areas of up to 1820 m2 g−1 and electrical conductivities of up to 285 S m−1. These features are amongst the highest reported thus far for any carboHIPEs, making the monoliths promising candidates for binderless EDLC electrode applications. It was demonstrated that symmetrical EDLC devices fabricated using rGO carboHIPEs outperformed other poly-Pickering-HIPE derived carbon foams, resulting in specific capacities of up to 26 F g−1, with open-cell rGO carboHIPE-based EDLCs exhibiting energy and power densities of 5.2 W h kg−1 and 280 W kg−1, respectively, maintaining a coulombic efficiency of ≥95%. This is superior to the performance of carboHIPEs derived from SiO2–polyHIPE composites, demonstrating the importance of the emulsifier used to stabilise the initial HIPE template from which the polyHIPEs were produced. By using rGO instead of the more standard SiO2 particles, carbon structures with better electrical conductivities, higher surface areas and improved EDLC device performance could be produced. Furthermore, the fine-tuning of carboHIPE pore-structures may be key to producing ideal EDLC behaviour and alleviating the diffusion-limited electrolyte ion transport found herein. This work offers a solid staging ground for the introduction of expanded dimensionalities so as to provide better porosities, enhanced surface area and potentially increased electrochemical performance from monolithic ‘as-produced’ electrodes.Experimental section
Materials
Hydrazine hydrate, divinylbenzene (DVB) (80%), CaCl2, azobisisobutyronitrile (AIBN) and ethanol were all purchased from Sigma-Aldrich. KOH was purchased from VWR, methanol was purchased from Fisher Scientific, Hypermer 2296 was kindly provided by Croda and Silver paint was purchased from Agar Scientific. All materials were used as received.Chemical reduction of graphene oxide
An aqueous graphene oxide (GO) suspension (10 mL, 10 mg mL−1) was added to a two-neck round bottom flask and diluted using distilled water until a solution of roughly 1 mg mL−1 was obtained (in this case total resulting volume was 100 mL). The sample was then reduced using a literature method32 and briefly outlined in this section: the round bottom flask containing the GO suspension was fitted with a reflux condenser before hydrazine hydrate (1 mL) was slowly added while stirring. The suspension started to change colour from orange/brown to a dark-brown/black within a few minutes after adding hydrazine hydrate. The suspension was then refluxed at 110 °C overnight while stirring. The resulting reduced graphene oxide (rGO) was collected by vacuum filtration and washed with water (5 × 100 mL) and methanol (5 × 100 mL) before being dried at 70 °C in a vacuum oven overnight.Production of rGO polyHIPEs
Reduced GO (15 mg, 5 mg mL−1) was weighed and added to DVB (3 mL) before the resulting mixture was placed in an ice bath and sonicated using a sonic probe for 2 h so that the rGO appeared to be well dispersed in DVB. AIBN (1 mol% with respect to monomer, 35 mg) was then quickly added and the mixture gently stirred before gradual addition of aqueous CaCl2 solution (9 mL, 10 g L−1). The aqueous phase was added at roughly 2 mL min−1 for 9 min while stirring slowly using a vortex mixer. After all the aqueous phase was added, the resulting emulsion was stirred much more vigorously using the vortex mixer for a further 5 min before the system was transferred into a 15 mL freestanding polypropylene (PP) centrifuge (Falcon®) tube and heated to 70 °C for 24 h in a convection oven. Afterwards, the resulting polyHIPE was removed from the PP tube and washed three times in an ethanol bath for 2 h at a time under slight agitation. The resulting structure was then dried in a vacuum oven at 110 °C overnight in order to remove any remaining water.Production of carboHIPEs
Prior to carbonisation, poly(DVB)-HIPEs were weighed and their volume measured. Samples were then heated at a ramp rate of 2 °C min−1 to 800 °C under a N2 atmosphere. Samples were held at 800 °C for 1 h before the furnace was allowed to cool to room temperature overnight (while still under a N2 atmosphere). After carbonisation, samples were weighed once more to determine carbon yield and their volume measured again to determine volume change.Characterisation of materials
All SEM images, with the exception of the highest magnification images, were taken on a variable pressure SEM (JEOL JSM 5610 LV (0.5–35 kV)). The high-resolution images were obtained using a high resolution field emission gun SEM (FEGSEM (5 kV, InLens detector)) (Leo Gemini 1525 coupled with SmartSEM software interface, Carl Zeiss NTS Ltd., UK). All polyHIPE samples were fixed on Al stubs (Agar Scientific Ltd., UK) using silver DAG paint used to attach samples securely and coated with gold (10 nm) prior to imaging. Image analysis, such as measuring average pore diameter of polyHIPEs was carried out using the image software ImageJ (version 1.48).42 The N2 isotherms of samples were measured using a porosity analyser (Micromeritics 3Flex) at −196 °C. Each sample (∼100 mg) was degassed under vacuum (0.2 mbar) at 120 °C overnight and then further degassed for 4 h (0.003 mbar) in situ at 120 °C prior to measurement. Surface areas were calculated using the Brunauer–Emmett–Teller (BET) method.37 The total volume of pores was calculated from the volume of N2 adsorbed at P/P0 = 0.97, while the micropore volume was determined using the t-plot method. Skeletal density ρs, was measured on powdered samples (≥0.5 g) using He (BOC, UK) displacement pycnometry (AccuPyc 1330, Micromeritics Ltd., U.S.A.) with fill pressure 19.5 psig. At least 10 measurements were taken and the average value recorded. The envelope densities ρe, were calculated using the mass m and volume V of cylindrical monoliths (ρe = m V−1). The percentage porosity P, for all samples was calculated using both envelope- and skeletal density (P = (1 − ρe/ρs) × 100%). Raman spectra were collected on a Renishaw inVia micro-Raman spectrometer with 532 nm (2.33 eV) DPSS diode (NA 0.8/100x, sample power 3.2 mW, WiRE 4.1 HF7241 software 2014 interface, Renishaw PLC, UK) in backscattered geometry, with relative laser spot diameters 0.8 μm with a spatial resolution of ca. 1 μm. Raman spectra were processed using WiRE software and were background subtracted (intelligent fitting polynomial 5) and normalised with respect to the G mode (1582 cm−1). In order to measure electrical conductivity poly(DVB)/carboHIPEs were placed between two flat headed probes (261e5109, RSComponents Ltd., UK) and the resistivity measured by impedance spectroscopy (IS) using a potentiostat (Gamry Instruments REF600-23011, initial frequency 10 kHz, final frequency 1 Hz, AC voltage 10 mV rms). Samples were painted using silver dag at either end in order to minimise contact resistance between the sample surface and the probes. The conductivity was calculated using the resistance measured in the linear region at frequency 100 Hz (R, U) through the sample, the monolith cross-sectional area and the monolith length.Supercapacitor assembly
Symmetric, two-electrode EDLC devices were assembled. The electrode's active material comprised cylindrical HIPE monoliths. Closed and open cell SiO2 composite carboHIPE monoliths featured 9.2 and 7.8 mm in diameter and 3.3 and 3.7 mm in thickness, respectively. Closed and open cell rGO-composite carboHIPE were 7.7 and 6.8 mm in diameter and had a thickness of 2.3 and 1.6 mm, respectively. The said monoliths were soaked overnight in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMI-TFSI, 99%, Iolitec). Each device comprised one square sheet of 60 μm thick cellulose paper (TF4060, NKK Nippon Kodoshi Corp.) of ∼2.3 cm2 of working area, which was fixed between the active areas of the two carboHIPE electrodes. This ensured separation and sufficient impregnation with the electrolyte. Each carboHIPE was placed over the edge of a rectangular strip of aluminium carbide-modified “nanowhisker” Al foil (product name: Toyal-Carbo®, provided by Toyo Aluminium K.K.), which acted as current collector and had dimensions of 10 cm in length and width matching the maximum diameter of the utilised carboHIPEs (∼0.8 cm). Once the aforementioned components were symmetrically fixed on top of one another, the resulting two-electrode device was enclosed into a PE/PA composite pouch. Afterwards, 0.5 mL of EMI-TFSI were pipetted into the said pouch which was then vacuum-sealed using a commercial sealer (Andrew James UK Ltd).Electrochemical testing
The performance of the fabricated supercapacitor pouch cells was evaluated by placing them in an isolating Faraday cage and connecting them, using a two electrode arrangement, to a Gamry Instruments, Inc. Reference 600™ potentiostat/galvanostat/ZRA instrument. The associated data analysis and fitting was carried out using the Gamry Framework EChem Analyst version 6.24. Cyclic voltammetry (CV) was completed at scan rates of 10, 100, 500 and 1000 mV s−1 with 10 mV step size and within the potential window of 0–3 V. Galvanostatic charge–discharge (GCD) testing was carried out at current densities of 0.01, 0.03, 0.06, 0.12, 0.3 and 0.6 A g−1 generating discharge curves (from 3 to 0 V) of potential vs. time. GCD-generated data of voltage vs. time, at the aforementioned currents, were used to obtain specific capacitance, energy and power densities (and thus the Ragone plot). Cyclic testing also involved GCD at peak current density of 0.6 A g−1 and repeated for 1000 cycles for all samples except the highest performing sample, rGO-CH2, which was cycled a total of 10000 times. Potentiostatic electrochemical impedance spectroscopy (EIS) was conducted from a maximum frequency of 1 MHz to a final frequency of 10 mHz with 10 points displayed per decade and AC voltage (RMS) of 10 mV. EIS was used determining the device's equivalent series resistance (ESR) as well displaying its resistive and capacitive response. A custom made three compartment electrochemical glass cell was used for the three electrode measurements. A potentiostat (Autolab, PGSTAT20) was used. The monolith was simply contacted by a Pt wire and immersed into the main compartment. The saturated calomel reference electrode was ionically connected to the main compartment of the electrochemical glass cell via a Luggin-Haber-capillary and a Pt counter electrode was ionically connected through a porous frit. The cell had a Teflon lid and was continuously purged with N2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Patricia Carry (Department of Chemical Engineering, Imperial College London) for assistance with skeletal density measurements. We also thank Cecilia Mattevi and Eslava Salvador (Department of Materials, Imperial College London) for supplying the initial graphene oxide suspensions. The authors acknowledge funding from the UK Engineering and Physical Sciences Research Council (EPSRC) through the projects “Graphene Three-Dimensional Networks” (EP/K01658X/1) and “Molecular Builders: Constructing Nanoporous Materials” (EP/J014974/1).References
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- D. D. L. Chung, Carbon, 2001, 39, 279–285 CrossRef CAS.
- F. Rodriguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef CAS.
- J. Yan, Q. Wang, T. Wei and Z. J. Fan, Adv. Energy Mater., 2014, 4, 1300816 CrossRef.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- S. M. Chen, R. Ramachandran, V. Mani and R. Saraswathi, Int. J. Electrochem. Sci., 2014, 9, 4072–4085 Search PubMed.
- J. Gamby, P. Taberna, P. Simon, J. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS.
- V. Ruiz, C. Blanco, R. Santamaria, J. M. Ramos-Fernandez, M. Martinez-Escandell, A. Sepulveda-Escribano and F. Rodriguez-Reinoso, Carbon, 2009, 47, 195–200 CrossRef CAS.
- A. Garcia-Gomez, P. Miles, T. A. Centeno and J. M. Rojo, Electrochem. Solid-State Lett., 2010, 13, A112–A114 CrossRef CAS.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- J. Lee, K. Sohn and T. Hyeon, J. Am. Chem. Soc., 2001, 123, 5146–5147 CrossRef CAS PubMed.
- J. Górka and M. Jaroniec, Carbon, 2011, 49, 154–160 CrossRef.
- N. R. Cameron, Polymer, 2005, 46, 1439–1449 CrossRef CAS.
- I. Pulko and P. Krajnc, Macromol. Rapid Commun., 2012, 33, 1731–1746 CrossRef CAS PubMed.
- H. F. Zhang and A. I. Cooper, Soft Matter, 2005, 1, 107–113 RSC.
- J. Jang and B. Lim, Adv. Mater., 2002, 14, 1390–1393 CrossRef CAS.
- S. B. Yoon, J. Y. Kim and J. S. Yu, Chem. Commun., 2001, 559–560 RSC.
- A. Szczurek, V. Fierro, A. Pizzi and A. Celzard, Carbon, 2014, 74, 352–362 CrossRef CAS.
- S. Ungureanu, M. Birot, H. Deleuze, V. Schmitt, N. Mano and R. Backov, Carbon, 2015, 91, 311–320 CrossRef CAS.
- M. Liu, L. Gan, F. Zhao, H. Xu, X. Fan, C. Tian, X. Wang, Z. Xu, Z. Hao and L. Chen, Carbon, 2007, 45, 2710–2712 CrossRef CAS.
- A. F. Gross and A. P. Nowak, Langmuir, 2010, 26, 11378–11383 CrossRef CAS PubMed.
- D. Wang, N. L. Smith and P. M. Budd, Polym. Int., 2005, 54, 297–303 CrossRef CAS.
- S. Israel, I. Gurevitch and M. S. Silverstein, Polymer, 2015, 72, 453–463 CrossRef CAS.
- A. Foulet, M. Birot, R. Backov, G. Sonnemann and H. Deleuze, Mater. Today Commun., 2016, 7, 108–116 CrossRef CAS.
- R. T. Woodward, F. De Luca, A. D. Roberts and A. Bismarck, Materials, 2016, 9, 776–785 CrossRef PubMed.
- R. T. Woodward, D. W. H. Fam, D. B. Anthony, J. Hong, T. O. McDonald, C. Petit, M. S. P. Shaffer and A. Bismarck, Carbon, 2016, 101, 253–260 CrossRef CAS.
- L. L. C. Wong, S. Barg, A. Menner, P. D. Pereira, G. Eda, M. Chowalla, E. Saiz and A. Bismarck, Polymer, 2014, 55, 395–402 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- V. O. Ikem, A. Menner, T. S. Horozov and A. Bismarck, Adv. Mater., 2010, 22, 3588–3592 CrossRef CAS PubMed.
- S. B. Yoon, J. Y. Kim and J.-S. Yu, Chem. Commun., 2001, 559–560 RSC.
- N. Brun, S. R. S. Prabaharan, C. Surcin, M. Morcrette, H. Deleuze, M. Birot, O. Babot, M. F. Achard and R. Backov, J. Phys. Chem. C, 2012, 116, 1408–1421 CAS.
- Y. H. Lin, T. Y. Wei, H. C. Chien and S. Y. Lu, Adv. Energy Mater., 2011, 1, 901–907 CrossRef CAS.
- E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- M.-X. Liu, L.-H. Gan, Y. Li, D.-Z. Zhu, Z.-J. Xu and L.-W. Chen, Chin. Chem. Lett., 2014, 25, 897–901 CrossRef CAS.
- W. T. Gu and G. Yushin, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 424–473 CrossRef CAS.
- T. Bordjiba, M. Mohamedi and L. H. Dao, J. Power Sources, 2007, 172, 991–998 CrossRef CAS.
- C. Lei, F. Markoulidis, Z. Ashitaka and C. Lekakou, Electrochim. Acta, 2013, 92, 183–187 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta09893f |
| This journal is © The Royal Society of Chemistry 2018 |