
Hierarchically porous sheath–core graphene-based fiber-shaped supercapacitors with high energy density†
 *ab
*ab
Abstract
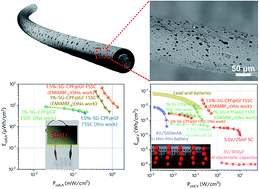
Owing to their high power density, fast charge/discharge rate, ultralong cycling life and safe operating conditions, all-carbon fiber-shaped supercapacitors (FSSCs) hold great promise for powering wearable electronics. However, their low energy density hinders them from practical application, due to the absence of effective approaches for highly electrically conductive fiber electrodes with high specific capacitance. Herein, we develop a scalable and cost-effective strategy towards hierarchically porous core–sheath graphene-based fiber electrodes with a rational PSD (88–97% micropores, 0–8.3% mesopores and 1.9–4.2% macropores) and high SSA (up to 416.4 m2 g−1). The hierarchical architecture is achieved by simply decorating graphene fiber with carbonized phenol formaldehyde (CPF) resin containing small-size (∼156 nm in diameter) graphene (SG), wherein the incorporation of CPF and SG synergistically provides ultrahigh micro-porosity with narrowed micro-/meso-PSDs and enhanced electrical conductivity, facilitating ion storage and transport. The assembled FSSCs exhibit an ultrahigh specific areal capacitance of 391.2 mF cm−2 in polyvinyl alcohol/H2SO4 electrolyte at 0.1 mA cm−2 in a two-electrode cell, which is 17 times that of graphene fibers. The entire-device energy density Ecell,A is 8.7 μW h cm−2 in aqueous electrolyte and 66.4 μW h cm−2 at an areal power density of 0.54 mW cm−2 in organic electrolyte. Moreover, the FSSCs also show ultralong cycling life (98.9% capacitance retention after 7000 cycles) and good flexibility. All these results have made it one of the best ever reported all-carbon FSSCs to date. This work may shed light on mass-manufacturing low-cost but high-performance wearable fiber-shaped energy storage devices.
 Please wait while we load your content...
Please wait while we load your content...

