
The Li-storage capacity of SiOC glasses with and without mixed silicon oxycarbide bonds
Magdalena
Graczyk-Zajac
a,
Dragoljub
Vrankovic
a,
Philipp
Waleska
b,
Christian
Hess
b,
Pradeep Vallachira
Sasikumar†
a,
Stefan
Lauterbach
c,
Hans-Joachim
Kleebe
c and
Gian Domenico
Sorarù
 *d
*d
aMaterialwissenschaft, Technische Universität Darmstadt, Otto Berndt Str. 2, 64287 Darmstadt, Germany
bEduard-Zintl-Institut für Anorganische Chemie und Physikalische Chemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 8, 64287 Darmstadt, Germany
cInstitute for Applied Geosciences, Technische Universität Darmstadt, Schnittspahnstr. 9, 64287 Darmstadt, Germany
dDipartimento di Ingegneria Industriale, Università di Trento, Via Sommarive 9, 38123 Trento, Italy. E-mail: soraru@ing.unitn.it
First published on 23rd November 2017
Abstract
In this work we investigate the electrochemical behaviour of two silicon oxycarbide (SiOC) glasses synthesized from the same starting precursor. In one case we perform pyrolysis in an Ar flow, while in the second case, the glass is synthesized under a CO2 flow. The microstructural characterization of the glasses unambiguously demonstrates that the Ar-pyrolyzed material (SiOC–Ar) is a SiOC/Cfree nanocomposite with mixed SiCxO4−x 0 ≤ x ≤ 4 units, whereas the CO2-pyrolyzed sample (SiOC–CO2) is a SiO2/Cfree nanocomposite with exclusively SiO4 units forming the amorphous network. Therefore, in this study we investigate two model systems, addressing the question as to whether the mixed SiCxO4−x units in the SiOC glass play an essential role regarding electrochemical performance. The UV-Raman analysis reveals that the sp2 carbon present in the mixed bond-containing sample is more disordered/has more defects than the one dispersed in the SiO2 matrix. Apart from the above dissimilarities, the materials present comparable microstructures and a similar amount of free carbon. Nevertheless, SiOC–Ar recovers almost twice higher reversible Li-ion storage capacity than SiOC–CO2 (325 vs. 165 mA h g−1, respectively). We rationalize this difference in terms of the enhanced Li-ion storage in the more disorder free carbon phase of SiOC–Ar, while this disorder is induced by the presence of the mixed-bond units.
1. Introduction
Li-ion batteries are widely used in portable electronics such as mobile phones and laptops and are now slowly entering the automotive market.1,2 Graphitic anodes provide stability and safety but their capacity does not exceed the theoretical value of 372 mA h g−1. However, in order to meet the consumer requirements for lighter, higher capacity and faster charge/discharge systems, there is strong interest in developing new materials, which can achieve such targets.3–5 Among many different Li-ion storage hosts, silicon oxycarbide glasses (SiOCs) have attracted much attention because they recover a capacity up to 900 mA h g−1 demonstrating excellent stability during high rate tests and prolonged cycling.6–12Silicon oxycarbides belong to the family of Polymer Derived Ceramics (PDC), which are obtained from preceramic polymers through a pyrolysis process in a controlled atmosphere.13 SiOC glasses are derived from crosslinked polysiloxanes. The polymer structure is based on a Si–O–Si tridimensional network with organic moieties, such as –CH3 or –C6H5 directly bonded to the silicon atoms.14,15 Upon pyrolysis at T ≥ 800 °C in an inert atmosphere these precursors form a silicon oxycarbide network made of Si-centered tetrahedral sites with O and C atoms sitting at the corners bridging 2 or more Si tetrahedra.16 Some of the C atoms, initially belonging to the organic moieties, are converted into a separate sp2 C phase, usually called the “free carbon” phase. The chemical composition of SiOC glasses can be expressed as follows: SiCxO2(1−x) + yCfree, where SiCxO2(1−x) represents the chemical composition of the amorphous network and yCfree the molar amount of the free carbon phase.17
When tested as Li-ion storage hosts, SiOC glasses have shown very high reversible capacities up to 900 mA h g−1 with a high rate capability, which allows recovery up to 200 mA h g−1 at 2C rate (charge/discharge in 30 minutes).18 The performance of composite materials consisting of SiOCs and graphene is even better with reversible capacities of ∼200 mA h g−1 stable over 1000 cycles at 5C rate (charge/discharge in 12 minutes).12,19 The drawback of SiOC anodes resides is the high first cycle irreversibility, which is around 30% in the best case, and the large hysteresis since the stored lithium ions are recovered at higher potentials thereby leading to the limited electrochemical performance of the cell.20–22 The mechanism of Li-storage in SiOCs has not been fully rationalized yet. Obviously, a better understanding of the Li storage sites in silicon oxycarbides will be of vital importance for overcoming the existing limitations of this electrode type while maintaining their advantages compared to commercial graphite anodes. In the literature different hypotheses to explain how lithium atoms are stored in SiOCs have been presented including the following: (i) intercalation between the sp2 C layers,8,23,24 (ii) in the edges and micropores of neighboring graphene layers23,25 and (iii) in the Si–O–C glass phase,26 and in particular in the mixed silicon oxycarbide units.27 According to the results of ab initio studies, mixed Si units impact Li insertion in SiOCs by lowering the chemical potential (energy levels) of unfilled carbon electronic states.28–30
Distinguishing between these hypotheses is not an easy task because the structure of silicon oxycarbide glasses is rather complicated and not yet fully understood, and it depends on many experimental parameters such as the composition of the precursor, the pyrolysis conditions (heating rate, maximum pyrolysis temperature, and atmosphere), etc.
Recently, the group of Narisawa at Osaka Prefecture University showed that a commercial polysiloxane resin, which upon pyrolysis in an argon flow would lead to the known silicon oxycarbide structure, namely SiCxO2(1x) + yCfree, upon pyrolysis in a CO2 flow it leads to a SiO2/Cfree nanocomposite in which no Si–C bonds are present.31 In other words, pyrolysis in a CO2 flow results in the complete cleavage of the Si–C bonds and at the end only SiO2 and free carbon are present in the ceramic residue.
These findings give us an excellent opportunity to investigate the role of Si mixed bonds in the electrochemical storage of Li ions. Accordingly, we have synthesized two silicon oxycarbides from the same starting precursor: in one case we performed the pyrolysis in an inert atmosphere, namely an Ar flow and in the second case the precursor was pyrolyzed in a CO2 flow with the aim to prepare two SiOC glasses having a similar amount and distribution of free carbon in a (i) silicon oxycarbide matrix and (ii) silica matrix. Furthermore, we rationalize the electrochemical performance of SiOC–Ar and SiOC–CO2 with respect to their microstructural properties, and in particular the presence/absence of mixed O–Si–C bonds.
2. Experimental part
2.1 Synthesis of the SiOC glasses
In this work the starting precursor has been prepared in the form of a highly porous polysiloxane colloidal aerogel in order to facilitate the diffusion and reaction of the flowing CO2 during the pyrolytic transformation. Indeed, it was shown by Narisawa et al. that, in order to have a complete reaction between the flowing gas and the preceramic polymer, the polysiloxane particle size must be below a few microns.32 Poly(methylhydrosiloxane) (PMHS, CAS # 63148-57-2) and divinylbenzene (DVB, 80%, mixture of isomers, CAS # 1321-74-0) were purchased from Alfa Aesar (USA). Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution ∼Pt 2% in xylene (CAS # 68478-92-2) was obtained from Sigma-Aldrich (USA). All reagents were used as received. PMHS and DVB were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 weight ratio in acetone with an 80% (vol/vol) dilution. In a typical synthesis 0.8 g of PMHS, 1.6 g of DVB, and 8 g of acetone were mixed together in a Teflon container, stirred for about 10 minutes and then 19 μl of Pt catalyst were added. The crosslinking reaction was performed in a pressure vessel at 120 °C for 6 h. After 6 h of curing, the gels were allowed to cool down to room temperature, carefully removed from the Teflon container and transferred to solvent (acetone) to remove the unreacted polymer and catalysts by changing the acetone twice a day for five days. Wet gels were then supercritically dried in CO2 using a home-made reactor at 50 °C and 100 bar.
:2 weight ratio in acetone with an 80% (vol/vol) dilution. In a typical synthesis 0.8 g of PMHS, 1.6 g of DVB, and 8 g of acetone were mixed together in a Teflon container, stirred for about 10 minutes and then 19 μl of Pt catalyst were added. The crosslinking reaction was performed in a pressure vessel at 120 °C for 6 h. After 6 h of curing, the gels were allowed to cool down to room temperature, carefully removed from the Teflon container and transferred to solvent (acetone) to remove the unreacted polymer and catalysts by changing the acetone twice a day for five days. Wet gels were then supercritically dried in CO2 using a home-made reactor at 50 °C and 100 bar.
The monolithic precursor gels were converted into the corresponding monolithic SiOC–Ar and SiOC–CO2 glasses through a pyrolysis process at 900 °C for 1 h, using an alumina tubular furnace (Lindbergh blue) with a heating rate of 5 °C min−1 and 100 ml min−1 of an Ar or a CO2 flow, respectively.
2.2 Structural and microstructural characterization
The amount of carbon present in the pyrolyzed SiOC samples was measured by hot-gas extraction, using a Leco-200 carbon analyzer. The amount of free carbon in the SiOC glasses was also estimated by thermogravimetric analysis (TGA) in flowing air. Thermogravimetric analysis (TGA) was performed on small aerogel fragments with a Netzsch STA 409 equipment (Netzsch Geraetebau Gmbh, Selb, Germany) at 5 °C min−1 in an air flow (30 cc min−1) up to 1300 °C.The specific surface area (SSA), porosity and pore size of the precursor and of the two SiOC aerogels were investigated by N2 physisorption using a Micromeritics 2010 ASAP instrument (Micromeritics, Norcross, GA, USA). N2 isotherms were collected at 77 K after degassing at 200 °C for 4 h. The specific surface area was determined from a BET (Brunauer, Emmet, and Teller) analysis in the P/P0 range of 0.05–0.30 using a molecular cross sectional area for N2 of 0.163 nm2 and a minimum of five data points. The pore size distribution (PSD) curves were evaluated using the BJH (Barrett, Joyner, and Halenda) analysis from the desorption isotherm. The bulk density was determined for bulk cylindrical samples measuring the mass with an analytical balance and the dimensions with a caliper.
Micro-Vis-Raman spectra were recorded with a confocal micro-Raman spectrometer (Horiba HR 800, Horiba, Japan), using an Ar-ion laser with a wavelength of 514.5 nm. Spectra were recorded in a Raman shift range from 0 to 4000 cm−1.
UV Raman Spectroscopy has been carried out using a tunable Ti:Sa solid state laser (Coherent, Indigo-S) and a triple stage spectrograph (Princeton Instruments, TriVista 555) with an attached CCD camera (Princeton Instruments, Spec10:2kBUV). The tunable laser system has been adjusted to a wavelength of 256.7 nm (THG). The spectral resolution of the spectrometer is 1 cm−1. All UV Raman spectra were collected under ambient conditions at room temperature (≈25 °C) using a laser power of 3.6 mW. Laser damage can be excluded, because the radiation is only softly focused on the surface of the sample creating a spot size of about 0.6 mm2.33 The acquisition time of each spectrum was ≥1 h to obtain a sufficient signal-to-noise.
The first order regime is comprised of the G band (perfect carbon lattice) and the D/T/D′′ band (Table 1), caused by deviation from the perfect graphitic lattice. Their respective appearance and intensity can provide important information about the molecular structure.34,35
| G band | ≈1580 cm−1 | In-plane breathing mode of hexagonal sp2-carbon rings (E2g symmetry) |
| D band (D1) | ≈1380 cm−1 | Disordered graphitic lattice, graphene layer edges/defects (A1g symmetry), turbostratic carbons |
| D2 band | ≈1620 cm−1 | Disordered graphitic lattice, (E2g symmetry), in UV-Raman this bands corresponds to C–C configurations with a wider electronic gap35,36 |
| D3 band | ≈1500 cm−1 | Amorphous carbon |
In order to prepare the aerogels for the TEM measurement, a small quantity of both SiOC powder samples was dispersed in ethanol using an ultrasonic bath. The suspension was transferred with a spray coater onto a thin amorphous carbon film, suspended on standard 300 mesh Cu TEM grids, using an ultrasonic vaporizer. The grid was dried under vacuum and cleaned in Ar-plasma for 20 s before it was transferred into a TEM column. No light carbon coating was applied, since one focus of the TEM investigations was to image the free-carbon phase. Phase contrast HRTEM was carried out employing a JEOL 2100 F (JEOL, Tokyo, Japan) transmission electron microscope equipped with a field emission gun, operated at 200 kV.
Chemical bonds present in the pyrolyzed SiOC samples were investigated by Fourier Transform Infrared (FT-IR) spectroscopy. FT-IR spectra were collected in transmission mode using a Varian 4100 FT-IR Excalibur Series equipment in the range 4000–400 cm−1 with KBr pellets. An average of 50 scans with a resolution of 2 cm−1 were recorded for each sample.
The local environment around the silicon atoms was investigated by 29Si solid state nuclear magnetic resonance (29Si SS-NMR). The analyses were carried out with a Bruker 300WB instrument operating at a proton frequency of 300.13 MHz. NMR spectra were acquired with a SP pulse sequence under the following conditions: 29Si frequency: 59.60 MHz, π/4 pulse length: 2.25 μs, recycle delay: 150 s, and 4k scans. The samples were packed in 4 mm zirconia rotors, which were spun at 5 kHz under an air flow. Q8M8 was used as an external secondary reference.
2.3 Electrochemical characterization
To prepare electrodes, the pyrolyzed SiOC samples were mixed with carbon black (Super P) and polyvinylidene fluoride binder (85:5:10 by weight) in N-methyl-2-pyrrolidone to form a slurry. This slurry was then coated onto a copper foil using a doctor blade and dried at 40 °C for 24 h. Electrodes of 10 mm diameter were cut out of the coated copper foil, and dried at 80 °C under vacuum in a Buchi oven for 24 h. Without further contact with air the dried electrodes were transferred to an argon-filled glovebox for cell assembly (Swagelok® type cell) using lithium foil as the counter electrode and QMA (Whatmann™, UK) as a separator. As the electrolyte 180 μl of 1 M LiPF6 dissolved in a commercial electrolyte EC:DMC/1:1 (Solvionic, France) was used. All cells were cycled at charging/discharging rates of 36 mA g−1 between 0.005 and 3 V versus Li/Li+.
3. Results and discussion
3.1 Electrochemical results
The first insertion/extraction of lithium ions into SiOC–Ar and SiOC–CO2 is depicted in Fig. 1. As a first observation we can say that the charge and discharge curves reveal only small differences. During the first lithiation, a continuous slope of the curve is found for SiOC–Ar. This slope is typical of silicon oxycarbide glasses, namely for SiOC glasses containing mixed SiCxO4−x 0 ≤ x ≤ 4 sites, and it has been reported and discussed by us elsewhere.9,11,37 In the lithiation of the CO2-treated aerogel a plateau-like slope at 0.25 V can be found. Since this sample is a SiO2/Cfree nanocomposite (as will be shown later on), we tentatively attribute this plateau to hardly reversible storage of lithium ions in the silica network. Due to the porous morphology of both samples (the corresponding experimental results of N2 adsorption will be addressed in the following paragraph) the losses related to a solid electrolyte interface (SEI) formation (0.6–2 V) are pronounced. Although the SSA of SiOC–CO2 is almost twice higher than that of SiOC–Ar, there is no significant difference in the charge lost in this potential range. This feature can be rationalized recalling that the CO2-treated sample is indeed a SiO2/C composite and silica does not contribute to the formation of the SEI. Moreover, HRTEM investigations (for details, see Fig. 8) revealed that for the Ar-treated samples the surface of the individual aerogel particles is covered by a continuous C film, while for the CO2-treated aerogel the C film is not continuous leading to the exposure of the SiO2 to the electrolyte. Therefore, the absolute surface areas efficiently covered by carbon are similar in these samples. | ||
| Fig. 1 First cycle of the galvanostatic insertion/extraction of lithium ions into SiOC–Ar and SiOC–CO2. | ||
Fig. 2 presents the prolonged galvanostatic cycling of the SiOC–Ar and SiOC–CO2 samples. Although the first lithiation capacities are similar (compare Fig. 1) and the charge recovered in the initial few cycles tends to fade, the stabilized capacity values of SiOC–Ar are almost twice as high as those of SiOC–CO2. This is the most important difference we found in the two studied samples and hereafter we will discuss and relate this finding with respect to the different chemical structures of the investigated glasses.
 | ||
| Fig. 2 Capacity recovered during the extended galvanostatic cycling of SiOC–Ar and SiOC–CO2. | ||
3.2 N2-adsorption measurements
We performed a careful structural and microstructural characterization of the two investigated materials to reveal the reasons for the pronounced difference in the electrochemical performance of SiOC–Ar and SiOC–CO2 glasses. Fig. 3 presents the results of the N2 physisorption analysis, while the relevant values are reported in Table 2. | ||
| Fig. 3 (a) Adsorption (solid circles)/desorption (empty circles) isotherms recorded for the SiOC aerogels; (b) cumulative pore volume (open circles) and pore size distribution (PSD, solid circles). | ||
| Sample | T (°C) | Density (g cm−3) | SSA (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|---|---|
| SiOC–Ar | 900 | 0.47 | 163 | 0.75 | 20–50 |
| SiOC–CO2 | 900 | 0.48 | 279 | 0.41 | 7–20 |
SiOC–Ar and SiOC–CO2 glasses show type IV adsorption/desorption isotherms typical of mesoporous materials (Fig. 3a) with a hysteresis loop in the 0.8–0.9 P/P0 range. SiOC–Ar reveals a total pore volume of 0.75 cm3 g−1 and a maximum of the pore size distribution curve in the range 30–40 nm (Fig. 3b). The pyrolysis in a CO2 flow leads to a mesoporous aerogel with a lower amount of total porosity (0.41 cm3 g−1) and smaller pore size with a maximum around 10–12 nm. Due to the contribution of the smaller pores, the specific surface area of SiOC–CO2 is higher compared to the sample treated in an Ar flow.
3.3 29Si solid-state NMR and FT-IR spectroscopy
The 29Si SS-NMR and FT-IR spectra recorded for the SiOC–Ar and SiOC–CO2 aerogels are shown in Fig. 4. | ||
| Fig. 4 (a) 29Si SS NMR and (b) FT-IR spectra recorded for SiOC–Ar and SiOC–CO2. | ||
The 29Si SS NMR spectrum of the Ar-treated sample shows the presence of resonances at −39, −71 and −110 ppm assigned to mixed C2SiO2, CSiO3 and SiO4 Si sites, respectively, which are typical of a silicon oxycarbide glass.38 In contrast, the spectrum of the sample pyrolyzed in a CO2 flow shows only resonance at −107 ppm, typical of SiO4 units of silica glass. Thus, the NMR study unambiguously proves that, as expected, the pyrolysis in a CO2 flow changed radically the structure of the SiOC glass by consuming all Si–C bonds and converting them into Si–O bonds. FT-IR spectra support the findings obtained by 29Si SS-NMR. The FT-IR spectra of both SiOC–Ar and SiOC–CO2 show peaks related to the presence of Si–O bonds in the range 1100–1000 cm−1 and at 450 and 800 cm−1. The main peak corresponding to the stretching of the Si–O–Si bonds is located at 1050 cm−1 for the SiOC–Ar sample and at 1075 cm−1 for the SiOC–CO2 aerogel. According to the literature, the shift toward lower wavenumbers (observed for SiOC–Ar) is associated with the insertion of C atoms into the silica network and a corresponding increase of the Si–O–Si bond angle above 150°.39 On the other hand, the peak at 1075 cm−1 for the stretching of the Si–O–Si bonds in SiOC–CO2 confirms the formation of a SiO2 network.31,40 In conclusion, both NMR and FT-IR spectroscopies clearly confirm that, upon pyrolysis in a CO2 flow, the silicon oxycarbide network is transformed into a silica network. The FT-IR spectra of both materials also show a peak around 1600 cm−1. It is assigned to the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of the Cfree phase.40,41 The peak at 1600 cm−1 is more intense in the case of the CO2-treated sample suggesting either a higher amount of Cfree or a better organized structure compared to the Ar-pyrolyzed aerogel. Finally, in the SiOC–Ar sample, the IR also reveals an absorption at 1700 cm−1 which indicates the presence of CO bonds.40,41 CO moieties could be formed, either during the pyrolysis process or after pyrolysis by exposing the aerogel to ambient conditions.42 Assuming that the CO bonds, observed in the Ar-treated sample, are formed after exposing the pyrolyzed aerogel to the laboratory atmosphere, then this would suggest that the Ar-pyrolyzed aerogels are more reactive compared to those pyrolyzed in CO2.
C bonds of the Cfree phase.40,41 The peak at 1600 cm−1 is more intense in the case of the CO2-treated sample suggesting either a higher amount of Cfree or a better organized structure compared to the Ar-pyrolyzed aerogel. Finally, in the SiOC–Ar sample, the IR also reveals an absorption at 1700 cm−1 which indicates the presence of CO bonds.40,41 CO moieties could be formed, either during the pyrolysis process or after pyrolysis by exposing the aerogel to ambient conditions.42 Assuming that the CO bonds, observed in the Ar-treated sample, are formed after exposing the pyrolyzed aerogel to the laboratory atmosphere, then this would suggest that the Ar-pyrolyzed aerogels are more reactive compared to those pyrolyzed in CO2.
Chemical analysis revealed that the total amount of C present in the SiOC aerogels amounts to 45 and 33 wt% for the SiOC–Ar and the SiOC–CO2 sample, respectively. Since for the CO2-treated sample C is present only in the Cfree phase, this amount (33 wt%) represents the amount of segregated carbon. For the SiOC–Ar sample the evaluation of the free carbon amount is not straightforward since some of the C atoms are engaged in forming Si–C bonds. We can only infer that the amount of segregated C in SiOC–Ar must be lower than 45 wt%. We also performed thermogravimetric analysis in air to assess the amounts of the free carbon phase. The corresponding TGA curves are depicted in Fig. 5.
 | ||
| Fig. 5 TGA of the SiOC–Ar and SiOC–CO2 aerogels in flowing air. | ||
Both curves show an initial small weight loss below 200 °C due to the evaporation of adsorbed water. For the SiOC–CO2 sample the weight is stable up to 450 °C. At ∼500 °C the Cfree is oxidized according to the reaction C + O2 → CO2, leading to a weight loss of 37 wt%.
Compared to the value of C measured by chemical analysis the result from TGA analysis seems to be overestimated. This discrepancy could be, at least partially, explained by considering that the sp2 C phase in the silicon oxycarbide glass, pyrolyzed at a relatively low temperature (900 °C), may still contain hydrogen, which also contributes to the measured weight loss. In conclusion, the amount of Cfree estimated by TGA for the CO2-treated sample (37 wt%) can be considered as an upper limit.
The TGA curve of the Ar-treated sample shows first a small increase (∼6 wt%) in the temperature range between 250 and 400 °C. This feature is related to the oxidation of Si–CHx (x = 1, 2) bonds leading to new Si–O/Si–OH bonds resulting in a corresponding net weight increase.40 For this sample oxidation of Cfree leads to a weight loss of 47% at temperatures between 400 and 650 °C. Also for SiOC–Ar, the amount of the sp2 C phase may be overestimated due to the likely presence of residual hydrogen in the pyrolyzed aerogel.
In summary, the TGA results confirm the absence of Si–C bonds in the CO2-pyrolyzed material and suggest that the SiOC–Ar sample contains slightly more free carbon than the CO2-treated aerogel.
3.4 UV/Vis-Raman spectroscopy
The Vis-Raman spectra are presented in Fig. 6. The D mode present at around 1335 cm−1 originates from disorder-induced breathing motions of six-fold aromatic rings, whereas the G mode at around 1590 cm−1 is induced by in-plane bond stretching of sp2 hybridized C atoms. The 2D bands at around 2700 cm−1 and 3200 cm−1 are attributed to second-order vibration modes of the D band. The Raman band at around 2940 cm−1 is the D + G combination mode induced by carbon disorder.35 The strong fluorescence at higher wavenumbers (beyond 2000 cm−1) registered for the SiOC–Ar sample is associated with the higher number of defects present in the sample or/and organic residues, e.g. radicals such as C˙ and CH˙.43 Nevertheless, the results of the carbon band fitting performed according to Sadezky et al. showed no significant difference in the carbon microstructure of the investigated aerogels.34 | ||
| Fig. 6 Vis-Raman spectra of the samples pyrolyzed under Ar and CO2. | ||
In order to assess subtle differences in the free carbon phase within the investigated aerogels, UV-Raman spectroscopy was performed. UV Raman, by exciting both π and σ-states, allows us to probe highly disordered/clustered carbons. The first order UV Raman spectra of SiOC–Ar and SiOC–CO2 together with the results of a peak-fit analysis are plotted in Fig. 7a and b, respectively. For a closer examination of the wavelength interval between 1000 cm−1 and 2000 cm−1 a fitting procedure considering the G-band and three D-bands (D, D2, and D3) was applied. The assignment of the bands to the related vibrations/carbon form is addressed in the Experimental section (see Table 1). The used line profiles were adopted from extensive studies on carbons by Sadezky et al. and Ferrari et al.34,35 (see Table 3). The D-band, which is related to the disordered graphitic lattice caused by defects on the edges of the turbostratic carbons, is very broad (although of low intensity). The G-band is comparably less pronounced confirming a low degree of graphitic order in the carbon phase. The D3-band indicates the presence of amorphous carbon. Its low intensity points out the turbostratic character of the free carbon phase. Both aerogels reveal a D2-band with the highest intensity, confirming the presence of a disordered graphitic lattice. Since this band is absent in the Vis-Raman spectra of the aerogels, we first try to assign it to the corresponding carbon form/hybridization. Sadezky et al. reported that in polycrystalline graphite the D2 band is usually of weak intensity; however, it becomes strong in soot-like materials. Castiglioni et al.36,44 investigated polyconjugated molecules, polycyclic aromatic hydrocarbons (PAHs), characterized by a planar network of sp2 carbon atoms with the same relative arrangement shown by carbon atoms in a graphite sheet. These molecules are considered as molecular models of graphitic ‘islands’ of a finite size, which are expected to occur in disordered and nanostructured carbon materials containing sp2 carbons. The presence of bands over 1600 cm−2 has been observed experimentally and modelled for these polyaromatic structures.36 We do not expect such organic structures in the aerogels pyrolyzed at 900 °C, but we rationalize the appearance of a strong D2 band for higher excitation energy (UV laser) by considering the presence of small, microcrystalline graphitic domains. Graphitic domains of a given size are characterized (in a similar way to that in the case of molecules) by a non-vanishing energy gap related to the electronic excitation localized on the domain. The frequency of the Raman bands is also size dependent (as shown by the study on molecules44) and therefore dependent on the energy gap. Raman experiments carried out at different excitation energies35,45,46 on a disordered sample containing a distribution of different graphitic domains extract the response of those domains, which satisfy the resonance conditions (Egap ≈ hνlaser). In other words, while changing the laser energy one probes different “confined” structures. Using the above reasoning, we conclude that the D2-band originates from small clusters of sp2 hybridized carbons characterized by a wide energy gap. Thus, those clusters can only by analysed using UV-excitation.
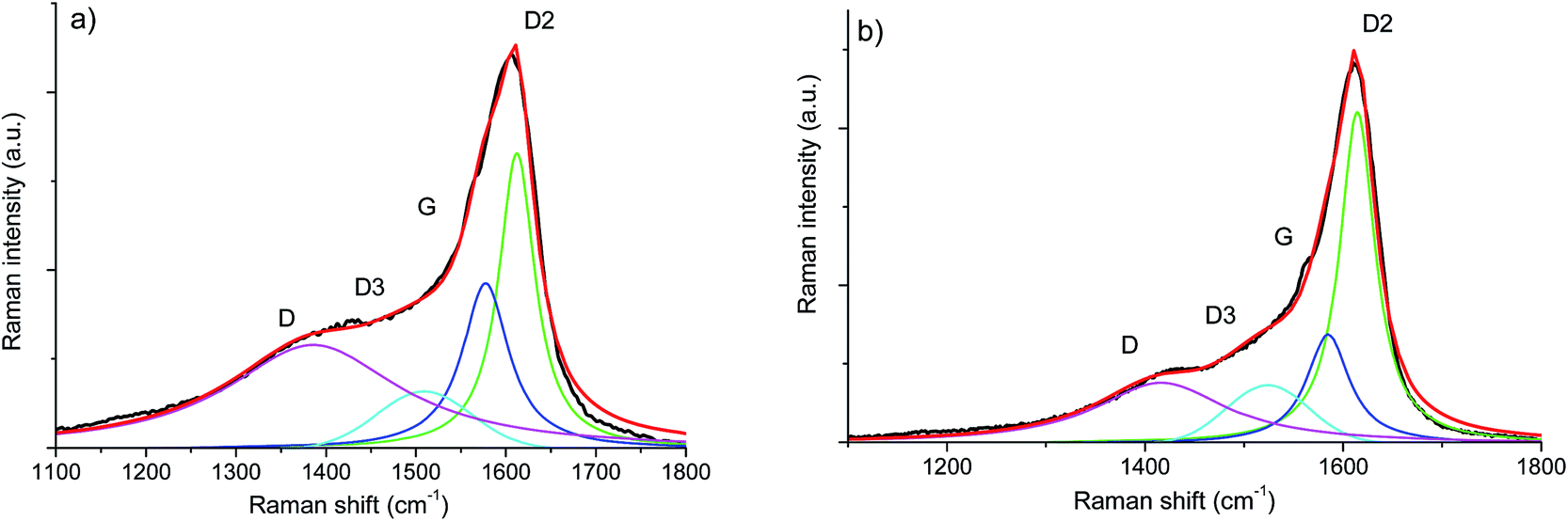 | ||
| Fig. 7 First order UV-Raman spectra of (a) SiOC–Ar and (b) SiOC–CO2 together with the results of peak-fitting analysis. | ||
| Raman band assignment first-order | SiOC–Ar | SiOC–CO2 | ||
|---|---|---|---|---|
| Position [cm−1] | Peak area contribution [%] | Position [cm−1] | Peak area contribution [%] | |
| D | 1385 | 44 | 1416 | 28 |
| D3 | 1509 | 9 | 1523 | 12 |
| G | 1577 | 20 | 1584 | 17 |
| D2 | 1612 | 27 | 1614 | 43 |
Fig. 8 presents the second order Raman spectra of SiOC–Ar and SiOC–CO2. At first glance, the second order spectrum of SiOC–Ar reveals a significant broadening around 2800 cm−1 in comparison to the spectra of the CO2 pyrolysed aerogel. For further analysis both spectra are fitted with Lorentzian shaped profiles according to Tyborski et al.47 The deconvolution of the Raman spectra yields five contributions located at ∼2800, ∼3000, 3140, 3190 and 3225 cm−1. With the exception of the signal at ∼3000 cm−1 all bands are assigned to overtones related to signals in the fundamental region of the Raman spectrum. In detail, the features at 3140 and 3190 cm−1 are assigned to the overtones of the G-band (E2g and E1u). The latter overtone belongs to an infrared-active fundamental. It becomes detectable in the Raman spectrum, because the decomposition of the direct product E1u × E1u contains always a fully symmetric representation.47 The two signals at 2800 and 3230 cm−1 are assigned to the overtones of the D and the D2-band, respectively. The broad and intense feature at around 3000 cm−1 is also related to the D band, as discussed by Tyborski et al. Accordingly, this feature is uniquely formed under UV excitation (4.83 eV) and originates from a double resonance effect involving an electronic transition as well as a resonance between two TO phonons at the M point of the phonon dispersion of graphite.47
 | ||
| Fig. 8 Second order UV Raman spectra of (a) SiOC–Ar and (b) SiOC–CO2 together with the results of peak-fitting analysis. A background subtraction was performed for all spectra. | ||
The contributions of the first/second order Raman signals are listed in Tables 3 and 4, respectively. These contributions represent a quantitative estimation of the amount of carbon with the corresponding microstructure. In the first order spectra, a significant difference is found in the areal contribution of D and D2 bands in dependence on the pyrolysis atmosphere. The contribution of the D band in the SiOC–aerogel is 44%, whereas for SiOC–CO2 the main contribution arises from the D2-band with 43%. The contributions of G and D3 bands are similar for both samples. In the second order spectra, the contribution of the defect overtones (2D bands) is higher for the SiOC–Ar in comparison to the SiOC–CO2 sample (74 vs. 68%, respectively) confirming the results of the analysis in the fundamental region.48 The contributions of the 2D2-band amount to 5 and 7% for SiOC–Ar and SiOC–CO2, respectively. This difference is much less pronounced than in the fundamental region. Nevertheless, one has to keep in mind that in general the intensity of the second order bands is low. Thus, the fitting and quantitative interpretation of those signals are less conclusive.
| Raman band assignment second-order | SiOC–Ar | SiOC–CO2 | ||||
|---|---|---|---|---|---|---|
| Position [cm−1] | Peak area contribution [%] | Position [cm−1] | Peak area contribution [%] | |||
| 2D | 2770 | 16 | 74 | 2812 | 18 | 68 |
| 2D (DR process) | 3000 | 58 | 3027 | 50 | ||
| E2g (2G band) | 3140 | 16 | 21 | 3140 | 20 | 26 |
| E1u (2G band) | 3189 | 5 | 3190 | 6 | ||
| 2D2 | 3224 | 5 | 3226 | 7 | ||
Taking into account the above analysis/discussion of the peak area contributions and the origin of the band the following conclusion on the microstructure of the carbon phase within the studied aerogels can be drawn:
(i) D band: SiOC–Ar is mostly (main contribution of the D band, both in the first and in the second order spectra) composed of disordered/high defect carbon (with traces of sp3) with defects mainly concentrated at the edges of the sp2 C planes. This implies that the carbon phase within the mixed-bond SiOC is less ordered and has more defects. The strong fluorescence, found in the SiOC–Ar Vis-Raman spectra, associated with the presence of defects/radicals and CO bonds as identified by means of FT-IR, supports the above statement. This conclusion is supported by both the fundamental and the overtone region.
(ii) D2-band: the carbon phase of the SiOC–CO2 aerogel consists mostly (43%) of very small clusters of sp2 hybridized carbon (possibly microcrystalline graphite). SiOC–Ar contains fewer small sp2 clusters (27%). This implies that, for the CO2-pyrolyzed aerogel, the carbon phase is better organized. This hypothesis is consistent with the analysis of the overtone region.
The formation of a more disordered Cfree for the Ar-pyrolyzed aerogel and conversely a more ordered free carbon phase for the CO2-treated material supports the findings by the NMR study, which indicate the formation of a SiO2 network without any Si–C bonds for the SiOC–CO2 sample. Therefore, since in the SiOC–CO2 sample the free carbon is not connected to the matrix via primary Si–C chemical bonds, it can more easily rearrange toward a more ordered carbon phase. On the other hand, in the Ar-treated material, Si–C bonds are present in the amorphous SiOC matrix. At this stage we do not have any experimental evidence that proves the existence of Si–C bonds at the SiOC/Cfree interface but, at the same time, we cannot exclude it. Accordingly, if Si–C bonds exist at the interface between the free carbon and the silicon oxycarbide matrix, then the rearrangement of the sp2 C planes into a more ordered free carbon cluster will be more difficult.
3.5 TEM investigation
Representative TEM micrographs of the Ar- versus the CO2-treated SiCO sample are depicted in Fig. 9. At first glimpse, both samples reveal a nearly identical microstructure: nanosized SiCO particles attached to each other with also a nanosized porosity. However, the sample pyrolyzed in Ar shows a slightly larger individual porosity, as compared to the sample treated in CO2 (Fig. 9a and b). | ||
| Fig. 9 TEM micrographs revealing an overview of (a) SiOC–Ar and (b) SiOC–CO2. The insets show characteristic powder particles. (c) and (d) are the corresponding images at a higher magnification, with a phase contrast typical of predominantly amorphous materials. (e) and (f) show Fourier-filtered HRTEM images from the boxed regions in (c) and (d), indicating the free-carbon phase at the outer surface of the SiOC matrix. Note that in the case of Ar-treatment, there is a continuous turbostratic carbon film visible, while in the CO2-treated material a partly discontinuous carbon film was observed. The inset in (e) shows the intensity profile (boxed area) of the outer free-carbon film, which is rather thick in this region (SiCO–Ar). | ||
At a higher magnification, the observed phase contrast for both samples is characteristic of predominantly amorphous materials (Fig. 8c and d). From the corresponding Fourier-filtered HRTEM images (Fig. 8e and f) it becomes obvious that there is no visible free-carbon phase present within the SiOC host matrix, independent of the glass nature, i.e., SiOC containing mixed SiCxO4−x sites versus a pure SiO2 glass matrix. This is consistent with earlier studies on SiOC, where the formation of the so-called basic structural units (BSUs) was reported to be visible via TEM imaging upon pyrolysis above 1000 °C.49,50 However, in both samples there is a thin film of turbostratic carbon present, being continuous in the case of Ar-treatment, while it is partly discontinuous in the SiOC–CO2 sample. It is assumed that the observed discontinuity originates from a surface reaction during pyrolysis of CO2 with the carbon layer: C + CO2 → 2CO. Note that the free-carbon film is rather thick, compared to the CO2-treated sample, as shown in the inset in Fig. 8e, revealing a spacing of the turbostratic carbon layers of approximately 0.23–0.24 nm, typical of disordered carbon.
The obtained TEM results clearly show that pyrolysis in different atmospheres does not significantly change the overall microstructure of SiOC, which is homogenous and predominantly amorphous in both cases. However, HRTEM confirms that the individual pore size is lower for the SiOC–CO2 material as compared to the SiOC–Ar sample. The local distribution and size of the Cfree phase are similar in both materials; i.e., a thin film of turbostratic carbon is present on the surface of the matrix particles. There is no evidence that pyrolysis in a CO2 atmosphere results in a local agglomeration of the free-carbon phase or phase separation of carbon within the matrix. The finding that the surface free carbon is partly discontinuous in the CO2-treated sample is consistent with the lower amount of free carbon measured with different techniques as well as the slightly higher surface film thickness observed in the SiCO–Ar sample (Fig. 9e).
4. Discussion
The 29Si MAS NMR results unambiguously demonstrated that the Ar-pyrolyzed material is a SiOC/Cfree nano-composite with mixed SiCxO4−x 0 ≤ x ≤ 4 units present in the silicon oxycarbide network. On the other hand, the CO2-pyrolyzed aerogel is a SiO2/Cfree nanocomposite with only SiO4 units forming the amorphous network. The amount of free carbon, estimated by TGA, is comparable in the two materials with a slightly higher amount in the Ar-treated SiOC (∼47 wt%) compared to the CO2-treated aerogel (∼37 wt%). The CO2 flow leads to a lower total pore volume with a smaller pore size. In terms of specific surface area, however, the smaller pores result into a higher value of the SSA (259 m2 g−1 for SiOC–CO2vs. 163 m2 g−1 for SiOC–Ar). The HRTEM study revealed for both samples a rather featureless microstructure, typical of predominantly amorphous materials. The free carbon phase, which is supposed to be finely dispersed in the amorphous matrix, is not detectable in both samples annealed at 900 °C. Therefore, the possibility that the pyrolysis in a CO2 flow will lead to a macroscopic phase separation of the free carbon phase within the SiO2 glass matrix can be excluded. Additionally, the TEM investigation revealed the presence of a continuous C film at the surface of SiOC–Ar particles, while the carbon film present on SiOC–CO2 is discontinuous. The UV-Raman analysis revealed that the sp2 C present in the mixed-bond containing samples is more disordered/has more defects than the one dispersed in the SiO2 glass. We could infer that the mixed SiCxO4−x 0 ≤ x ≤ 4 units, which are probably present not only in the amorphous SiOC network but also at the interface between the SiOC and the Cfree layers, hinder the organization of the free carbon phase.Until now, three electrochemically active sites for Li-ion storage have been experimentally identified within carbon-rich SiOC by 7Li-MAS-NMR measurements.23,51–53 Accordingly, the major Li-ion host sites are interstitial spaces and edges of graphene and carbon layers within the free carbon phase. A minor storage contribution is assigned to less ionic Li species that are stored in micropores and to diamagnetic Li species that are directly or indirectly stored in the mixed SiOC network. Thus, let us first consider the sp2 carbon as the only phase, which can reversibly store Li ions. The higher capacity observed for the SiOC–Ar samples might be attributed to the higher amount of Cfree. However, the SiOC–Ar aerogel contains only ∼10 wt% more free carbon than the SiOC–CO2 sample (∼47 wt% for the SiOC–Ar vs. 37 wt% for the SiOC–CO2) but displays an almost doubled Li-storage reversible capacity. In our former work9 we clearly showed that for SiOC glasses containing more than 20 wt% of the free carbon phase, the increasing amount of carbon hardly affects the Li-storage capacity. Therefore, the higher Cfree content (wt%) itself cannot rationalize the higher Li-storage capacity of the SiOC–Ar. As a consequence, we must assume that either (i) the capacity of the free carbon phase of the two composites is different (and in particular the capacity of the sp2 C of the SiOC–Ar is higher than the one of SiOC–CO2) or (ii) the glass matrix itself provides additional sites to reversibly store lithium ions. The Raman spectroscopy study suggests that, indeed, the free carbon phase in the SiOC–Ar samples has a higher degree of disorder/defects compared to the “micrographitic” like Cfree of the CO2-treated samples. The more disordered carbon allows a higher capacity to store lithium, not only between the sp2 C layers but also at the edge of the layers.37,54 In this case, the role of the mixed SiCxO4−x units present in the glassy matrix would lead to an indirect increase in the capacity by inducing the formation of a more disordered C phase.
5. Conclusions
In this work we discussed the role of mixed SiCxO4−x 0 ≤ x ≤ 4 sites with respect to their potentially high reversible Li-ion storage capacity. Two model systems were studied, a SiOC matrix with mixed units (SiOC–Ar) and a material with no such mixed structural units (SiOC–CO2), where in the latter sample the matrix converted to pure SiO2 upon pyrolysis. The mixed bonds present in the SiOC–Ar material induce (i) the formation of a more disordered/high defect carbon phase, which thereby has a higher capacity for reversible storage of Li ions and/or (ii) directly provides reversible storage sites at the interface between the amorphous network and the free carbon. Hence, it can be concluded that there is indeed a pronounced effect of mixed units within the SiOC glass matrix on the Li-storage capacity of the material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Emanuela Callone is gratefully acknowledged for performing the 29Si SS NMR analyses using the equipment of the “Klaus Müller” NMR Lab. of the Industrial Engineering Department at the University of Trento. Mrs Ababo Gudisa is gratefully acknowledged for the synthesis of the precursor and ceramic aerogels. Dr Peter Kroll is gratefully acknowledged for the very helpful discussion.References
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. M. Tarascon, Philos. Trans. R. Soc., A, 2010, 368, 3227–3241 CrossRef PubMed.
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed.
- S. Goriparti, E. Miele, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 CAS.
- W. Xing, A. M. Wilson, K. Eguchi, G. Zank and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2410–2416 CrossRef CAS.
- D. Ahn and R. Raj, J. Power Sources, 2011, 196, 2179–2186 CrossRef CAS.
- M. Graczyk-Zajac, L. M. Reinold, J. Kaspar, P. V. W. Sasikumar, G.-D. Soraru and R. Riedel, Nanomaterials, 2015, 5, 233–245 CrossRef CAS PubMed.
- J. Kaspar, M. Graczyk-Zajac, S. Choudhury and R. Riedel, Electrochim. Acta, 2016, 216, 196–202 CrossRef CAS.
- M. Graczyk-Zajac, L. Toma, C. Fasel and R. Riedel, Solid State Ionics, 2012, 225, 522–526 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, Solid State Ionics, 2012, 225, 527–531 CrossRef CAS.
- L. David, R. Bhandavat, U. Barrera and G. Singh, Nat. Commun., 2016, 7, 9792 Search PubMed.
- P. Colombo, G. Mera, R. Riedel and G. D. Soraru, J. Am. Ceram. Soc., 2010, 93, 1805–1837 CAS.
- G. M. Renlund, S. Prochazka and R. H. Doremus, J. Mater. Res., 1991, 6, 2716–2722 CrossRef CAS.
- H. Zhang and C. G. Pantano, J. Am. Ceram. Soc., 1990, 73, 958–963 CrossRef CAS.
- G. M. Renlund, S. Prochazka and R. H. Doremus, J. Mater. Res., 1991, 6, 2723–2734 CrossRef CAS.
- G. D. Soraru, G. D'Andrea, R. Campostrini, F. Babonneau and G. Mariotto, J. Am. Ceram. Soc., 1995, 78, 379–387 CrossRef CAS.
- M. Wilamowska, V. S. Pradeep, M. Graczyk-Zajac, R. Riedel and G. D. Soraru, Solid State Ionics, 2014, 260, 94–100 CrossRef CAS.
- L. David, K. M. Shareef, M. A. Abass and G. Singh, RSC Adv., 2016, 6, 53894–53902 RSC.
- H. Konno, T. Morishita, S. Sato, H. Habazaki and M. Inagaki, Carbon, 2005, 43, 1111–1114 CrossRef CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, Chem. Lett., 2009, 38, 86–87 CrossRef CAS.
- D. Ahn and R. Raj, J. Power Sources, 2010, 195, 3900–3906 CrossRef CAS.
- H. Fukui, O. Hisashi, T. Hino and K. Kanamura, ACS Appl. Mater. Interfaces, 2010, 4, 998–1008 Search PubMed.
- V. S. Pradeep, M. Graczyk-Zajac, R. Riedel and G. D. Soraru, Electrochim. Acta, 2014, 119, 78–85 CrossRef CAS.
- H. Fukui, Y. Harimoto, M. Akasaka and K. Eguchi, ACS Appl. Mater. Interfaces, 2014, 6, 12827–12836 CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, J. Electrochem. Soc., 2013, 160, A1276–A1281 CrossRef CAS.
- P. E. Sanchez-Jimenez and R. Raj, J. Am. Ceram. Soc., 2010, 93, 1127–1135 CrossRef CAS.
- P. Kroll, MRS Online Proc. Libr., 2011, 1313, 1–6 Search PubMed.
- N. Liao, B. Zheng, M. Zhang and W. Xue, J. Mater. Chem. A, 2016, 4, 12328–12333 CAS.
- H. Sun and K. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 35001–35009 CAS.
- M. Narisawa, F. Funabiki, A. Iwase, F. Wakai and H. Hosono, J. Am. Ceram. Soc., 2015, 98, 3373–3380 CrossRef CAS.
- M. Narisawa, A. Iwase, S. Watase, K. Matsukawa and T. Kawai, in Innovative Processing and Manufacturing of Advanced Ceramics and Composites II, John Wiley & Sons, Inc., 2014, pp. 79–84, DOI:10.1002/9781118771464.ch7.
- P. S. Waleska and C. Hess, J. Phys. Chem. C, 2016, 120, 18510–18519 CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Poeschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- C. Castiglioni, M. Tommasini and G. Zerbi, Philos. Trans. R. Soc., A, 2004, 362, 2425–2459 CrossRef CAS PubMed.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, J. Power Sources, 2013, 244, 450–455 CrossRef CAS.
- S. J. Widgeon, S. Sen, G. Mera, E. Ionescu, R. Riedel and A. Navrotsky, Chem. Mater., 2010, 22, 6221–6228 CrossRef CAS.
- A. Grill and D. A. Neumayer, J. Appl. Phys., 2003, 94, 6697–6707 CrossRef CAS.
- M. Narisawa, K. Terauds, R. Raj, Y. Kawamoto, T. Matsui and A. Iwase, Scr. Mater., 2013, 69, 602–605 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 2004 Search PubMed.
- E. Zera, W. Nickel, S. Kaskel and G. D. Soraru, J. Eur. Ceram. Soc., 2016, 36, 423–428 CrossRef CAS.
- H. Bréquel, J. Parmentier, S. Walter, R. Badheka, G. Trimmel, S. Masse, J. Latournerie, P. Dempsey, C. Turquat, A. Desmartin-Chomel, L. Le Neindre-Prum, U. A. Jayasooriya, D. Hourlier, H. J. Kleebe, G. D. Sorarù, S. Enzo and F. Babonneau, Chem. Mater., 2004, 16, 2585–2598 CrossRef.
- C. Castiglioni and M. Tommasini, Opt. Pura Apl., 2007, 40, 169–174 Search PubMed.
- A. C. Ferrari and J. Robertson, Mater. Res. Soc. Symp. Proc., 2000, 593, 299–304 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- C. Tyborski, F. Herziger, R. Gillen and J. Maultzsch, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 041401 CrossRef.
- F. Roth, P. Waleska, C. Hess, E. Ionescu and N. Nicoloso, J. Ceram. Soc. Jpn., 2016, 124, 1042–1045 CrossRef CAS.
- H.-J. Kleebe, C. Turquat and G. D. Soraru, J. Am. Ceram. Soc., 2001, 84, 1073–1080 CrossRef CAS.
- G. Gregori, C. Turquat, H.-J. Kleebe and G. D. Soraru, Key Eng. Mater., 2002, 206, 2061–2064 CrossRef.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, J. Power Sources, 2011, 196, 371–378 CrossRef CAS.
- H. Fukui, K. Eguchi, H. Ohsuka, T. Hino and K. Kanamura, J. Power Sources, 2013, 243, 152–158 CrossRef CAS.
- M. Haaks, J. Kaspar, A. Franz, M. Graczyk-Zajac, R. Riedel and M. Vogel, Solid State Ionics, 2016, 287, 28–35 CrossRef CAS.
- L. M. Reinold, Y. Yamada, M. Graczyk-Zajac, H. Munakata, K. Kanamura and R. Riedel, J. Power Sources, 2015, 282, 409–415 CrossRef CAS.
Footnote |
| † Present address: Laboratory for High Performance Ceramics, EMPA, Dübendorf, Switzerland. |
| This journal is © The Royal Society of Chemistry 2018 |