
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal structure and surface characteristics of Sr-doped GdBaCo2O6−δ double perovskites: oxygen evolution reaction and conductivity†
Stevin S.
Pramana
 *ab,
Andrea
Cavallaro
a,
Cheng
Li
a,
Albertus D.
Handoko
cd,
Kuang Wen
Chan
c,
Robert J.
Walker
a,
Anna
Regoutz
a,
Jason S.
Herrin
e,
Boon Siang
Yeo
c,
David J.
Payne
a,
John A.
Kilner
af,
Mary P.
Ryan
a and
Stephen J.
Skinner
*a
*ab,
Andrea
Cavallaro
a,
Cheng
Li
a,
Albertus D.
Handoko
cd,
Kuang Wen
Chan
c,
Robert J.
Walker
a,
Anna
Regoutz
a,
Jason S.
Herrin
e,
Boon Siang
Yeo
c,
David J.
Payne
a,
John A.
Kilner
af,
Mary P.
Ryan
a and
Stephen J.
Skinner
*a
aDepartment of Materials, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: s.skinner@imperial.ac.uk; stevin.pramana@newcastle.ac.uk
bSchool of Engineering, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
cDepartment of Chemistry, Faculty of Science, National University of Singapore, 3 Science Drive 3, 117543, Singapore
dAgency for Science, Technology and Research (A*STAR), Institute of Materials Research and Engineering, Innovis Building, 2 Fusionopolis Way, 138634, Singapore
eFacility for Analysis, Characterization, Testing and Simulation/Earth Observatory of Singapore, Nanyang Technological University, Nanyang Avenue, 639798, Singapore
fInternational Institute for Carbon Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
First published on 4th December 2017
Abstract
A cheap and direct solution towards engineering better catalysts through identification of novel materials is required for a sustainable energy system. Perovskite oxides have emerged as potential candidates to replace the less economically attractive Pt and IrO2 water splitting catalysts. In this work, excellent electrical conductivity (980 S cm−1) was found for the double perovskite of composition GdBa0.6Sr0.4Co2O6−δ which is consistent with a better oxygen evolution reaction activity with the onset polarisation of 1.51 V with respect to a reversible hydrogen electrode (RHE). GdBa1−xSrxCo2O6−δ with increasing Sr content was found to crystallise in the higher symmetry tetragonal P4/mmm space group in comparison with the undoped GdBaCo2O6−δ which is orthorhombic (Pmmm), and yields higher oxygen uptake, accompanied by higher Co oxidation states. This outstanding electrochemical performance is explained by the wider carrier bandwidth, which is a function of Co–O–Co buckling angles and Co–O bond lengths. Furthermore the higher oxygen evolution activity was observed despite the formation of non-lattice oxides (mainly hydroxide species) and enrichment of alkaline earth ions on the surface.
1. Introduction
Single layered perovskites (ABO3−x; A = rare earth and alkaline earth, B = transition metal) have been well documented as excellent intermediate temperature (∼600 to 800 °C) solid oxide fuel cell (SOFC) cathodes due to their high oxygen surface exchange, and excellent ionic and electronic conductivity.1–7 Interestingly, this class of material can also perform as an oxygen evolution catalyst.8–11 More recently, transformation of single perovskites with randomly occupied A site into ordered perovskites (AA′BB′O6−δ; A = rare earth, A′ = alkaline earth, B and B′ = transition metals) has shown improved oxygen transport properties and may potentially replace the conventional single layered perovskites for a range of technological applications.12–14 Whilst the oxygen evolution yield of double perovskites is scarcely documented, Grimaud et al.,15 Wu et al.16 and Zhao et al.17 reported the prospect of (ABa)Co2O6−δ (A = Pr, Sm, Gd and Ho) and PrBa1−xSrxCo2O6−δ and PrBa0.5Sr0.5Co1.5Fe0.5O6−δ, respectively to become active catalysts for water oxidation in alkaline solution. This was postulated to be related to the filling of 3d eg orbital occupancy to unity and to the location of the oxygen p-band with respect to the Fermi level, similar to the band structure found in Ba0.5Sr0.5Co0.8Fe0.2O3−δ single perovskite.9 In addition, Matsumoto et al.8 reported that the σ* band formation and higher transition metal oxidation state contributed to higher oxygen evolution catalytic activity in perovskite oxides. In another double perovskite system, PrBa0.85Ca0.15MnFeO6−δ was stable and catalytically active for both oxygen reduction and evolution reactions.18Double perovskites with the general formula AA′BB′O6−δ afford flexibility in the cation coordination environment and thus offer the opportunity to tune the band structure by varying the nominal composition. The stoichiometric δ = 0 composition leads to the B cation being octahedrally connected to the oxygen ions, whilst BO5 square pyramids are formed for δ = 1 due to the disappearance of the octahedral apical oxygen ion. This oxygen vacancy ordering is directly linked to the A and A′ cation occupancies in the unique crystallographic sites along the z crystallographic axis, leading to doubling in the c lattice parameter, when compared to the parent single perovskite. This cation ordering varies with temperature and A/A′ ionic radii.19,20 Three commonly reported crystal structure configurations for AA′B2O6−δ (A = Gd, A′ = Ba and B = Co) are presented in Fig. 1: tetragonal P4/mmm, orthorhombic Pmmm (formula unit per unit cell, Z = 1) and Pmmm (Z = 2) space groups. The difference between the last two structure types is that where Z = 2 there is further ordering in the y crystallographic axis, in addition to z, whilst for Z = 1, only ordering in z is observed. The oxygen vacancies in these ordered double perovskites were often reported to lie in the rare-earth layers hence lowering the coordination number from 12 to 8 for the smaller rare-earth ions and keeping the same coordination for the larger alkaline earth metals.21,22 This influences oxide ion migration where fast diffusion pathways were found to be in the ab planes in the ordered structure, as predicted by molecular dynamics simulation, rather than isotropic conduction in three dimensions in the disordered single perovskite phase.12 Thus, oriented samples in the form of either single crystals or epitaxially grown films will enhance the observation of this anisotropic behaviour.
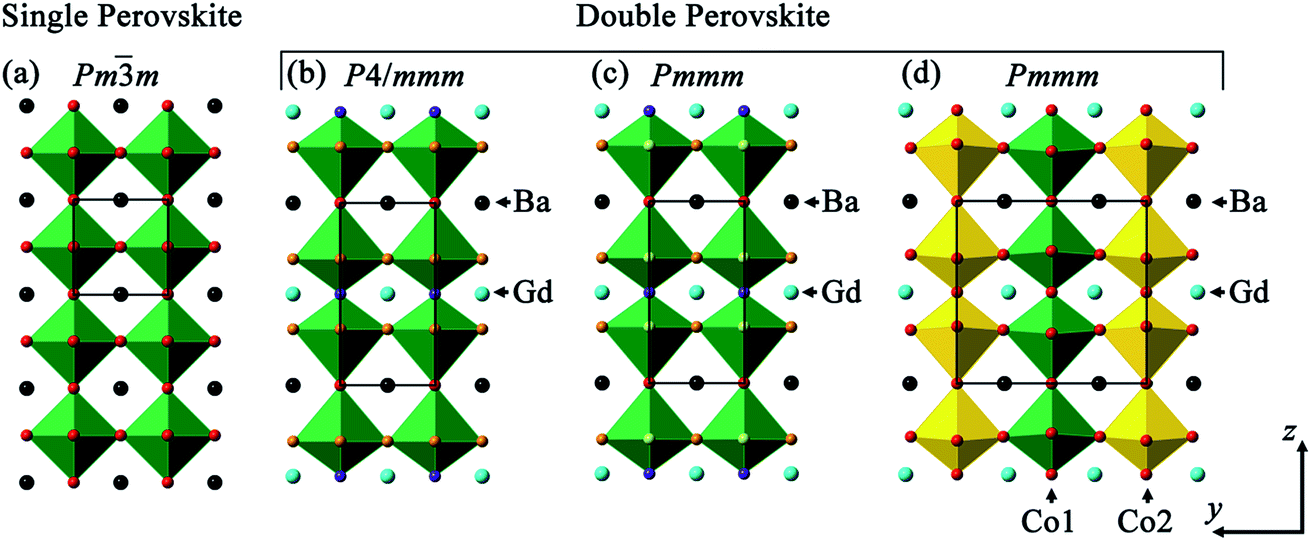 | ||
| Fig. 1 Crystal structure representation of (a) single ABO3 perovskite where A is represented in black and BO6 polyhedra in green; and (b–d) double perovskite GdBaCo2O6 where Gd3+ (blue) and Ba2+ cations (black) are ordered in z direction with (b) tetragonal P4/mmm, (c) orthorhombic Pmmm and (d) Pmmm with additional ordered cations along b depicted by Co1O6 (green) and Co2O6 (yellow) octahedra ordering. | ||
Depending on the oxygen stoichiometry, GdBaCo2O6−δ single crystals were reported to crystallise in the tetragonal (0 < δ < 0.40), orthorhombic with doubling along b (Z = 2; 0.40 < δ < 0.55) and back to tetragonal symmetry (0.55 < δ ≤ 1) at room temperature (RT).23 However, using RT powder X-ray diffraction (XRD), Roy et al.24 described the GdBaCo2O5.45 using Pmmm space group (Z = 2) whilst Pmmm (Z = 1) for GdBaCo2O5. Introduction of a Sr dopant into the Ba lattice site in GdBa1−xSrxCo2O6−δ leads to the higher symmetry tetragonal P4/mmm structure (0.2 ≤ x ≤ 0.6) with another transformation to a structure with Pnma symmetry observed as Ba was fully substituted by Sr (GdSrCo2O6).25 Higher Sr dopant in PrBa1−xSrxCo2O6−δ led to better oxygen evolution activity with the highest reported for the PrBa0.25Sr0.75Co2O6−δ composition.16 The complex ordering in this type of perovskite as a function of oxygen content, temperature and dopant level has resulted in various space groups being reported, although the correlation between crystal structure and electrochemical performance has rarely been probed.
The bulk chemistry and structure explained previously is often different from the surface, defined as the interface between gas–solid phase. Lee et al.26 reported that the surface segregation on perovskite oxides was mainly driven by the elastic and electrostatic interactions. As oxygen reduction and evolution reactions between the gas phase and ions primarily occurs at the surface, the surface characteristics are unequivocally required to be understood. Using low energy ion scattering (LEIS),27,28 which is sensitive to the atomic composition at the outermost surface, Sr and Ba were found to segregate to the surface in polished pellets (subjected to annealing at 1000 °C for 12 h in 200 mbar O2) of polycrystalline La0.6Sr0.4Co0.2Fe0.8O3−δ and GdBaCo2O6−δ, respectively with transition metal enrichment in the sub-surface region (2–10 nm).29,30 In this work, we investigate the chemical and electronic speciation at the surface of strontium doped GdBaCo2O6−δ double perovskites and relate these with the bulk composition, crystal structure, and electrochemical properties.
2. Experimental methods
2.1. Synthesis
Powders with nominal compositions of GdBa1−xSrxCo2O6−δ (x = 0, 0.1, 0.2, 0.3, 0.4 and 0.5) were synthesised using the solid state reaction of Gd2O3 (Alfa Aesar, 99.99%), BaCO3 (Alfa Aesar, 99.8%), SrCO3 (Alfa Aesar, 99.99%) and Co3O4 (Alfa Aesar, 99.7%). Stoichiometric amounts of starting powders were manually ground in an agate mortar, sintered at 1100 °C for 60 h in static air in alumina crucibles with intermediate grinding without any pre-calcination step.2.2. Characterisation methods
The oxygen content calculated from the oxidation state of cobalt was determined using iodometric titration. ∼70 mg of sample was dissolved in 20 ml hydrochloric acid (12 M) by stirring in an argon gas flushed flask, covered by 0.1 M KI. 0.01 M Na2S2O3 solution was used to titrate the iodine generated with starch used as an indicator.Laboratory powder X-ray diffraction (XRD) patterns were collected at room temperature from 10–120° 2θ using a PANalytical X'Pert Pro MPD utilising CuKα source radiation (λaverage = 1.54 Å) with a step size of 0.016° and accumulating for 0.8 s per step. The crystal structures of GdBa1−xSrxCo2O6−δ were refined by Rietveld analysis31,32 using a fundamental parameter peak shape profile33 implemented in TOPAS Version 4.1.34 The starting models used the P4/mmm,19Pmmm (Z = 1)19 and Pmmm (Z = 2)35 atomic positions of GdBaCo2O6−δ with Sr occupying the same site as Ba. For each data set, a five-coefficient Chebyshev polynomial and 1/x background, a zero error, unit–cell parameters, scale factors and crystal size were refined sequentially. X-ray form factors for Gd3+, Ba2+, Sr2+, Co3+ and O2− were used.
Samples for low energy ion scattering (LEIS), collected using Qtac100 spectrometer, IonTOF GmbH, Germany, were uniaxially pressed, and followed by sintering at 1100 °C for 12 h. The achieved density was 85–95% of theoretical density, measured by the Archimedes method. The pellets were polished to a final particle size of 1/4 μm and subjected to subsequent heat treatment annealing at 600 °C and 800 °C for 12 h in static air. The sample labelling is listed in Table 1. The primary beam used was 5 keV Ne+ normal to the surface with the collection angle of 145° with an analysis area of 500 μm × 500 μm whilst the sputter beam of 0.5 keV Ar+ ions at 59° incidence angle was utilised with a sputter area of 1000 μm × 1000 μm. The larger sputter area ensures that the analysis area is within the sputtered area. The spectra were fitted using SurfaceLab 6 software (IonTOF GmbH, Germany) with Gaussian peaks of different elemental components with the in-depth signal described by error functions.36,37 The estimated depth was calibrated using interferometric (Zygo NewView 200 interferometer) measurement and the assumption of a constant sputter rate throughout the analysis was made.
| Label | Description | Nominal composition | Measured composition |
|---|---|---|---|
| GBCO | As polished | GdBaCo2O6−δ | Gd1.04(1)Ba1.01(2)Co1.95(1)O6−δ |
| GBCO-600C | Polished and heat-treated at 600 °C | GdBaCo2O6−δ | Gd1.02(4)Ba1.01(2)Co1.96(3)O6−δ |
| GBCO-800C | Polished and heat-treated at 800 °C | GdBaCo2O6−δ | Gd1.03(2)Ba1.00(1)Co1.98(3)O6−δ |
| GB8S2CO | As polished | GdBa0.8Sr0.2Co2O6−δ | Gd1.01(2)Ba0.82(2)Sr0.20(2)Co1.97(3)O6−δ |
| GB8S2CO-600C | Polished and heat-treated at 600 °C | GdBa0.8Sr0.2Co2O6−δ | Gd1.02(2)Ba0.81(2)Sr0.19(2)Co1.97(2)O6−δ |
| GB8S2CO-800C | Polished and heat-treated at 800 °C | GdBa0.8Sr0.2Co2O6−δ | Gd1.02(3)Ba0.80(2)Sr0.20(2)Co1.98(2)O6−δ |
| GB7S3CO | As polished | GdBa0.7Sr0.3Co2O6−δ | Gd1.01(2)Ba0.71(3)Sr0.28(5)Co1.99(5)O6−δ |
| GB7S3CO-600C | Polished and heat-treated at 600 °C | GdBa0.7Sr0.3Co2O6−δ | Gd1.01(2)Ba0.71(2)Sr0.29(2)Co1.99(2)O6−δ |
| GB7S3CO-800C | Polished and heat-treated at 800 °C | GdBa0.7Sr0.3Co2O6−δ | Gd1.02(2)Ba0.71(2)Sr0.29(2)Co1.98(2)O6−δ |
| GB6S4CO | As polished | GdBa0.6Sr0.4Co2O6−δ | Gd1.01(3)Ba0.62(2)Sr0.40(2)Co1.97(3)O6−δ |
| GB6S4CO-600C | Polished and heat-treated at 600 °C | GdBa0.6Sr0.4Co2O6−δ | Gd1.02(2)Ba0.61(2)Sr0.39(3)Co1.98(2)O6−δ |
| GB6S4CO-800C | Polished and heat-treated at 800 °C | GdBa0.6Sr0.4Co2O6−δ | Gd1.02(2)Ba0.61(3)Sr0.39(4)Co1.98(4)O6−δ |
The average bulk chemical quantification of the samples used for LEIS analysis over 30–40 data points was determined using electron probe microanalysis (EPMA) in a JEOL JXA-8530F instrument equipped with five wavelength-dispersive spectrometers (WDS). The surface was carbon coated to minimise the effect of charging. The accelerating voltage and current used were 15 kV and 20 nA, respectively. Standards of gadolinium metal (Gd), BaSO4 (Ba), SrSO4 (Sr) and cobalt metal (Co), obtained from Astimex, were employed. The peak, low background and high background counting time were 20 s, 10 s and 10 s, respectively.
The pellets were fractured in a glove bag under N2 atmosphere to minimise exposure to air and moisture. The exposed fresh pellet surface was characterised using X-ray photoelectron spectroscopy (XPS). The spectra were recorded on a Thermo Scientific K-Alpha+ X-ray photoelectron spectrometer system operating at 2 × 10−9 mbar base pressure. This system incorporates a monochromated, microfocused Al Kα X-ray source (hν = 1486.6 eV) and a 180° double focusing hemispherical analyser with a 2D detector. The X-ray source was operated at 6 mA emission current and 12 kV anode bias and a flood gun was used to minimise sample charging. Data were collected at 20 eV pass energy for core level spectra (Ba 4d, Ba 3d, Gd 4d, Sr 3d, Co 2p, O 1s and C 1s), and at 15 eV pass energy for valence band spectra using an X-ray spot size of 400 μm2. All data were analysed using the Thermo Scientific Avantage software package.
2.3. Electrochemistry measurement
The total direct current (DC) conductivity was measured by a standard four-point probe method using a Solartron Modulab on a uniaxially pressed rectangular block (33 mm × 4 mm × 4 mm) sintered at 1100 °C (∼85 to 95% theoretical density). Silver paint was used as a contact with Ag wires. The measurement was conducted under ambient conditions and a DC current of 200 mA was applied.Double perovskite catalyst inks were prepared by mixing 2 mg of the as-synthesised catalyst, 0.1 ml propan-2-ol (Fisher, Analytical Reagent), 0.4 ml deionised H2O (Barnstead, ≥ 18.2 MΩ cm) and 60 μL of Nafion 117 solution (Sigma Aldrich). The suspensions were sonicated at 20 °C for 30 minutes to ensure homogeneous mixing. 5 μL of the catalyst ink was then drop-cast onto polished glassy carbon electrodes (3 mm diameter, CH Instruments). 17.9 μg of catalyst was typically loaded onto each electrode. The electrochemical measurements were performed in a glass H-cell separated with a porous glass plug in the 3-electrode configuration without rotation. A Hg/HgO (in 0.1 M KOH) electrode (CH Instruments) and a platinum wire were used respectively as reference and counter electrodes. The electrolyte was a 0.1 M KOH solution (Alfa Aesar, 99.98%). The reference was calibrated against RHE (Hydroflex, Gaskatel GmbH) in the same electrolyte. A Gamry Reference 600 potentiostat was used to control and measure potentials/currents. A real-time current interrupt iR drop compensation was applied to the measurements. All current densities were normalised to the geometric surface area of the working electrode.
3. Results and discussion
3.1. Bulk composition and crystal structure
The average bulk composition of double perovskites measured using electron probe microanalysis, within an 800 nm interaction volume as calculated by Monte Carlo simulation of electron trajectory in solids (Casino),38 is close to the nominal composition within the standard deviation (Table 1). However, a closer inspection of backscattered electron images shows that there are brighter regions which were quantified to be a single perovskite of composition Gd1.00(3)Co0.97(3)O3 (Fig. 2 and S1†) and the area density of this secondary phase was more pronounced as the nominal Sr content in the GBCO crystal increased. The oxygen content, determined from iodometric titration, increased from 5.21(1) to 5.55(1) as the nominal Sr dopant level increased from GBCO (x = 0) to GB5S5CO (x = 0.5) (Table 2). | ||
| Fig. 2 Back-scattered electron (BSE) images of (a) GBCO, (b) GBCO-600C, (c) GBCO-800C, (d) GB7S3CO, (e) GB7S3CO-600C, (f) GB7S3CO-800C with an inset of enlarged area within the box. Brighter regions marked by red arrows were quantified as a single perovskite Gd1.00(3)Co0.97(3)O3 phase. | ||
| Compositions | Space group | a (Å) | b (Å) | c (Å) | Volume (3) | Oxygen content (6 − δ) |
|---|---|---|---|---|---|---|
| GBCO | Pmmm | 3.87844(7) | 7.8279(2) | 7.5350(1) | 228.763(8) | 5.21(1) |
| GB9S1CO | Pmmm | 3.87015(5) | 7.8065(1) | 7.53891(8) | 227.769(6) | 5.28(1) |
| GB8S2CO | Pmmm | 3.8685(1) | 3.8833(1) | 7.5421(2) | 113.300(6) | 5.38(1) |
| GB7S3CO | Pmmm | 3.8634(1) | 3.8673(1) | 7.5454(2) | 112.735(6) | 5.51(1) |
| GB6S4CO | P4/mmm | 3.8580(1) | =a | 7.5429(2) | 112.270(7) | 5.54(1) |
| GB5S5CO | P4/mmm | 3.8512(1) | =a | 7.5376(2) | 111.794(7) | 5.55(1) |
In order to understand the crystal structure of these double perovskite phases, Rietveld refinement was performed on powder XRD patterns.31,32 The GBCO sample was found to crystallise in the Pmmm space group (Z = 2) with a = 3.87844(7) Å, b = 7.8279(2) Å, c = 7.5350(1) Å. The doubling in c shows that the ordered double perovskite was successfully synthesised (2θ001 = 11.74°) (Fig. 3). In addition, the b lattice parameter was also doubled, as indicated by the presence of the (010) reflection at 2θ = 11.29°, in addition to other minor reflections such as (130) and (131) at 2θ = 41.69 and 43.46° respectively. This discounted the possibility of either the P4/mmm or Pmmm (Z = 1) space groups existing. Introducing preferred orientation and anisotropic peak broadening corrections led to the reliability factors of Rwp = 0.029 and RBragg = 0.007.
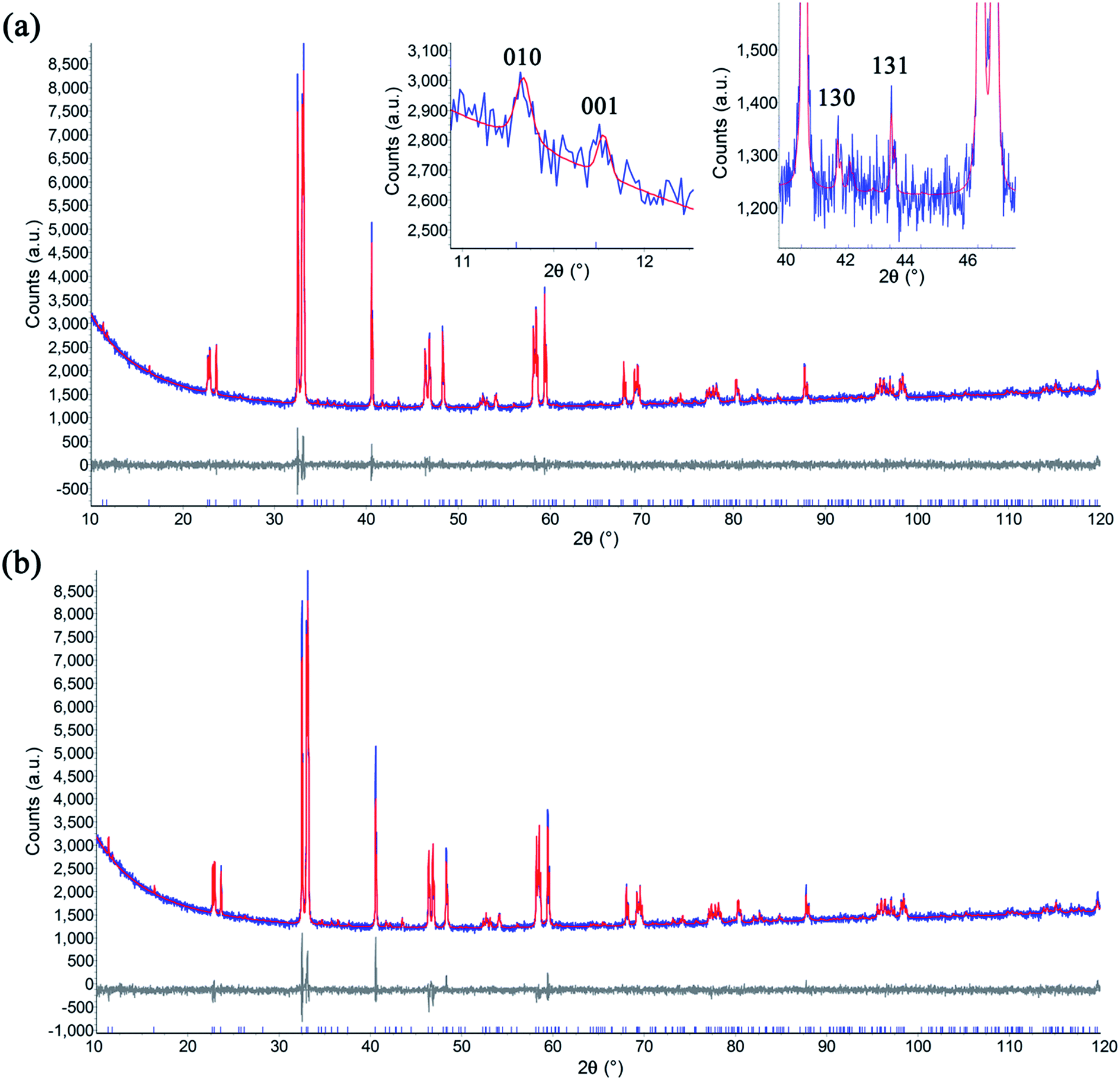 | ||
| Fig. 3 Rietveld plot of the XRD data of GBCO collected at room temperature, (a) with (Rwp = 0.029, RBragg = 0.007) and (b) without (Rwp = 0.033, RBragg = 0.020) preferred orientation and anisotropic peak broadening correction. The existence of 010, 130 and 131 reflections at 2θ = 11.29, 41.69 and 43.46° signify the doubling in b (inset). The observed and calculated intensity are shown by blue and red line, respectively, with the differences plotted beneath. Vertical markers indicate the Bragg reflections. | ||
Introducing 10 at% of Sr to the Ba site (GB9S1CO) did not change the symmetry (Fig. S2†). However, the addition of 20 at% Sr (GB8S2CO) diminished the ordering along the y crystallographic axis, yielding a structure that adopted the Pmmm space group with Z = 1 (a = 3.8685(1) Å, b = 3.8833(1) Å, c = 7.5421(2)) whilst retaining the z ordering. Tetragonal P4/mmm was discounted because the (200) reflection (2θ = 47°) was observed to split into two peaks, assigned as (200) and (020), leading to an orthorhombic Pmmm space group assignment (Fig. S3,†Rwp = 0.028, RBragg = 0.005). This was accompanied by the formation of 0.9(2) wt% single perovskite GdCoO3 phase, consistent with the EPMA results and the back scattering electron (BSE) micrographs. GB7S3CO crystallised with the same space group as GB8S2CO (Fig. S4†). GB6S4CO and GB5S5CO, however, could be indexed with a tetragonal structure (P4/mmm space group) verified by the presence of a single (200) reflection. The impurity of GdCoO3 increased to 3.0(3) wt% for GB5S5CO as Sr substituted onto the Ba lattice site. Overall, the a, b and unit cell volume decreased as a function of Sr content as the ionic radius of Sr2+ in 12 coordination (1.44 Å) is smaller than that of Ba2+ (1.61 Å).39 In contrast, c shows an anomaly which increased slightly up to GB7S3CO and decreased as Sr level increased (Fig. 4).
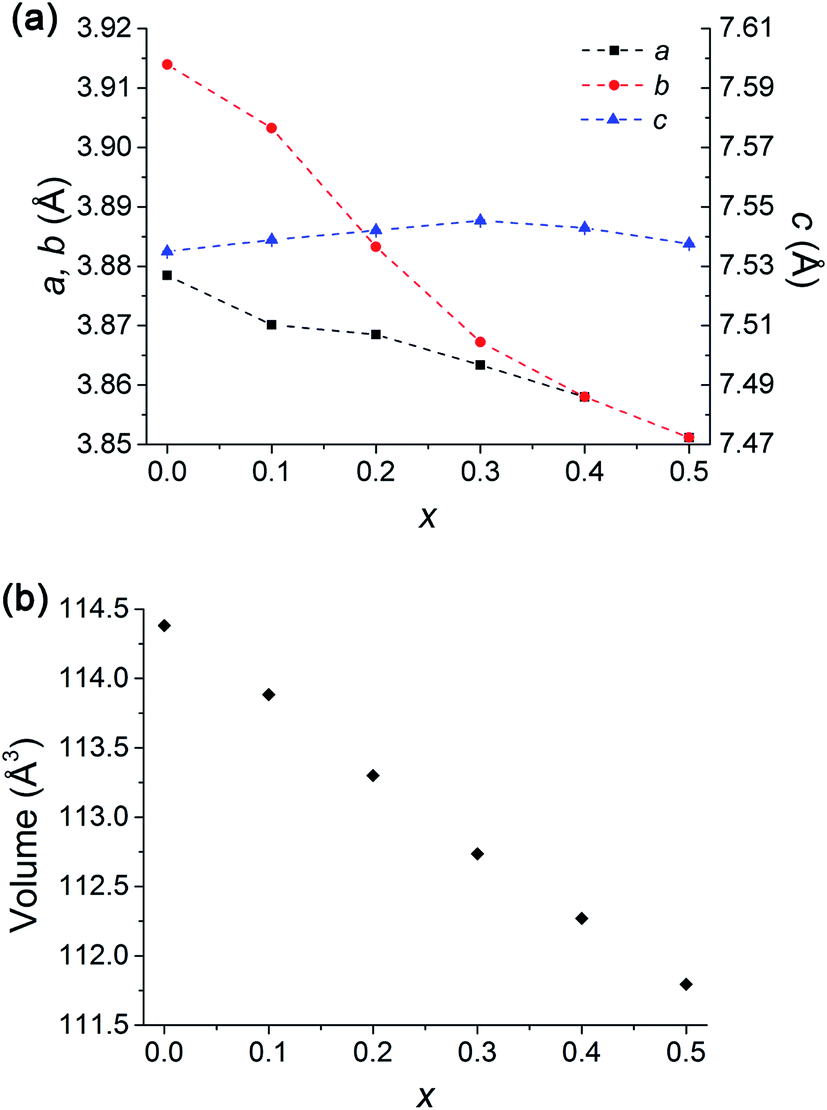 | ||
| Fig. 4 (a) Lattice parameters of GdBa1−xSrxCo2O6−δ showing a decrease in a and b (for x = 0 and 0.1, 1/2b is used for comparison) parameters with increasing Sr dopant whilst c increases to x = 0.3 and decreases with x > 0.3. Dashed lines are plotted as a guide to the eye. (b) Volume decreases as x increases consistent with smaller ionic radius of Sr2+ than Ba2+. Error bars are smaller than the symbols. Note transition to tetragonal at x = 0.4. | ||
The average equatorial bond lengths of Co–O ((i) Pmmm, Z = 2: Co1–O6,7 and Co2–O5,7, (ii) Pmmm, Z = 1: Co1–O2,3, (iii) P4/mmm: Co1–O2) are relatively constant for GBCO, GB9S1CO and GB8S2CO, whilst with further increase in Sr content, a decrease in equatorial bond length was observed, in agreement with the smaller ionic radius of Sr2+ (Fig. S7a†). An interesting observation is that the axial Co–O bond length in the Ba plane increased slightly when Sr ≥ 40 at%. Bond valence sum (BVS) analysis,40,41 which indicates the bond strength in ionic compounds, was calculated taking into account the vacancy in the oxygen site that is located on the Gd plane (z = 0.5). For all compounds the BVS of cobalt was found to be in good agreement with the oxidation state obtained from the iodometric titrations (Tables S7–S9†).
3.2. Surface electronic structure and chemistry
The freshly fractured surfaces of GBCO, GB8S2CO, GB7S3CO, and GB6S4CO pellets were characterised using XPS. A consistent trend in the Ba and Sr core level intensities was found following the changes in nominal composition (Fig. 5a and b). The Ba 4d core level spectra show two chemical environments. The dominant lower binding energy (BE) feature with peak maxima at 87.7–88.1 eV can be assigned to the Ba–O environment in the perovskite, in agreement with previous reports e.g. on YBa2Cu3O7.42 The BE of this peak varies as Sr is introduced in the BaO plane of the GBCO structure, and consequently the local chemical environment of Ba changes. The Gd 4d core level (Fig. 5b) shows the broad line shape typical for rare-earths with partially filled 4f shells, which is due to a complicated intrinsic line structure from multiplet splitting.43,44 The Gd 4d5/2 binding energy position is 141.4 eV with a spin–orbit-split (SOS) of 5.5 eV in good agreement with a Gd oxide environment.45 The Sr 3d core line, which is at the lower BE side of the Gd 4d line, is dominated by the bulk oxide contribution. The BE position of Sr 3d5/2 is 132.4 eV, consistent with values reported for SrFeO3−δ and La0.8Sr0.2CoO3−δ in the literature.46,47 The Sr core level also shows a slight change in BE with a change in doping level of about 200 meV. Again this can be attributed to a change in local environment in the Ba/SrO plane of the GBCO structure. In contrast, no changes in BE position are found for the Gd core level as Gd is located in a separate AO plane with no direct interaction with Ba or Sr, confirming the existence of ordered A cations of GdO and Ba/SrO layers in the double perovskite.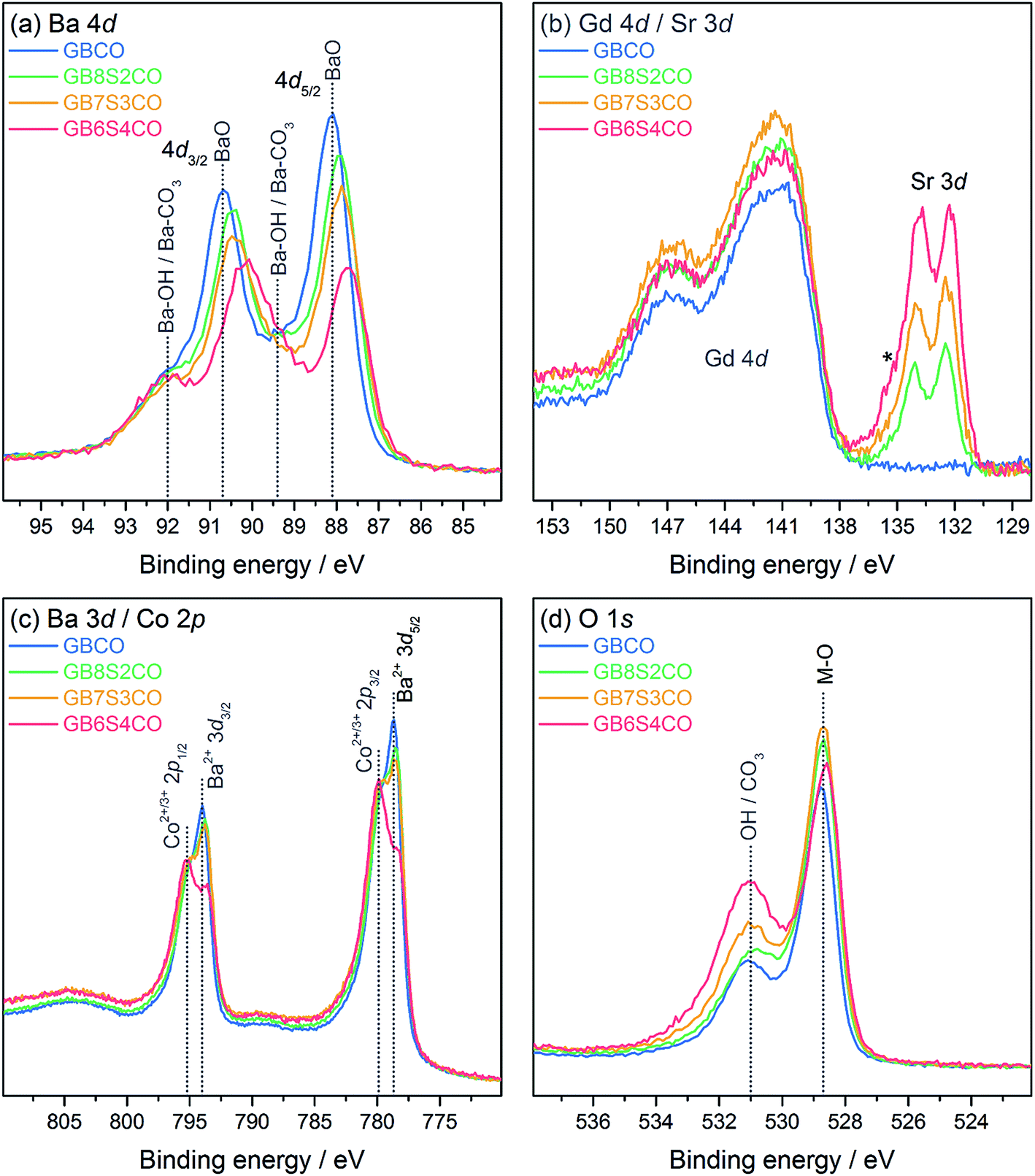 | ||
| Fig. 5 X-ray photoelectron spectra of GBCO, GB8S2CO, GB7S3CO and GB6S4CO, including (a) Ba 4d, (b) Gd 4d and Sr 3d (* = Sr–OH/Sr–CO3), (c) Ba 3d and Co 2p, and (d) O 1s core level spectra. | ||
In addition to the perovskite-related peaks, both Ba 4d and Sr 3d core levels show higher BE contributions, for example, the peak at 89.4 eV in the Ba 4d spectra, which are due to hydroxide or carbonate environments. The presence of this second environment is further confirmed by the O 1s core line (Fig. 5d), where a BE component at higher BE (531.0 eV) compared to the contribution from the perovskite at 528.7 eV is observed. From the O 1s spectrum it is not possible to distinguish between hydroxide and carbonate environments as their BEs are very close to each other.48 However, the C 1s line confirms that this contribution stems predominantly from hydroxide contributions as the amount of carbonate species detected at 289.3 eV is minimal (Fig. S8†). Carbonate formation, which is often found on Ba and Sr oxide surfaces, is connected strongly to the sample quality and surface preparation,42 and is most likely minimal in the present case due to the careful sample preparation for XPS measurements.
The overlap with the Ba 3d core line made it impossible to determine Co 2p BE positions accurately enough to assess the oxidation state of Co, as the chemical shift between Co2+/Co3+ is only around 0.3–0.4 eV.49 As the main oxidation state of Co in GdBa1−xSrxCo2O6−δ is 3+ (δ = 1 leads to Co2.5+, whereas δ = 0 leads to Co3.5+), however, due to the presence of satellite structures on the Co 2p core level in the present case, a mixed Co2+/Co3+ state was found. These satellites only occur in Co2+ due to its paramagnetic nature, whilst no satellites are found for Co3+.49 The accurate Co bulk oxidation state was determined using the titration method. For GB6S4CO for example, an average oxidation state of +3.04 was found, which suggests a small contribution from Co4+ to balance the Co2+ population found in XPS.
In addition to core level spectra, the valence bands (VBs) of the pellets were measured (Fig. 6). The wide VBs show several shallow core levels including Gd 4f (at 7.9 eV), Ba 5p (5p3/2 at 12.4–12.9 eV), overlapping Sr 4p and Gd 5p (between 16.7 and 24.2 eV), and O 2s (26.8 eV), which are in good agreement with previously reported VBs.50,51 As in the deeper core levels the changes in Ba![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sr ratios are reflected clearly in the changes in line intensities of the shallow Ba and Sr core levels here. Fig. 6b shows an expanded view of the valence band maximum VBmax relative to the Fermi energy EF. The exact position of the VBmax determined from XPS, and in particular the spectral weight at EF, has been the subject of previous work51,52 and the data collected here for GBCO agree with these reports showing negligible spectral weight at EF. Upon Sr-doping of GBCO a clear shift of the VBmax towards EF of the order of 200 meV is observed. At the highest Sr-doping level in GB6S4CO a clear spectral contribution at EF is visible.
:Sr ratios are reflected clearly in the changes in line intensities of the shallow Ba and Sr core levels here. Fig. 6b shows an expanded view of the valence band maximum VBmax relative to the Fermi energy EF. The exact position of the VBmax determined from XPS, and in particular the spectral weight at EF, has been the subject of previous work51,52 and the data collected here for GBCO agree with these reports showing negligible spectral weight at EF. Upon Sr-doping of GBCO a clear shift of the VBmax towards EF of the order of 200 meV is observed. At the highest Sr-doping level in GB6S4CO a clear spectral contribution at EF is visible.
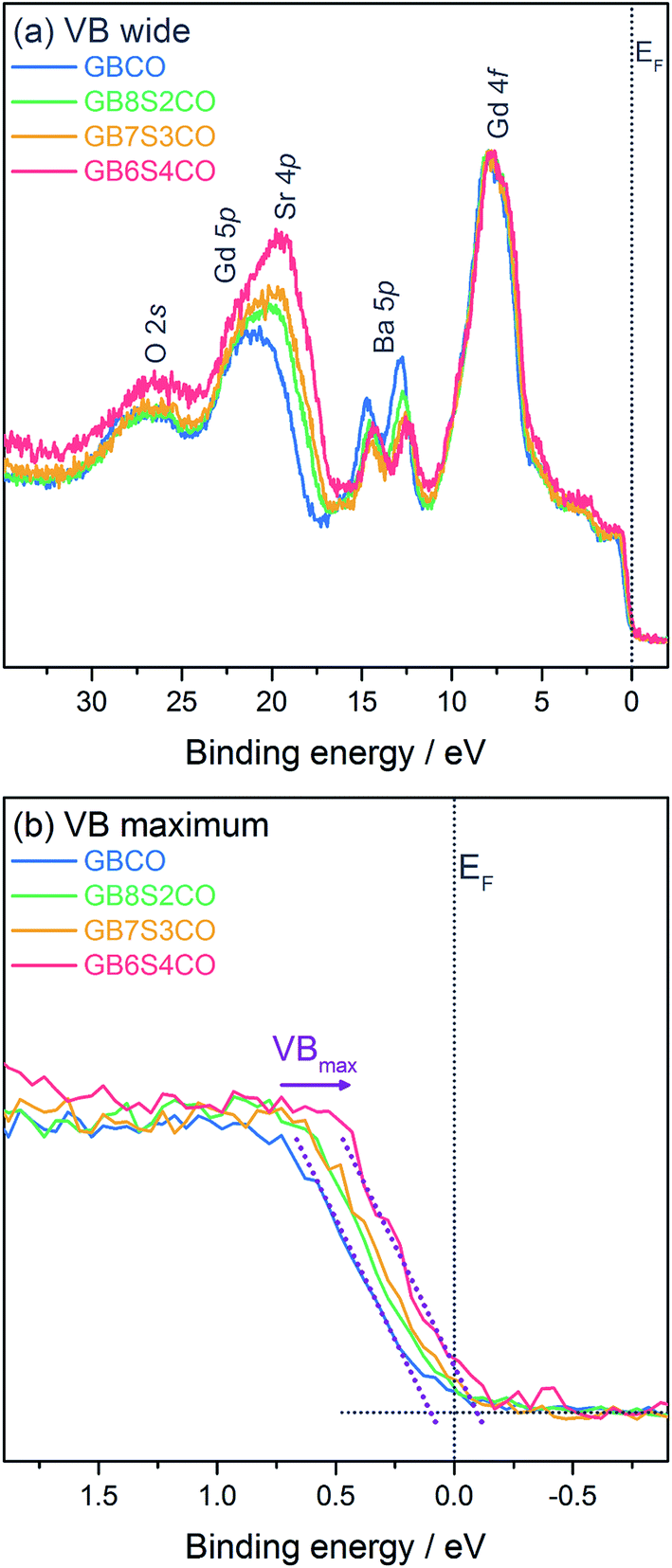 | ||
| Fig. 6 X-ray photoelectron spectra of GBCO, GB8S2CO, GB7S3CO, and GB6S4CO, including (a) wide valence region, and (b) an expanded view around the valence band maximum VBmax. Linear fits to the background and VBmax are shown as dotted lines in (b). The dotted line at 0 eV indicates the position of the Fermi energy EF. | ||
LEIS depth profile spectra are presented against the normalised number of cations where cation compositions in the bulk are normalised to unity (Fig. 7 and S9†). The LEIS spectra of GBCO samples annealed at 600 °C and 800 °C show depletion of Gd at the surface and the depth profile does not show any significant variation between the two annealing temperatures (Fig. 7a). It is also clear that there was Ba enrichment at the surface regardless of the annealing temperature, and that the Ba content decreased towards the bulk composition with increasing sputtering time. A higher annealing temperature led to Ba enrichment to a greater depth (∼4 nm). The surface Co content was shown to be depleted after annealing at 600 °C. In the sub-surface region, Co content was observed to increase beyond the ratio suggested by the bulk composition before decreasing to reach the bulk composition. In contrast, annealing at 800 °C yielded an ideal (bulk-like) Co composition at the surface, followed by Co enrichment in the sub-surface region, similar to what was observed with lower temperature annealing but with slightly lower cation counts than 600 °C at 0.7 nm < depth < 5 nm away from the surface. The ratio of the [A] site, which is occupied by the rare earth and alkaline earth ions, to the [B] site tenanted by Co, shows the enrichment of Co in the sub-surface layer (Fig. 7d), aligned with the previously published results.29 The diffusion of alkaline earth ions rather than rare earth or transition metal species to the surface has been reported to prevent the electrostatic surface energy diverging and hence stabilises the surface, i.e. creating a non-polar surface.53,54
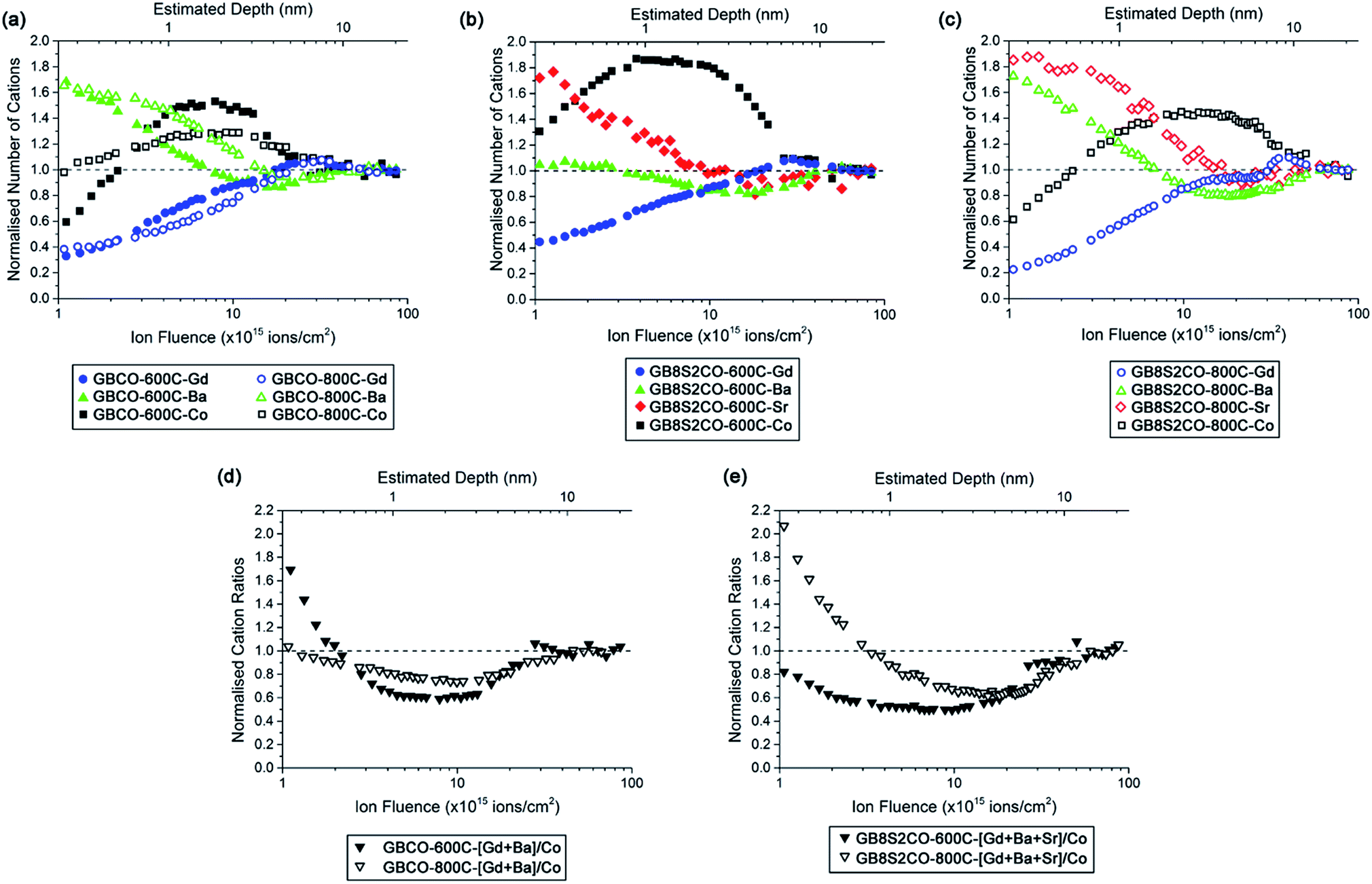 | ||
| Fig. 7 Cation depth profiles obtained from low energy ion scattering (LEIS) spectroscopy (a) individual cations in GBCO, (b) individual cations in GB8S2CO annealed at 600 °C and (c) individual cations in GB8S2CO annealed at 800 °C, whilst (d, e) [Gd + Ba + Sr]/Co ratio in GBCO and GB8S2CO, respectively. | ||
Doping the Ba site with 20 at% Sr (GB8S2CO) resulted in similar depletion of Gd at the surface and increased with respect to the increasing Ar+ sputter ion fluence (Fig. 7b and c). Surface barium enrichment was only found on the sample annealed at 800 °C, whilst the Ba content was close to the bulk composition throughout the whole sputtered depth for the sample annealed at 600 °C. The strontium depth profile was observed to be similar in nature to the Ba profile in the GBCO sample which suggests that Sr more actively segregated to the surface than Ba in the GB8S2CO. Supported by the XPS results, the enrichment of the alkaline earth at the surface is accompanied with the formation of hydroxide compounds. A published report on the segregation behaviour of La0.8(Ca,Sr,Ba)0.2MnO3 shows that Ba tends to segregate to the surface due to a larger dopant size mismatch to the La host.26 In this work, Sr substitution into the Ba site leads to Sr segregation to the sample surface. Another interesting finding is that in the near-surface region Co is more enriched for samples annealed at lower temperature. The LEIS spectra for GB7S3CO and GB6S4CO annealed at the same temperatures show similar trends with respect to sputtered ion fluence (Fig. S9†). For all Sr-doped GBCO, the [A] to [B] cation ratio for the sample annealed at 800 °C is greater than 600 °C and the sub-surface layer is dominated by Co. The only anomaly is at higher Sr levels (GB6S4CO, Fig. S9c†), where the surface Sr content was found to be not heavily enriched for the sample annealed at 600 °C, which requires further investigation.
3.3. Conductivity and electrochemical performance
In GBCO (relative density of 85%), there are three phase transitions observed, which are at 70 °C, 100 °C and 360 °C with activation energies (Ea) typical of a semiconductor (0.19 eV (T < 70 °C), 1.23 eV (70 °C < T < 100 °C) and 0.02 eV (100 °C < T < 360 °C)) (Fig. 8). Above 360 °C, a metallic characteristic is observed. This metallic transition is reported to be accompanied by an orthorhombic Pmmm to tetragonal P4/mmm phase transition.55 A sharp electrical conductivity drop at 470 °C is also observed. As Sr replaced Ba, the transition temperature was found to be lower. In GB8S2CO (relative density of 93%), Ea of 0.22 eV (T < 70 °C) and 0.02 eV (70 °C < T < 320 °C) was observed, whilst Ea of 0.01 eV (T < 250 °C) was found in GB6S4CO (relative density of 87%). These are followed by a transition to metallic behaviour at temperatures above 320 °C for the former and above 250 °C for the latter. At 130 °C, the conductivity of GB6S4CO reached as high as 980 S cm−1, whilst 890 S cm−1 was obtained for GB8S2CO and 720 S cm−1 for GBCO. The higher oxygen content, confirmed by iodometric titration, leads to an increase in electronic holes which can be formulated in the Kröger–Vink notation as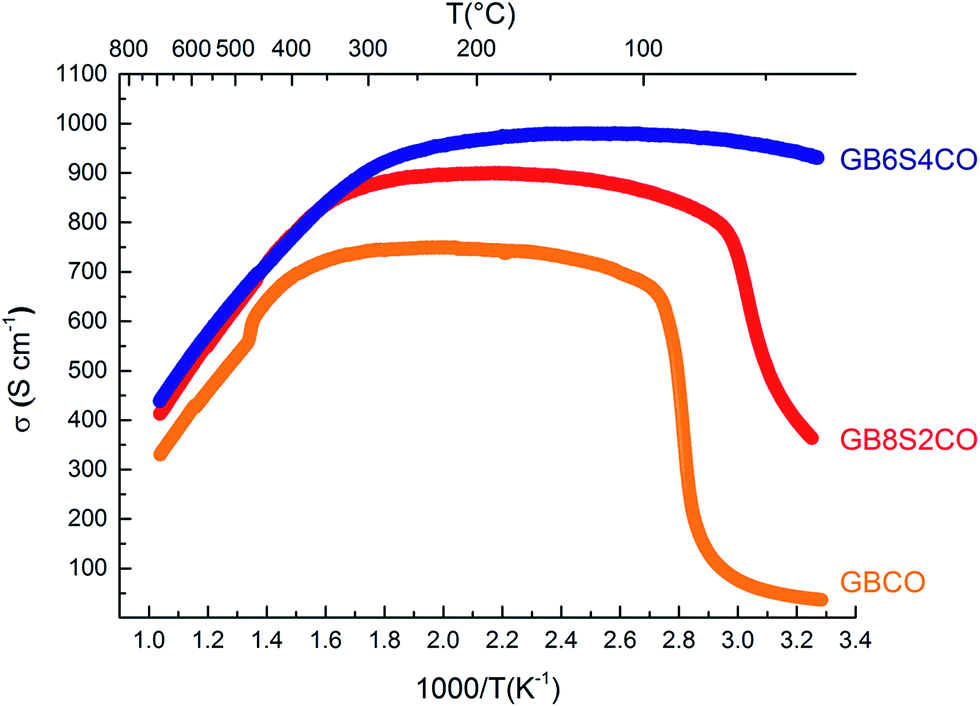 | ||
| Fig. 8 Total DC electrical conductivity of GBCO (orange), GB8S2CO (red) and GB6S4CO (blue) as a function of temperature showing higher conductivity was achieved for higher Sr-doped sample. | ||
Oxidation of Co2+ to Co3+ is represented by
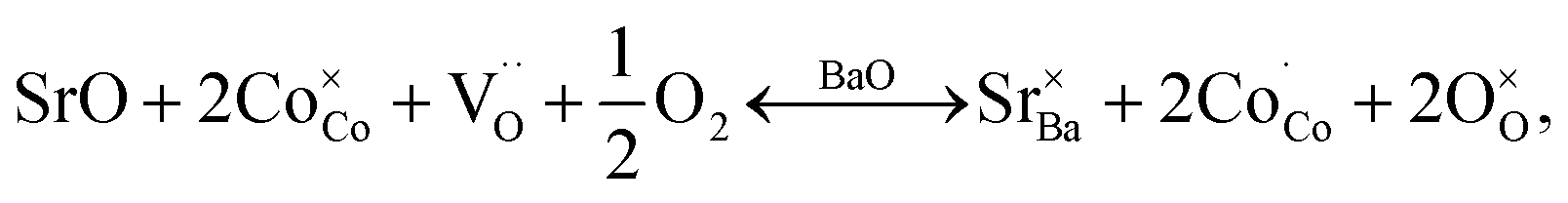
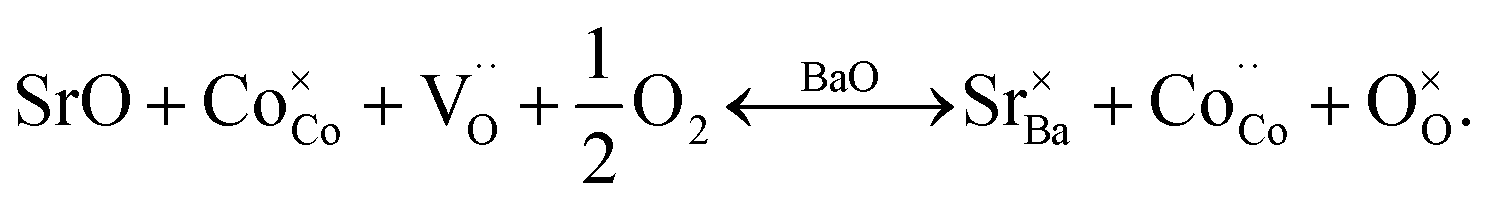
Furthermore, when the structure is less distorted (based on consideration of the Goldschmidt tolerance factor56) which is close to the ideal cubic perovskite, the conductivity tends to be higher.25 This is due to a wider charge carrier bandwidth and overlap between Co 3d and O 2p orbitals which is a function of the buckling angle of the Co–O–Co bonds and Co–O bond lengths (Tables S7–S9†). Within the tight-binding approximation, the carrier bandwidth (w) is given as:57
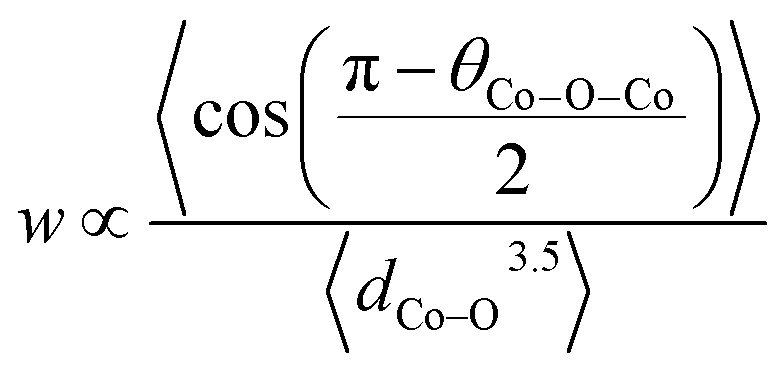 | (1) |
Combining the effect of average axial and equatorial buckling angles (θCo–O–Co) and bond lengths (dCo–O), w increases with Sr dopant leading to higher electronic conductivity (Fig. S7b†), consistent with earlier reports.58,59
The onset potential for oxygen evolution in Sr doped GBCO was found to be ∼1.50 to 1.52 V with respect to the reversible hydrogen electrode (RHE). This is approximately 50–70 mV earlier than that for the undoped GBCO (1.57 V) as determined by linear sweep voltammetry with scanning rate of 1 mV s−1 performed on static electrodes (Fig. 9a). No significant improvement in the onset potential was observed as more Sr was introduced; however, a much higher oxygen evolution current density was achieved when GBCO was doped with 40 at% Sr (GB6S4CO). Notably, the Tafel slope also decreased, from close to 100 mV dec−1 for the undoped GBCO, to around 80 mV dec−1 for Sr doped GBCO which is closer to the reported 60 mV dec−1 for (ABa)Co2O6−δ (A = Pr, Sm, Gd and Ho) in O2-saturated 0.1 M KOH electrolyte.15 The smaller Tafel slope seen in the doped sample indicates a possible shift in the rate limiting step of the oxygen evolution reaction from M-OH formation to M-OO formation or O2(g) desorption, where M represents a site on the surface.60
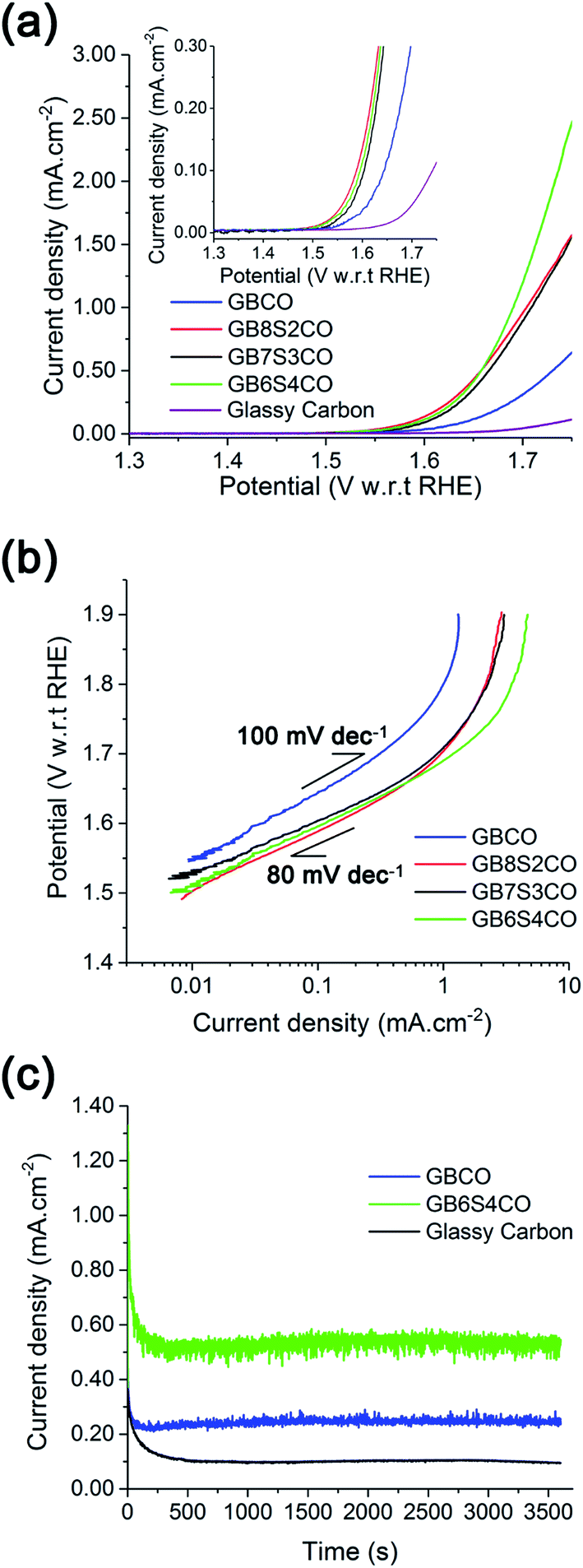 | ||
| Fig. 9 (a) Linear sweep voltammetry at the 6th cycle and the corresponding (b) Tafel plot (at 1 mV s−1 scan rate) in 0.1 M KOH electrolyte. (c) Chronoamperometry measurements of different GBSCO compounds at 1.65 V (η = 420 mV). Electrochemical measurements were iR compensated using current interrupt method. Higher performance was achieved with higher Sr dopant level. | ||
We note that the OER activity of this double perovskite is comparable with previously reported LaCoO3−δ, better than LaCrO3−δ and LaMnO3−δ single perovskites, but lower than Ba0.5Sr0.5Co0.8Fe0.2O3−δ.9 The OER performances of selected double perovskites are listed in Table S10.† The higher current density exhibited by the Sr doped GBCO was also sustainable, as demonstrated by chronoamperometric measurements, with its activity after one hour at 1.65 V (η = 420 mV) maintained at 0.53 mA cm−2, which is ∼2.5 times higher compared to the undoped GBCO (Fig. 9c).
4. Conclusions
Strontium-doped double perovskites (GdBa1−xSrxCo2O6−δ) possess improved oxygen evolution catalytic performance and higher electrical conductivity compared to undoped GdBaCo2O6−δ. XRD measurements together with Rietveld analysis showed that materials with higher Sr dopant content crystallised with higher symmetry, transforming from orthorhombic Pmmm with doubling in b and c (GBCO and GB9S1CO), transiting to the same Pmmm space group with doubling only in c (GB8S2CO and GB7S3CO) and finally to tetragonal P4/mmm (GB6S4CO and GB5S5CO). This transformation was accompanied by oxygen uptake, lowering the oxygen vacancy content. A combination of higher oxygen concentration, higher Co oxidation state and higher carrier bandwidth (which depends on the Co–O–Co buckling angles and Co–O bond lengths) explains the higher electronic conductivity. The surface enrichment of Ba and Sr, which are mainly in the form of hydroxides, was observed by LEIS and XPS. However, this does not make the surface inactive as shown by higher oxygen evolution current density observed with higher Sr dopant level. Hence, a thorough understanding of the crystallographic occupancy ordering and surface characteristics is essential for optimising the electrical conductivity, ionic mobility and catalytic performance, critical to designing efficient devices.Author's contributions
S. S. P. and S. J. S. conceived and designed this work. S. S. P performed the XRD experiments and analysed the crystal structure of materials. A. C. performed the electrical measurements. S. S. P., A. C., C. L. and J. A. K carried out the low energy ion scattering spectroscopy measurements and analysed the spectra. R. J. W, A. R. and D. J. P. were involved in the X-ray photoelectron spectroscopy measurements and analyses. Electron probe microanalyses were conducted by S. S. P and J. S. H.; A. D. H., K. W. C., B. S. Y and M. P. R conducted the electrochemistry experiments and analysed the results. All authors co-wrote and reviewed the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to gratefully acknowledge the support of the EPSRC (EP/M014142/1). Additionally, we acknowledge the support of King Abdullah University of Science and Technology, who partially funded this work (S. S. P. and A. C.). We acknowledge the Facility for Analysis, Characterization, Testing and Simulation, Nanyang Technological University, Singapore, for use of their electron microscopy. D. J. P. acknowledges support from the Royal Society for his University Research Fellowship (No. UF100105). D. J. P. and A. R. acknowledge support from the EPSRC (No. EP/M013839/1, EP/M028291/1, EP/M014142/1 and EP/M014304/1). Data supporting this publication is openly available under an ‘Open Data Commons Open Database License’. Additional metadata are available at: http://dx.doi.org/10.17634/154300-55. Please contact Newcastle Research Data Service at http://rdm@ncl.ac.uk for access instructions.References
- S. J. Skinner, Int. J. Inorg. Mater., 2001, 3, 113–121 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1263–B1268 CrossRef CAS.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- S. Carter, A. Selcuk, R. J. Chater, J. Kajda, J. A. Kilner and B. C. H. Steele, Solid State Ionics, 1992, 53–56, 597–605 CrossRef CAS.
- J. Richter, P. Holtappels, T. Graule, T. Nakamura and L. Gauckler, Monatsh. Chem., 2009, 140, 985–999 CrossRef CAS.
- Y. Chen, W. Zhou, D. Ding, M. Liu, F. Ciucci, M. Tade and Z. Shao, Adv. Energy Mater., 2015, 5, 1500537 CrossRef.
- S. Tao, J. T. S. Irvine and J. A. Kilner, Adv. Mater., 2005, 17, 1734–1737 CrossRef CAS.
- Y. Matsumoto, S. Yamada, T. Nishida and E. Sato, J. Electrochem. Soc., 1980, 127, 2360–2364 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- J.-I. Jung, S. Park, M.-G. Kim and J. Cho, Adv. Energy Mater., 2015, 5, 1501560 CrossRef.
- D. Parfitt, A. Chroneos, A. Tarancon and J. A. Kilner, J. Mater. Chem., 2011, 21, 2183–2186 RSC.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Appl. Phys. Lett., 2005, 86, 091910 CrossRef.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Prog. Solid State Chem., 2007, 35, 481–490 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed.
- Z. Wu, L.-P. Sun, T. Xia, L.-H. Huo, H. Zhao, A. Rougier and J.-C. Grenier, J. Power Sources, 2016, 334, 86–93 CrossRef CAS.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- B. Hua, Y.-F. Sun, M. Li, N. Yan, J. Chen, Y.-Q. Zhang, Y. Zeng, B. Shalchi Amirkhiz and J.-L. Luo, Chem. Mater., 2017, 29, 6228–6237 CrossRef CAS.
- L. Lo Presti, M. Allieta, M. Scavini, P. Ghigna, L. Loconte, V. Scagnoli and M. Brunelli, Phys. Rev. B: Condens. Matter, 2011, 84, 104107 CrossRef.
- P. S. Anderson, C. A. Kirk, J. Knudsen, I. M. Reaney and A. R. West, Solid State Sci., 2005, 7, 1149–1156 CrossRef CAS.
- G. King and P. M. Woodward, J. Mater. Chem., 2010, 20, 5785–5796 RSC.
- M. Bahout, S. S. Pramana, J. M. Hanlon, V. Dorcet, R. I. Smith, S. Paofai and S. J. Skinner, J. Mater. Chem. A, 2015, 3, 15420–15431 CAS.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Phys. Rev. B: Condens. Matter, 2005, 71, 134414 CrossRef.
- S. Roy, M. Khan, Y. Q. Guo, J. Craig and N. Ali, Phys. Rev. B: Condens. Matter, 2002, 65, 064437 CrossRef.
- J.-H. Kim, F. Prado and A. Manthiram, J. Electrochem. Soc., 2008, 155, B1023–B1028 CrossRef CAS.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- H. H. Brongersma, in Characterization of Materials, ed. E. N. Kaufmann, John Wiley & Sons, Inc., 2002, pp. 2024–2044 Search PubMed.
- H. H. Brongersma, M. Draxler, M. de Ridder and P. Bauer, Surf. Sci. Rep., 2007, 62, 63–109 CrossRef CAS.
- J. Druce, H. Tellez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 CAS.
- H. Tellez, J. Druce, J. A. Kilner and T. Ishihara, Faraday Discuss., 2015, 182, 145–157 RSC.
- H. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- R. W. Cheary and A. Coelho, J. Appl. Crystallogr., 1992, 25, 109–121 CrossRef CAS.
- Bruker AXS, TOPAS Version 4.1, Madison, Wisconsin, USA, 2008 Search PubMed.
- C. Frontera, J. L. García-Muñoz, A. Llobet and M. A. G. Aranda, Phys. Rev. B: Condens. Matter, 2002, 65, 180405 CrossRef.
- D. Primetzhofer, M. Spitz, E. Taglauer and P. Bauer, Surf. Sci., 2011, 605, 1913–1917 CrossRef CAS PubMed.
- A. Rafati, R. Veen and D. G. Castner, Surf. Interface Anal., 2013, 45, 1737–1741 CrossRef CAS PubMed.
- D. Drouin, A. R. Couture, D. Joly, X. Tastet, V. Aimez and R. Gauvin, Scanning, 2007, 29, 92–101 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- R. P. Vasquez, J. Electron Spectrosc. Relat. Phenom., 1994, 66, 241–255 CrossRef CAS.
- K. Wandelt and C. R. Brundle, Surf. Sci., 1985, 157, 162–182 CrossRef CAS.
- S. P. Kowalczyk, N. Edelstein, F. R. McFeely, L. Ley and D. A. Shirley, Chem. Phys. Lett., 1974, 29, 491–495 CrossRef CAS.
- Y. Uwamino, T. Ishizuka and H. Yamatera, J. Electron Spectrosc. Relat. Phenom., 1984, 34, 67–78 CrossRef CAS.
- H. Falcón, J. A. Barbero, J. A. Alonso, M. J. Martínez-Lope and J. L. G. Fierro, Chem. Mater., 2002, 14, 2325–2333 CrossRef.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y.-L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 6081–6088 CAS.
- P. Nachimuthu, Y. J. Kim, S. V. N. T. Kuchibhatla, Z. Q. Yu, W. Jiang, M. H. Engelhard, V. Shutthanandan, J. Szanyi and S. Thevuthasan, J. Phys. Chem. C, 2009, 113, 14324–14328 CAS.
- J. Haber and L. Ungier, J. Electron Spectrosc. Relat. Phenom., 1977, 12, 305–312 CrossRef CAS.
- W. R. Flavell, A. G. Thomas, D. Tsoutsou, A. K. Mallick, M. North, E. A. Seddon, C. Cacho, A. E. R. Malins, S. Patel, R. L. Stockbauer, R. L. Kurtz, P. T. Sprunger, S. N. Barilo, S. V. Shiryaev and G. L. Bychkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 224427 CrossRef.
- T. C. Koethe, PhD thesis, University of Cologne, 2007.
- Z. Hu, W. Hua, T. C. Koethe, S. N. Barilo, S. V. Shiryaev, G. L. Bychkov, C. Schüßler-Langeheine, T. Lorenz, A. Tanaka, H. H. Hsieh, H. J. Lin, C. T. Chen, N. B. Brookes, S. Agrestini, Y. Y. Chin, M. Rotter and L. H. Tjeng, New J. Phys., 2012, 14, 123025 CrossRef.
- P. W. Tasker, J. Phys. C: Solid State Phys., 1979, 12, 4977 CrossRef CAS.
- N. Claudine, J. Phys.: Condens. Matter, 2000, 12, R367 CrossRef.
- A. Tarancón, D. Marrero-López, J. Peña-Martínez, J. C. Ruiz-Morales and P. Núñez, Solid State Ionics, 2008, 179, 611–618 CrossRef.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- M. Medarde, J. Mesot, P. Lacorre, S. Rosenkranz, P. Fischer and K. Gobrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 9248–9258 CrossRef CAS.
- K. T. Lee and A. Manthiram, J. Electrochem. Soc., 2005, 152, A197–A204 CrossRef CAS.
- Z. Chen, T. A. Tyson, K. H. Ahn, Z. Zhong and J. Hu, J. Magn. Magn. Mater., 2010, 322, 3049–3052 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta06817d |
| This journal is © The Royal Society of Chemistry 2018 |