
Prussian blue analogues derived porous nitrogen-doped carbon microspheres as high-performance metal-free peroxymonosulfate activators for non-radical-dominated degradation of organic pollutants†
 *a
Ping
Xu
a
and
*a
Ping
Xu
a
and
Abstract
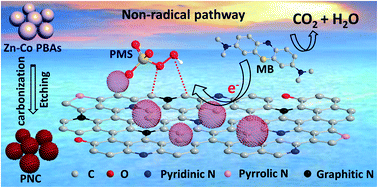
Nitrogen-doped carbon materials are becoming a new type of metal-free heterogeneous catalysts in advanced oxidation processes (AOPs) for wastewater treatment and environmental remediation. In this study, porous nitrogen-doped carbon (PNC) microspheres derived from Zn–Co Prussian blue analogues (Zn–Co PBAs) are employed as heterogeneous catalysts in peroxymonosulfate (PMS) activation. The unique configuration of the metal centers/clusters bound by cyanide groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) of Zn–Co PBAs offers the PNC microspheres abundant porosity, a high graphitization degree, and rich nitrogen substitution, which lead to improvements in the catalytic performance. PNC-800 (pyrolyzed at 800 °C) exhibits better performance than other common carbon materials and homologous nitrogen-doped carbocatalysts derived from ZIF-8/ZIF-67. Based on radical quenching and trapping experiments, a non-radical pathway is proposed to dominate methylene blue (MB) degradation; and the high graphitization degree and rich surface graphitic N sites of PNC-800 are two key factors that induce the non-radical pathway. Several influential factors, including catalyst dosage, PMS concentration, pH value and reaction temperature, are investigated in detail. Notably, MB degradation over PNC-800 is almost completely insusceptible to common ions and natural organic matter, and maintains its catalytic efficiency under the background conditions of several real water samples. More interestingly, this non-radical pathway and the good catalytic performance of the PNC-800/PMS system are universal in the degradation of other typical organic pollutants. We believe that these PNC microspheres may be a promising green heterogeneous catalyst for the degradation of organic pollutants, and this study can be used for the design of high-performance carbocatalysts in non-radical systems in the future.
N) of Zn–Co PBAs offers the PNC microspheres abundant porosity, a high graphitization degree, and rich nitrogen substitution, which lead to improvements in the catalytic performance. PNC-800 (pyrolyzed at 800 °C) exhibits better performance than other common carbon materials and homologous nitrogen-doped carbocatalysts derived from ZIF-8/ZIF-67. Based on radical quenching and trapping experiments, a non-radical pathway is proposed to dominate methylene blue (MB) degradation; and the high graphitization degree and rich surface graphitic N sites of PNC-800 are two key factors that induce the non-radical pathway. Several influential factors, including catalyst dosage, PMS concentration, pH value and reaction temperature, are investigated in detail. Notably, MB degradation over PNC-800 is almost completely insusceptible to common ions and natural organic matter, and maintains its catalytic efficiency under the background conditions of several real water samples. More interestingly, this non-radical pathway and the good catalytic performance of the PNC-800/PMS system are universal in the degradation of other typical organic pollutants. We believe that these PNC microspheres may be a promising green heterogeneous catalyst for the degradation of organic pollutants, and this study can be used for the design of high-performance carbocatalysts in non-radical systems in the future.
 Please wait while we load your content...
Please wait while we load your content...

