
Pressure-induced chemistry for the 2D to 3D transformation of zeolites†
 *ac
Sharon E.
Ashbrook,
a
Ángel
Morales-García,
cd
Petr
Nachtigall,
c
J. Paul
Attfield,
b
Russell E.
Morris
ac
*ac
Sharon E.
Ashbrook,
a
Ángel
Morales-García,
cd
Petr
Nachtigall,
c
J. Paul
Attfield,
b
Russell E.
Morris
ac
Abstract
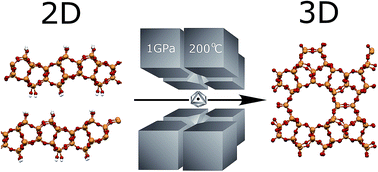
ADOR, an unconventional synthesis strategy based on a four-step mechanism: assembly, disassembly, organization, and reassembly, has opened new possibilities in zeolite chemistry. The ADOR approach led to the discovery of the IPC family of materials with tuneable porosity. Here we present the first pressure-induced ADOR transformation of 2D zeolite precursor IPC-1P into fully crystalline 3D zeolite IPC-2 (OKO topology) using a Walker-type multianvil apparatus under a pressure of 1 GPa at 200 °C. Surprisingly, the high-pressure material is of lower density (higher porosity) than the product obtained from simply calcining the IPC-1P precursor at high temperature, which produces IPC-4 (PCR topology). The sample was characterized by PXRD, 29Si MAS NMR, SEM, and HRTEM. Theoretical calculations suggest that high pressure can lead to the preparation of other ADOR zeolites that have not yet been prepared.
- This article is part of the themed collections: In memory of Petr Nachtigall, Celebrating our 2019 Prize and Award winners and Advances in Solid State Chemistry and its Applications
 Please wait while we load your content...
Please wait while we load your content...

