
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guiding 3D cell migration in deformed synthetic hydrogel microstructures†
Miriam
Dietrich
 ab,
Hugo
Le Roy
c,
David B.
Brückner
d,
Hanna
Engelke
e,
Roman
Zantl
b,
Joachim O.
Rädler
*a and
Chase P.
Broedersz
*d
ab,
Hugo
Le Roy
c,
David B.
Brückner
d,
Hanna
Engelke
e,
Roman
Zantl
b,
Joachim O.
Rädler
*a and
Chase P.
Broedersz
*d
aFaculty of Physics and Center for NanoScience, Ludwig-Maximilians-University, Munich, Germany. E-mail: raedler@lmu.de
bibidi GmbH, Martinsried, Germany
cÉcole Normale supérieure Paris-Saclay, France
dArnold-Sommerfeld Center for Theoretical Physics and Center for NanoScience, Ludwig-Maximilians-University, Munich, Germany. E-mail: C.Broedersz@lmu.de
eDepartment of Chemistry and Center for NanoScience, Ludwig-Maximilians-University, Munich, Germany
First published on 29th March 2018
Abstract
The ability of cells to navigate through the extracellular matrix, a network of biopolymers, is controlled by an interplay of cellular activity and mechanical network properties. Synthetic hydrogels with highly tuneable compositions and elastic properties are convenient model systems for the investigation of cell migration in 3D polymer networks. To study the impact of macroscopic deformations on single cell migration, we present a novel method to introduce uniaxial strain in matrices by microstructuring photo-polymerizable hydrogel strips with embedded cells in a channel slide. We find that such confined swelling results in a strained matrix in which cells exhibit an anisotropic migration response parallel to the strain direction. Surprisingly, however, the anisotropy of migration reaches a maximum at intermediate strain levels and decreases strongly at higher strains. We account for this non-monotonic response in the migration anisotropy with a computational model, in which we describe a cell performing durotactic and proteolytic migration in a deformable elastic meshwork. Our simulations reveal that the macroscopically applied strain induces a local geometric anisotropic stiffening of the matrix. This local anisotropic stiffening acts as a guidance cue for directed cell migration, resulting in a non-monotonic dependence on strain, as observed in our experiments. Our findings provide a mechanism for mechanical guidance that connects network properties on the cellular scale to cell migration behaviour.
Introduction
Multicellular organisms consist of a composite of cells and the extracellular matrix (ECM) that forms the scaffolding in which cells live and move. In animal tissue, the ECM is composed of a complex network of various biopolymers, including collagen and proteoglycans. The migration of cells in this environment is important to a variety of physiological processes, including the immune response, embryogenesis, and cancer metastasis.1–3 To navigate such a complex environment, cells employ a multitude of biochemical signalling pathways. However, cells also make use of the available mechanical information, by probing the surrounding matrix via integrin-mediated adhesions. Integrins couple the ECM to the acto-myosin machinery of the cell, thereby enabling the transmission of forces between the cell and its environment.4 This coupling equips the cell with mechanosensitive capability. Indeed, the structure of focal adhesions and the cytoskeleton can be altered by changing the ECM stiffness, with the formation of larger adhesion complexes and more pronounced actin structures on stiffer substrates.5 This mechanosensitivity also affects migration: most cells typically migrate from the soft to the stiff side of a substrate, a phenomenon called durotaxis.6,7 While much is known about the mechanosensitive signalling and response pathways in cells,8 it is still unclear how mechanical cues such as deformations and heterogeneities in the matrix affect migration in 3D substrates.9–11Synthetic hydrogels have been introduced to study cell migration in 3D environments with highly controllable mechanical properties. These hydrogels can be composed of polyethylene glycol (PEG) with exact molecular composition such that the mesh size and the mechanical properties of the matrix can be precisely tuned for bioengineering applications and in vitro experiments.12–16 For example, the viability of primary cells and specific signalling pathways important for angiogenesis can be enhanced by altering the concentration and availability of integrin binding sequences containing the RGD-peptide-motif in synthetic hydrogels.17,18 Furthermore, to enable cell migration in gels with small mesh sizes, the presence of proteinase-sensitive cross-linkers in PEG-based hydrogels and adhesion mediating peptide sequences are crucial.19,20 Even though the physical properties of these hydrogels can be tailored, this often only applies to the macroscopic properties. However, cells interact with their surrounding on a scale of a few tens of micrometers.21 It is therefore important to not only tune the macroscopic properties of the matrix, but to also consider the network properties on the microscale and below.9
Macroscopic deformations in the ECM could alter the network arrangement on the cellular scale. Interestingly, cells can reorient to strains applied to the substrate.22–24 Indeed, a number of studies with 2D or 3D substrates show that the cell orientation direction varies when changing the matrix composition and dimensionality or when using different strain patterns.11 In collagen gels, for example, the protease activity varies when the fibres are strained25 and cells embedded in deformed collagen matrices orient parallel to the strain direction.26 The response of cells to a static external strain in biopolymer gels like collagen has been attributed to the induced alignment of fibres in the direction of the strain27–29 or to the strain stiffening behaviour of these extracellular fibre networks.22,30 However, cell reorientation and directed migration in a strain field was also observed in synthetic hydrogels, which do not exhibit strong macroscopic strain stiffening.6,31 Furthermore, highly cross-linked synthetic hydrogels do not contain large fibres, which could align to a strain. Thus, the underlying mechanism of how cells sense and react to strain in synthetic hydrogels remains unclear.
Here we investigate, using both experiments and theoretical modelling, how deformations in matrix metalloproteinases (MMP) degradable and RGD functionalized PEG-based hydrogels affect the migration of embedded motile cells. We fabricate small strips of hydrogel photo-polymerized inside a microchannel slide, which results in anisotropic swelling in the direction of the strip width, straining the network uniaxially. We find that HT-1080 cells embedded in such gel strips exhibit a preferred migration direction parallel to the strain direction. However, the anisotropy of the cell migration reveals a non-monotonic dependence on the magnitude of the strain. To understand this striking phenomenon, we introduce a computational model of a proteolytically active cell, which performs durotactic migration in a strained 2D network. The experimentally observed migration behaviour is reproduced by our model and can be explained by a local stiffening mechanism at the cellular scale. This anisotropic stiffening thereby provides a physical mechanism to explain the non-monotonicity of anisotropic cell migration with strain. Our study demonstrates that the microscopic properties of cell matrices are crucial to elucidate how mechanical cues can manipulate cell migration behaviour.
Results
A synthetic hydrogel with embedded cells serves as a model system for cell matrices with tuneable degradability
To analyse cell migration in a simplified and highly controllable environment, we use a synthetic PEG-based material to encapsulate HT-1080 cells in a thick slab of hydrogel. These HT-1080 cells represent a well characterised fibrosarcoma cell line that expresses matrix metalloproteinases (MMPs) to digest the ECM, and is widely used in 3D migration experiments. The gel consists of 4-armed PEG-norbornene (PEG-NB), which is cross-linked by a peptide sequence that is cleavable by MMPs, as depicted in Fig. 1A. We add the peptide sequence CRGDS to promote cell adhesion via integrins to the otherwise bio-inert PEG backbone. A radical photo-initiator is included in the pre-polymer solution to initiate the thiol–ene polymerization reaction. To obtain isotropically swollen gels, we polymerize the hydrogel by homogeneous illumination of the entire polymer solution and allow the gel to float after polymerization. The floating gel swells isotropically and is subsequently immobilised on a micro-well surface.32 The photo-induced polymerization is biocompatible and yields matrices with a storage modulus of 20–70 Pa (ESI,† Fig. S1) and mesh sizes of a few tens of nm's after swelling.20,33–35 Because of this small mesh size, cells can only migrate through the network if they are able to digest the cross-links with MMPs.19,36 We did not observe significant deformations of the cell body when it squeezes through the mesh, which was observed in prior work.37,38 Note, however, that the pores in our hydrogels are orders of magnitude smaller than the minimal cell diameter that can be achieved by HT-1080 cells, and therefore matrix digestion by MMPs is necessary for cell migration in these hydrogels.37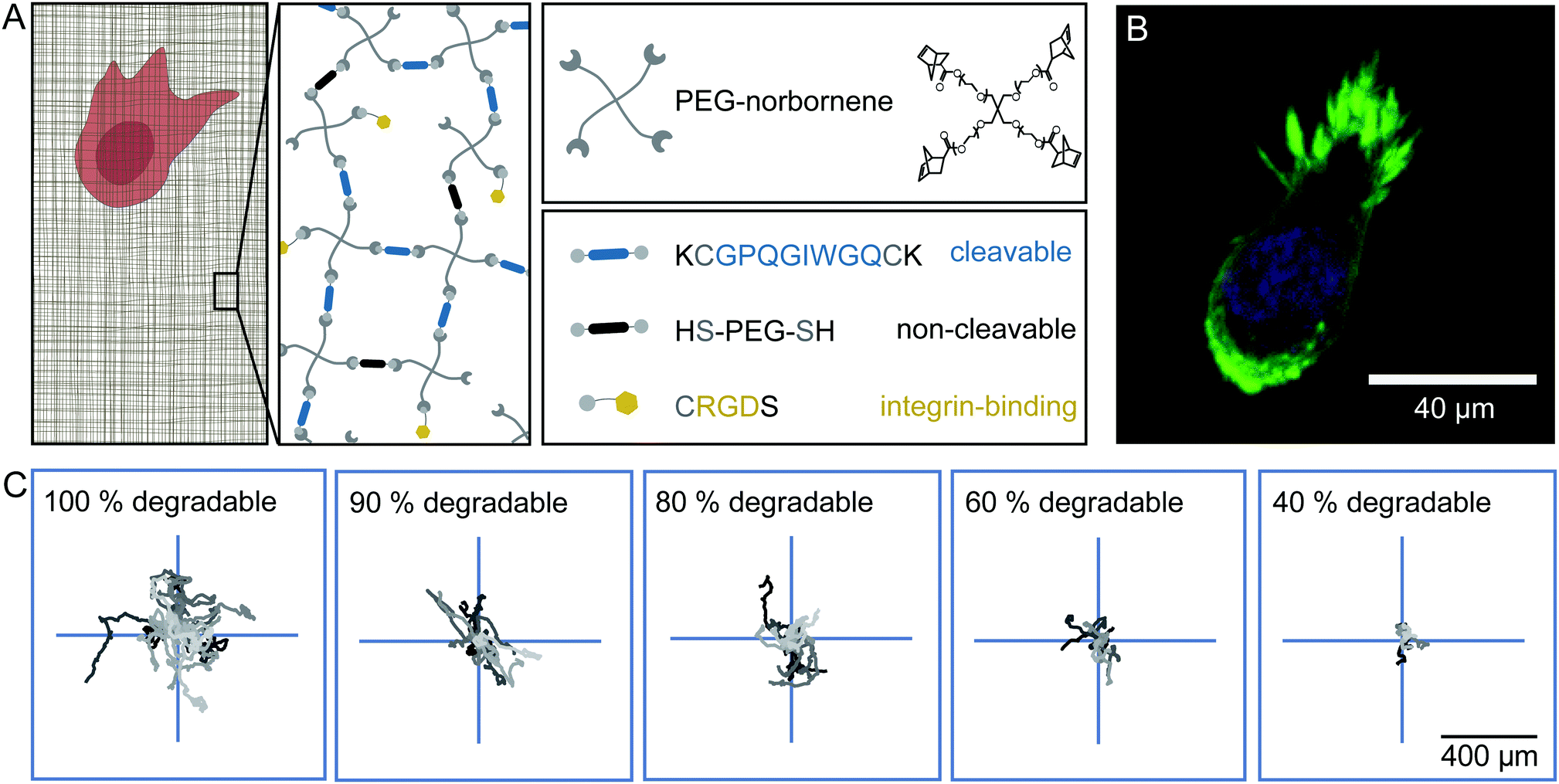 | ||
| Fig. 1 Experimental set-up of the synthetic hydrogel. (A) Schematic presentation of the synthetic hydrogel and its components. Norbornene functionalized 4-armed PEG is used as a monomer. For cross-linking, a peptide sequence (displayed in blue) that can be cleaved by cell-secreted matrix-metalloproteinases is used to create a migratable hydrogel. Substituting the cleavable cross-linker with a bio-inert dithiol-PEG (displayed in black) renders the gel non-migratable. RGD containing peptide sequences (displayed in yellow) enable cells to interact with the hydrogel through integrins. (B) Confocal image of a HT-1080 LifeAct-TagGFP2 cell in an isotropically swollen gel, 20 h after encapsulation. The actin structures are displayed in green and the nucleus is stained in blue. (C) Centred trajectories of HT-1080 cells migrating in hydrogels of different degradability for 24 h. | ||
We observe that cells move through the hydrogels with a rounded morphology and small protrusions at the leading edge (Fig. 1B), as previously described for HT-1080 cells embedded in synthetic hydrogels and dense collagen networks.34 The trajectories of the cells inside the hydrogel appear to be random and isotropic (Fig. 1C). We can influence the migratory behaviour of these cells by substituting a fraction of the MMP cleavable peptide cross-linker by a non-cleavable PEG-dithiol linker to reduce the overall degradability of the gel. Even in gels where only 40% of the cross-links are cleavable, the cells still migrate, but their overall displacement decreases significantly in comparison to cells migrating in a completely degradable gel for the same amount of time, as illustrated in Fig. 1C (see also ESI,† Movies S1–S3). Under all these conditions, we observe that the cells migrate isotropically through the gels (ESI,† Fig. S2).
Uniaxial strain by anisotropic swelling of confined microstructured hydrogels induces anisotropic cell migration
Next, we sought to investigate how network deformations affect cell migration. To induce a strain in 100% degradable gels, we form hydrogel microstructures under confinement. Small strips with a high aspect ratio are polymerized inside 400 μm high channel slides by photolithography, as illustrated in Fig. 2A. After flushing the system with cell culture media, the hydrogel strips swell. We only analyse hydrogel swelling and cell migration in the middle 20% of the longitudinal section of the strips to avoid edge effects of the strip ends.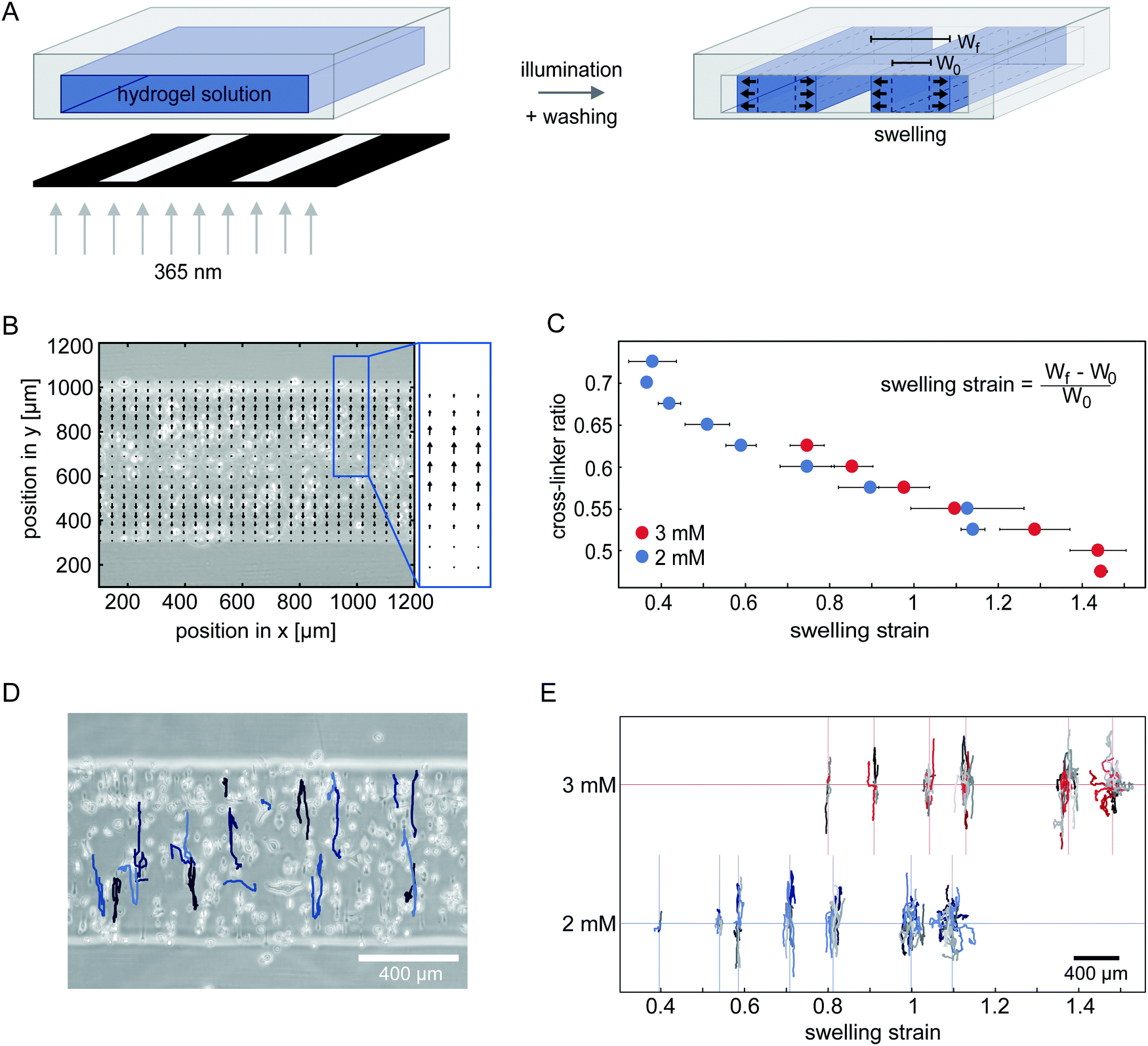 | ||
| Fig. 2 Applying uniaxial strain to the network by anisotropic swelling of microstructured hydrogels. (A) Schematic representation of the photo-lithographic structuring of hydrogel strips inside channels. After polymerization, unpolymerized hydrogel precursor solution is flushed out of the system by washing with cell culture media. Structures swell after washing. (B) Accumulated velocity field derived from PIV analysis of the swelling process, using fluorescent tracer beads suspended in 2 mM gels with a cross-linker ratio of 0.65. (C) The strain induced in the gels with embedded cells by swelling can be varied by changing the cross-linker to monomer ratio in the hydrogel mixture. Two different concentrations of PEG-NB monomer are tested. (2 mM PEG-NB gels displayed in blue, 3 mM PEG-NB gels displayed in red.) (D) Example trajectories of HT-1080 cells migrating in a 100% degradable 2 mM hydrogel with a cross-linker ratio of 0.6 for 24 h. (E) Centred trajectories of HT-1080 cells in hydrogel strips of different swelling strain with 2 mM PEG-NB (blue) and 3 mM PEG-NB (red). | ||
To investigate the direction of swelling in our confined geometry in detail, we embed small fluorescent beads in a range of hydrogels polymerized with different compositions. To control the hydrogel composition, we vary both the overall PEG-NB monomer concentration and the cross-linker to monomer ratio. We monitor the movement of the tracer beads inside the hydrogel throughout the swelling process (see ESI,† Movie S4) and analyse their trajectories with particle image velocimetry (PIV) to obtain velocity fields that quantify the swelling behaviour. After 2 h no further bead movement in the gel is detectable, indicating a stable swelling of the hydrogel structures. The accumulated velocity fields of tracer beads within the first 2 h is displayed in Fig. 2B, showing bead movement mostly in the direction along the short axis of the strip. This anisotropic swelling behaviour is present in all the gels we tested (ESI,† Fig. S3 and S4). For smaller cross-linker ratios, we measure higher overall velocities demonstrating a higher degree of swelling. Hence, by varying the cross-linker ratio of the hydrogel, we can tune the swelling and thereby the uniaxial strain induced in the gel.
To quantify the strain in the hydrogel due to the anisotropic swelling, we compare the width of hydrogel strips with embedded cells after completed swelling (Wf) with the initial strip width of 400 μm (W0).
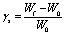 | (1) |
To analyse how cells migrate in a uniaxially strained network, we embed HT-1080 cells in hydrogel strips and monitor their migration for 24 h starting 3 h after encapsulation (see ESI,† Movies S5 and S6). With increasing cross-linker ratio, the percentage of migrating cells in the strips decreases to the point where motility is completely inhibited (ESI,† Fig. S5). To illustrate the migratory behaviour of cells in hydrogels, we show a phase-contrast image of the analysed hydrogel area overlaid with tracked cell trajectories in Fig. 2D. Cells in this gel exhibit a highly anisotropic migration, with the main migration direction oriented parallel to the swelling direction. This observation is consistent with prior experiments showing that fibroblasts preferentially migrate parallel to an applied static strain inside 3D substrates.31 Interestingly, however, when we compare the trajectories of cells migrating in hydrogels with different strains in our experiments, we observe a gradual shift from anisotropic migration in networks with moderate strains to a more isotropic mode of migration with higher strains (Fig. 2E and ESI,† Fig. S6). This migration behaviour is surprising, because the migration anisotropy decreases with increasing strain anisotropy. Our observation suggests that the strain in the hydrogel is perhaps not the only relevant factor for cell guidance.
Model of a durotactic motile cell with proteolytic activity in an elastic network
To elucidate the basic principles of cell migration in strained networks, we develop a simple theoretical model. Specifically, we aim to capture the basic physical processes of mechanosensing and cell migration using a system-level description of a cell moving through an elastic network. In our model, we explicitly describe the matrix through which the cell moves using a coarse-grained model of a triangular spring network.39 To introduce the intrinsic disorder of a real hydrogel, we randomly delete a fraction of the bonds in this network. The cell can mechanically interact with the network and move between lattice nodes.To develop a simple description of how the cell interacts with the polymer meshwork, we briefly summarise the key aspects of cell migration in such environments. In general, cells adhere to and contract the matrix, which allows the cell to mechanically probe its surroundings and generate a force to move the cell body as a whole. Cells typically move from the soft side of a substrate towards stiffer regions – a phenomenon called durotaxis.6 Furthermore, to move through a dense 3D network, cells have to digest the matrix using proteinases. Interestingly, experiments have revealed that cells do not digest the matrix where cell adhesion and force-generation occurs.40,41 Instead, matrix proteolysis is locally separated from force generation and is mostly localised behind the leading edge and near the cell body for HT-1080 cells in collagen networks,40 as illustrated schematically in Fig. 3A.
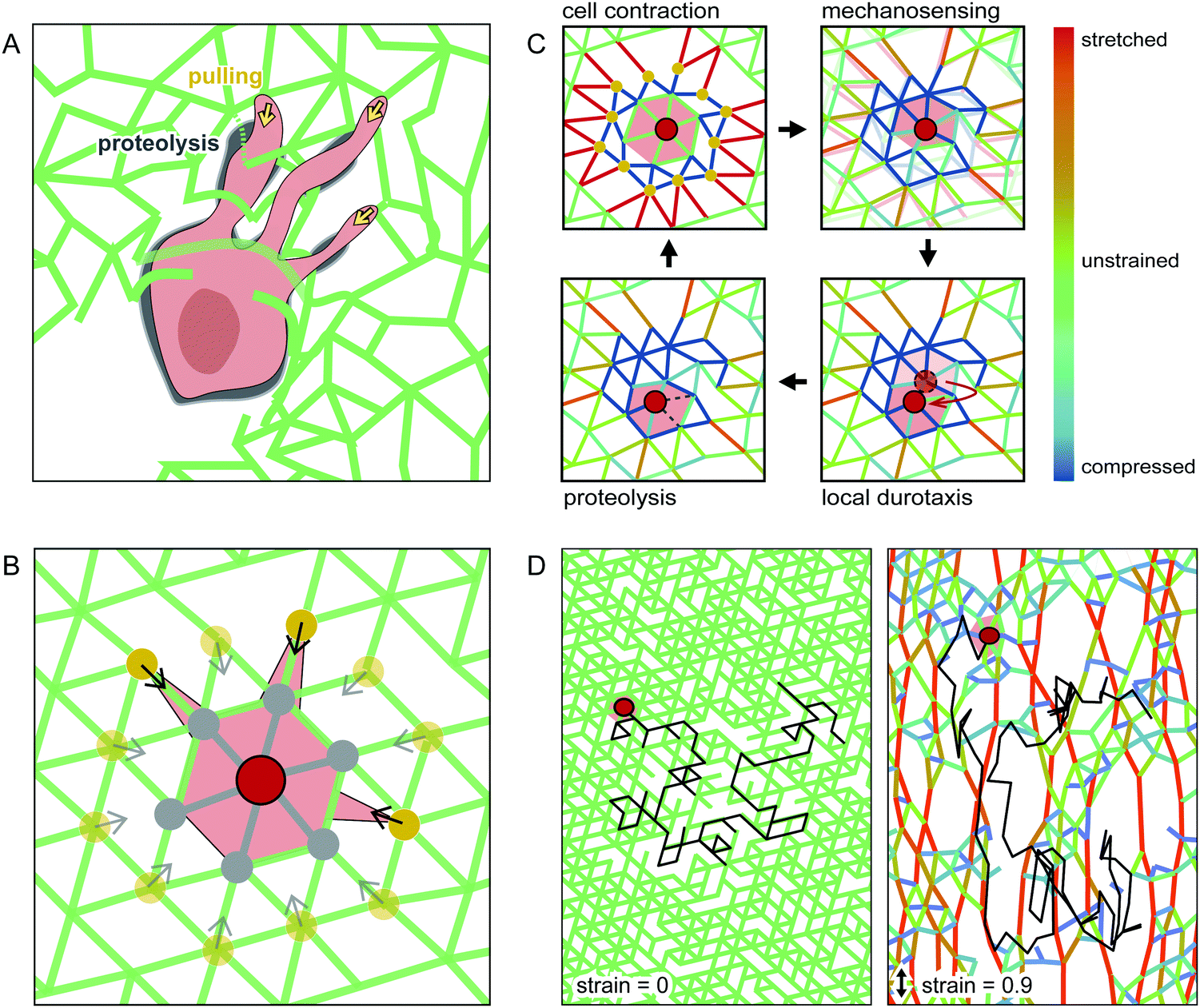 | ||
| Fig. 3 Schematic of our computational model. (A) Illustration of a contractile cell in an inhomogeneous network, with adhesions to the matrix at the cell front. Proteolysis is located slightly behind the leading edge and close to the cell body. (B) Cell centre (red dot) on a triangular spring lattice interacts with its second neighbours (yellow dots) by pulling on them. In every computational iteration, bonds to its first neighbours (grey dots) are cleaved. (C) Illustration of the cell migration cycle assumed for our model. (D) Example trajectory (black line) of a simulated cell moving in an unstrained (left) and a strained network (right). | ||
To capture these aspects of proteolytic cell migration, we propose a minimal model with the following steps: (i) Contraction: the cell pulls on the nearby lattice nodes in the network (yellow dots in Fig. 3B), thereby deforming the network. (ii) Mechanosensing: we calculate the local stiffness of the deformed nodes on which the cell pulls. (iii) Local durotaxis: the cell centre moves to the neighbouring node (shown in grey in Fig. 3B) with the highest local stiffness. (iv) Proteolysis: to capture MMP activity, we allow the cell to digest lattice bonds at a fixed rate. Importantly, this MMP activity only acts locally. Therefore, only bonds near the cell body can be cleaved by the cell (marked in grey in Fig. 3B). By repeating these four basic steps of this cell migration cycle (Fig. 3C), we simulate cell movement on a 2D lattice. For simplicity, we do not include cell polarization in our migration model (see ESI† for a model extension with time-averaged mechanosensing as polarization factor). To model externally deformed matrices, we stretch the spring network uniaxially up to a given strain under fixed boundary conditions before cell migration is simulated. Examples of simulated cell trajectories in strained and unstrained networks are shown in Fig. 3D and ESI,† Movies S7–S9.
Our simple model enables us to simulate proteolytic cell migration on a 2D elastic lattice. To verify our model, we first compare the migratory behaviour of cells in our simulation with experimental data in isotropic networks. In both the model and our experiments, we observe that the mean squared displacement (MSD) increases with time as an approximate power law with an exponent of roughly 1.4–1.8 (Fig. 4A). This dependence indicates super-diffusive behaviour. Interestingly, if we increase the proteolysis rate in the model, the exponent of the apparent power law increases. This suggests that the migration of the cell becomes more persistent with increasing proteolytic activity (see ESI,† Fig. S7 for MSD comparison in strained systems). To further quantify the statistics of cell migration, we determine the velocity autocorrelation function (VACF) of the migration velocity. In our experiments, we find that the VACF decays with a characteristic time that appears to decrease only weakly with the degree of degradability of the hydrogels. Note, the simulated VACF is qualitatively similar to the experimental result even though we did not include cell polarization in this model. Cell polarization would imply an intrinsic persistence time for the migratory behaviour. By contrast, the persistence of cell migration in our model is an emergent phenomenon, which derives from the dilution of the network due to proteolytic digestion. Cells digest the network, which renders the local stiffness softer in the wake of the cell. As a result, durotactic cells tend to move away from their previous locations giving rise to persistent motion. For all further simulations, we set the proteolytic rate rp in the model to 1, for which we observe a reasonable agreement with the experimental migration statistics of cells in isotropic networks.
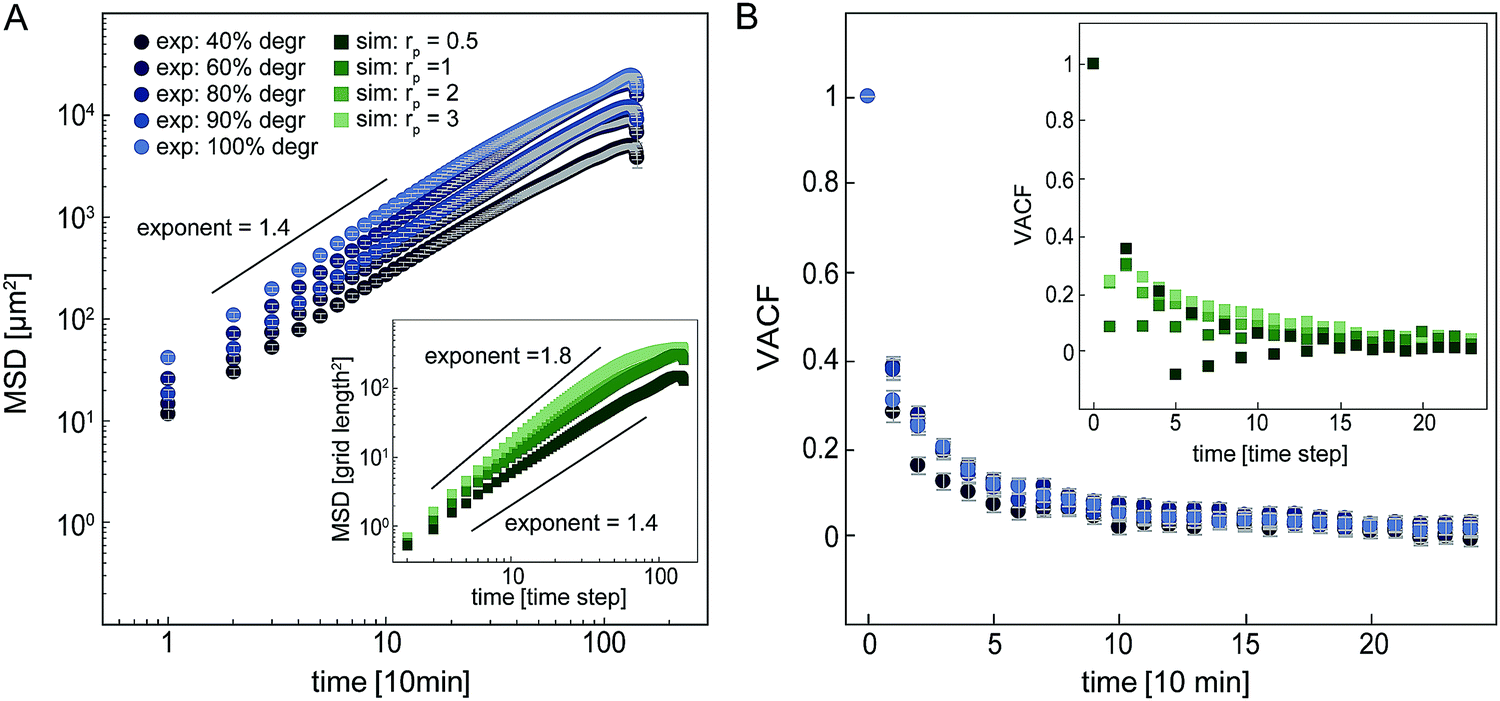 | ||
| Fig. 4 Analysis of cell migration in unstrained networks with varying degradability in experiments and different proteolytic activities in the simulations. (A) Mean squared displacement (MSD). Experimental data displayed with circles, simulated data in inset displayed as squares. (B) Normalized velocity autocorrelation function (VACF). Experimental data is displayed with circles, simulated data in the insets is displayed as squares. | ||
Non-monotonic response of cell migration to external strain can be explained by anisotropic geometric strain-stiffening on the microscale
After verifying our simple migration model in isotropic systems, we next sought to investigate cell migration in uniaxially strained networks by comparing simulated and experimental cell trajectories. To quantify the degree of anisotropy of the cell trajectories, we calculate the Anisotropic Migration Index (AMI), by comparing the total distance travelled by cells parallel, D‖, and perpendicular, D⊥, to the main strain direction,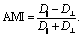 | (2) |
Experimentally, we observe that the AMI of cells in isotropically swollen 2 mM PEG-NB gels is close to zero, indicating isotropic migration (Fig. 5A). As the strain is increased, the AMI increases to values as high as 0.6, indicating highly anisotropic cell migration along the strain direction. However, when the strain is increased further, the migration of cells becomes more isotropic again, as was already suggested by the raw trajectories in Fig. 2E. This non-monotonic response of the anisotropy of cell migration to an increasing strain is surprising and suggests that the strain triggers an additional mechanism in the matrix that guides cell migration depending on the strain magnitude.
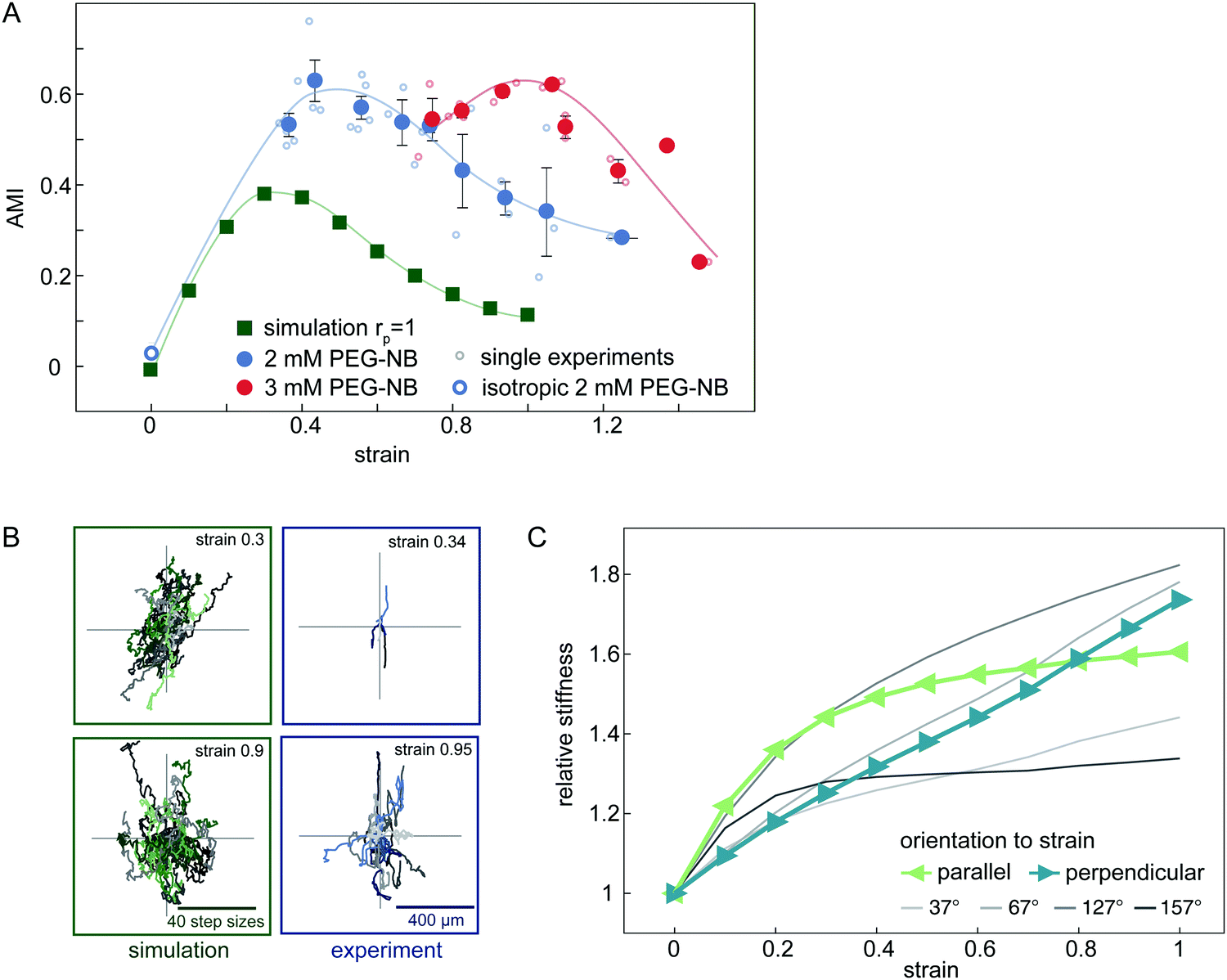 | ||
| Fig. 5 Analysis of the anisotropic migration in uniaxially strained networks. (A) Anisotropic Migration Index (AMI) of experimental and simulated data. Simulated data is shown by green squares. Each single experiment is displayed by small circles and binned data by filled circles. (2 mM PEG-NB gels in blue, 3 mM PEG-NB gels in red). Error bars are standard error of the mean. (B) Centred trajectories of simulated and experimental data for strains of 0.9 and 0.3. (C) Local relative network stiffness on the cellular scale in different orientations to the strain direction in a simulated network. Directions parallel and perpendicular to the strain are depicted in green and blue respectively. | ||
In our model, an externally applied strain similarly leads to an anisotropic migration oriented preferentially parallel to the strain. Furthermore, we also observe a non-monotonic relationship between anisotropic migration and strain amplitude in the simulations, in accord with our experimental results. However, the overall anisotropy and corresponding AMI values are smaller in the simulated data (Fig. 5A). Nonetheless, our model is able to qualitatively capture the non-monotonic anisotropic migration response of cells migrating in deformed hydrogels. Furthermore, by including a simple cell polarization mechanism in the migration model, we can quantitatively reproduce the maximal AMI observed in our experiments (ESI,† Fig. S9).
To understand the origins of the non-monotonic dependence of the anisotropic cell migration on external strain, we use our model to investigate the local matrix stiffness. Recall, in our model the cell performs local durotactic migration and is therefore guided by local stiffness differences in the matrix, always moving in the direction of highest local stiffness. To investigate the local “stiffness landscape”, we analyse the node stiffness in different orientations relative to the external strain direction. Even though the springs in our network model are linear, we observe that the local stiffness depends on the external strain, but in a distinct way for different orientations (Fig. 5C). This stiffness is measured before cell migration in the network is simulated, therefore representing an intrinsic matrix property (see also ESI,† Fig. S8).
The initial local matrix stiffness perpendicular to the deformation axis increases linearly with strain amplitude (Fig. 5C). This perpendicular stiffening is a direct result of the tension in the springs along the strained directions. Conceptually, this is a simple geometric effect similar to the greater stiffness experienced when plucking a string under increasing tension. By contrast, the local stiffness parallel to the deformation axis exhibits a fast initial stiffening, followed by saturation to a constant value at higher strains. This stiffening mechanism has previously been observed for the macroscopic response of the network.42–44 Briefly, intrinsic heterogeneities in the network with a reduced local connectivity introduce softer elastic modes in the system, which get pulled out by the macroscopic strain. Thus, even though the springs that describe the network elasticity are linear, geometric effects induce stiffening of the local environment. In addition, the mean node stiffness measured by cells, which migrate through strained networks is also affected by the proteolytic digestion of the matrix by the cell (see ESI,† Fig. S8). The nonlinear effects described above may thus be enhanced by the proteolytic digestion of matrix bonds, which lowers the local network connectivity and thus introduces heterogeneity.
Geometric stiffening effects lead to a local anisotropy in network stiffness that depends non-monotonously on the strain magnitude. Because of this anisotropic stiffening, we expect that at small strains a durotactic cell will migrate preferentially parallel to the deformation axis, where the node stiffness is highest, while at higher strain values, the cell will tend to steer away from the deformation axis. Thus, the local orientation-dependent stiffening of the matrix can account for the non-monotonic behaviour of the anisotropic cell migration in gels with increasing strain.
Discussion
Here we use a hydrogel system34,45 to investigate how uniaxial deformations in synthetic hydrogels influence the migration of embedded HT-1080 cells. In this system, cell migration is dependent on the proteolytic digestion of matrix cross-links, because the mesh size of the gel is on the order of tens of nanometres, considerably smaller than the cell diameter.19 We confirmed the importance of proteolytic activity by showing that cell migration is hampered when the fraction of degradable cross-links in the gel is too low (Fig. 1C). The linear elastic properties of the PEG-based matrix, as well as the defined composition and adjustability of the matrix properties provide a simplified, controllable environment for embedded cells.46,47 Defined matrix properties enable the detection of fundamental guidance principles in such systems.48,49 By using uniaxially swollen PEG-based hydrogels and our minimal cell migration model, we showed that HT-1080 cells preferentially migrate parallel to the main strain direction with the degree of anisotropy of migration depending non-monotonically on the strain magnitude.To analyse cell migration in deformed matrices, we established a new set-up to induce strain in synthetic, photo-polymerizable hydrogels by microstructuring strips into channels. These hydrogels are confined in the z-direction of the channel. Because of this axial confinement and the high aspect ratio of the strips, the inherent swelling of the hydrogel only occurs in the direction of the short axis of the strip. Since the strain is induced by this uniaxial swelling process and not through mechanical stretching, no compression in the direction perpendicular to the strain occurs. The resulting uniaxial strain field offers an advantage over other straining devices where a mechanical stretch often results in complex strain fields, complicating the interpretation of experimental results.23,24,50,51 Another advantage of our system is the excellent optical accessibility due to the use of commercially available channel slides of high optical quality. However, a draw-back of our system is that we regulate the degree of swelling and therefore the strain of our system by changing the cross-linker amount in the matrix (see Fig. 2). Thus, both the rigidity and the mesh size of the gel differ for different deformations generated in the matrix.
Several studies have analysed how cells respond to external strains in the matrix, depending on the temporal and spatial properties of the imposed strain as well as the rigidity and composition of the matrix.11,24,26,31 However, the underlying mechanism for the observed cell alignment remains unclear.24,31 In naturally derived gels such as collagen or Matrigel, the strong strain stiffening response of these non-linear elastic materials can generate a macroscopic stiffness anisotropy in the gels.52,53 Furthermore, multiaxial rheological experiments revealed that under strain, biopolymer matrices not only stiffen when strained but also show a softening in the compressed direction.54 Such stiffness anisotropies can provide durotactic cues to cells, which together with the alignment of fibres in strained collagen networks has been suggested as possible mechanism to explain the preferential orientation of cell trajectories along the main strain direction.28,55 However, cell alignment was also observed in synthetic hydrogels that show linear macroscopic elastic properties when stretched.6,31 To explain the cell alignment in direction of the external strain, a theoretical model was introduced,56 showing the alignment of static cells to the main strain direction, but the actual proteolytic migration of cells in deformed networks was not considered in this model.
Our experimental analysis of cell migration directionality in strained matrices shows a preferred migration of HT-1080 cells parallel to the external strain direction. Importantly, this preferred migration along the deformation axes shows a non-monotonic dependence on the strain magnitude, with the strongest alignment at intermediate strain levels. To further elucidate cell behaviour in strained matrices, we developed a minimal model of a cell migrating in a strained network. In our model, the cell is assumed to migrate in the direction of highest local stiffness and randomly dilutes cross-links locally around the cell body. The modelled cell migration captures our experimental observation: cells preferentially migrate along the deformation axis, but the degree of alignment along this axis depends non-monotonically on the strain magnitude. Furthermore, our model reveals a mechanism that gives rise to such a non-monotonic dependence: the network of linear springs in our model locally stiffens due to the network strain, but this nonlinear effect is itself anisotropic. Indeed, the stiffness of the network nodes probed in different orientations to the strain direction depend in different ways on the strain magnitude (Fig. 5C). This nonlinear anisotropy can therefore account for the non-monotonicity of the cell migration behaviour with applied strain (Fig. 5A).
The overall anisotropy of the migration directionality in the model is smaller than our experimental results. This small quantitative discrepancy could arise because we neglect the effects of cell polarization in our model. Nonetheless, cell polarization will not affect the magnitude of migration anisotropy, unless the polarization itself is strain or stiffness sensitive. Indeed, if cells migrate more persistently along the stiffer, strained direction this would result in higher values of the AMI. We explored this idea by considering an extension of our model where mechanosensing is performed based on time-averaging local stiffness measurements. In this extended model, the cell performs durotactic migration that is not only based on the current stiffness gradient, but also on a limited number of previously encountered gradients. Such a sensing memory can increase the maximal anisotropic migration in our model, achieving a better quantitative agreement with the observed experimental AMI values (see ESI,† Fig. S9). Furthermore, the proteolysis step in our model is assumed to be random and thus does not depend on the amount of strain applied to a cross-link. In experiments, however, the susceptibility of strained collagen fibrils to digestion by MMPs or other collagenases was observed to be influenced by applied strains.25,57–59 Therefore, the strain in our experiments may affect matrix proteolysis as well as mesh size differences in different directions of the matrix, thereby favouring the migration parallel to the external strain and leading to higher AMI values. Thus, an important future direction is to understand how these factors compete with the nonlinear anisotropic effects in guiding cell migration in strained 3D environments.
Conclusion
By combining an experimental approach to study cell migration within a reduced 3D matrix with theoretical modeling, we have shown that cells in uniaxially strained hydrogels migrate preferentially parallel to the external strain direction, but with the degree of alignment depending non-monotonically on the strain magnitude. The non-monotonicity of the Anisotropic Migration Index (AMI) can be explained by a model of durotactic cell migration, in which the local anisotropic geometric stiffening of the matrix guides cell migration. Multiple studies on naturally derived gels, such as collagen and Matrigel, have suggested that stiffness acts as a guidance cue for cell migration in strained 2D and 3D matrices.52,53 Here, we propose that local strain stiffening also occurs in cross-linked synthetic hydrogels, and that the resulting stiffness anisotropies in the matrix influence cell migration directionality. Such local changes of the mechanical properties of synthetic hydrogels should therefore also be considered when using similar hydrogels in implants or as tissue substitutes. Indeed, the non-monotonic local stiffening of the network indicated by our model may act as a regulator for cell migration directionality in synthetic hydrogels.Materials and methods
Cell culture
HT-1080 cells (DSMZ) are cultured in normal growth medium consisting of Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% foetal bovine serum (Sigma). For experiments 1% penicillin/streptomycin (Sigma) is added to the normal culture medium. HT-1080 LifeAct-TagGFP2 cells (ibidi) are cultured in normal growth medium with addition of 0.75 mg ml−1 Geneticin (Gibco) as selective antibiotic to maintain transgene expression. All cultures are incubated at 37 °C and 5% CO2.Preparation of the pre-polymer solution
Pre-polymer solution is prepared in PBS containing 2–3 mM 20 kDa 4-armed PEG-norbornene (PEG-NB, JenKem Technology), an off-stoichiometric amount of dithiol-containing, MMP-degradable cross-linking peptide (KCGPQGIWGQCK, Iris Biotech), 1 mM CRGDS-peptide (Iris Biotech) and 3 mM of the photo-initiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, synthesized as previously described35,60). To decrease the degradability of the network, parts of the peptide cross-linker are substituted by a 1 kDa PEG chain that contains a thiol group at each end (PEG-dithiol, Sigma). To encapsulate cells in the gel, HT-1080 cells, suspended in PBS, are added to the pre-polymer solution at a final concentration of 6.7 × 105 cells per ml.To tune the gel composition, we can vary the amount of PEG-NB monomer, as well as the amount of cross-linker. The cross-linker ratio rc is defined according to eqn (3), by comparing the ratio of functional groups of the cross-linker (two thiol groups in each cross-linker) to the concentration of functional groups of the PEG-NB monomer (4 norbornene groups on each monomer) in the pre-polymer solution.
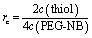 | (3) |
Preparation of freely swollen hydrogel
Small amounts of pre-polymer solution and air are alternately aspirated with a pipette and injected into a silicon tubing (Tygon) with an inner diameter of 1.6 μm to form small gel slabs. The tube is illuminated with a collimated 365 nm LED light source (Rapp OptoElectronic) of 10 mW cm−2 for 30 s. The polymerized gels are pushed out of the tube into normal growth media by air pressure. They float in cell culture media and are allowed to swell for 2 h under standard conditions (37 °C, 5% CO2, 100% humidity).To enable long-time microscopic observation of cell migration inside the gel, without displacement of the gel itself, the hydrogel has to be fixed to a surface after swelling. Therefore, the bottom of an uncoated μ-slide angiogenesis (ibidi) is functionalized with PEG-NB. A mixture of 5 mM PEG-NB with 3 mM of the photo-initiator 4-benzoyl-benzylamine hydrochloride (Fluorochem) is illuminated through the slide bottom with 302 nm light (Blak-Ray XX-15M, UVP) for 30 min. After washing the surface with PBS, a mixture of 20 mM PEG-dithiol and 10 mM LAP was illuminated with 365 nm light for 20 s, which yields a thiol presenting surface. After washing with PBS, 1 μl of 0.5 mM LAP in PBS is added to the functionalized surface and a gel slab is placed on top of the droplet. Illumination with 365 nm for 5 s covalently binds the gel to the surface. The wells are washed with culture media after illumination.
High resolution microscopy
To retrieve high resolution images of HT-1080 cells embedded in synthetic hydrogel, gel slabs are prepared as described in the Section “Preparation of freely swollen hydrogel” with one exception. Wild type HT-1080 cells are substituted by HT-1080 LifeAct-TagGFP2 to visualize the actin structure of the cells. A Zeiss Cell Observer SD equipped with a Zeiss Plan Apochromat 63× oil objective is used for spinning disc confocal microscopy. While imaging, samples are kept at 37 °C and 5% CO2 atmosphere. z-Stacks with a distance of 1 μm are recorded over the whole cell body height and projected with the image processing program ImageJ (ImageJ 1.50b) to a single image using the Max projection option.Preparation of hydrogel microstructures inside channel slides
To microstructure hydrogels in confinement, pre-polymer solution containing HT-1080 cells is injected into the channels of a μ-slide VI 0.4 uncoated (ibidi) and illuminated at 10 mW cm−2 for 20 s with collimated 365 nm light through a custom-made chrome mask (structures: 400 μm strips width, 600 μm spacing, 5 mm strip length, channel height 400 μm). After polymerization, the channels are washed with culture medium and incubated under standard conditions (37 °C, 5% CO2, 100% humidity).Particle image velocimetry analysis to visualize the swelling behaviour
Hydrogel strips are polymerized as described in the Section “Preparation of hydrogel microstructures inside channel slides”. However, in addition fluorescent latex beads with a diameter of 1.1 μm (Sigma) are added to the pre-polymer solution at a final concentration of 9 × 108 beads per ml. Directly after illumination, slides are mounted on a Nikon Eclipse Ti-E inverted microscope. Channels are washed with PBS and time-lapse imaging with 2 min interval for 3 h is started directly afterwards. Particle image velocimetry analysis (PIV) of the data is performed with the MatPIV toolbox for MatLab (J Kristian Sveen: http://folk.uio.no/jks/matpiv/, GNU general public license) with a slightly customized script. Changes in the script include a smallest interrogation window size of 64 × 64 pixels with a 50% overlap, a filtering process with signal-to-noise ratio filter, a global histogram operator and a local filter. All vectors which are removed by the filtering process are replaced by a linear interpolation from the neighbouring vectors if at least 5 surrounding vectors remain. Setting this minimal limit ensures a localization of the vector field inside the gel strip and prevents the propagation of the field beyond the strip edges.Measurement of the swelling strain
Hydrogel microstructures of various composition are prepared as described in the paragraph “Preparation of hydrogel microstructures inside channel slides”. Completely swollen gels with embedded cells are imaged 3 h after polymerization on an Olympus CKX41 inverted microscope equipped with a gas incubation and heating system (ibidi) to maintain standard incubation conditions while imaging. To determine the swelling strain, γs, the strip width in the middle of the longitudinal section of the structure is measured with ImageJ. The swelling strain is defined by comparing the structure width after swelling (Wf) with the initial structure width (W0), according to eqn (1). The initial structure width after polymerization is 400 μm. Results are displayed as mean value with standard deviation.Migration studies of HT-1080 cells inside gels
Hydrogel strips or openly swollen gel slabs are prepared as described in the paragraph “Preparation of hydrogel microstructures inside channel slides” or “Preparation of freely swollen hydrogel”, respectively. The strips are imaged on an Olympus CKX41 inverted microscope and a Nikon Eclipse Ti-E inverted microscope, respectively. Both are equipped with a gas incubation and heating system (ibidi) to maintain standard incubation conditions. The medium in the reservoirs of the μ-slide VI 0.4 is overlaid with Anti-Evaporation Oil (ibidi) as described in the slide instructions, to avoid medium evaporation. Time-lapse series with 10 min intervals for 24 h are recorded starting 3 h after polymerization. Cells are tracked with the ImageJ plug-in ‘Manual Tracking’. Cells which migrated a distance smaller than 40 μm are considered non-migrating. For each condition 3–5 biological replicates are performed with 2–3 positions each. 25 cells per position are randomly selected for analysis. For the hydrogel slabs, a static structure in the gel is tracked for every position to correct cell migration tracks, due to slight overall movements of the gel.Analysis of the cell migration behaviour in isotropic hydrogels
To verify the theoretical model, an analysis of the basic migration parameters is performed for the experimental and simulated data of cells moving in an isotropic network. The autocorrelation function of the cell migration velocity (VACF) is given byVACF(t) = 〈![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0076_20d1.gif) (τ + t)·(τ)〉τ, (τ + t)·(τ)〉τ, | (4) |
(t) are the velocity vectors of a cell at times t, and the brackets indicate a time average at fixed τ. The Mean Squared Displacement (MSD) is calculated usingMSD(t) = 〈(![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) (τ + t) − (τ))2〉τ, (τ + t) − (τ))2〉τ, | (5) |
(t) are the position of the cell at time t, and the brackets indicate a time average at fixed τ.
Quantification of the anisotropic cell migration
For each tracked cell the coordinates at every time point are recorded (xt, yt). The direction of uniaxial strain in the hydrogel strips, as well as in our simulations, is parallel to the y-direction. The cumulated covered distance (D⊥ and D‖, respectively) is calculated separately for the x and y direction according to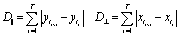 | (6) |
For simulated data, the displacement parallel to the strain is corrected by the applied strain γ (eqn (7)) before the AMI is calculated. Without correcting for the changed node to node distance upon deformation of the modelled system, the retrieved AMI would be biased towards positive values.
| D‖,simulated = D‖,measured/(γ + 1) | (7) |
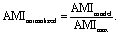 | (8) |
Theoretical modelling and local stiffness calculations
A description of the theoretical model can be found in the ESI,† together with the calculation of the local node stiffness in the simulated network.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. D. thanks Lea Tomášová for useful discussions and Daniel Rüdiger for his help with bulk stiffness measurements. This work was supported by grants from the German Science Foundation (DFG) through the Collaborative Research Center (SFB) 1032 (Projects B01, B11 and B12). D. B. B. is supported by a DFG fellowship within the Graduate School of Quantitative Biosciences Munich (QBM).References
- D. E. Koser, A. J. Thompson, S. K. Foster, A. Dwivedy, E. K. Pillai, G. K. Sheridan, H. Svoboda, M. Viana, L. da, F. Costa, J. Guck, C. E. Holt and K. Franze, Nat. Neurosci., 2016, 19, 1592–1598 CrossRef CAS PubMed.
- H. Wiig and M. a Swartz, Physiol. Rev., 2012, 92, 1005–1060 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- J. C. Friedland, M. H. Lee and D. Boettinger, Science, 2009, 323, 642–644 CrossRef CAS PubMed.
- B. Geiger, J. P. Spatz and A. D. Bershadsky, Nat. Rev. Mol. Cell Biol., 2009, 10, 21–33 CrossRef CAS PubMed.
- C. M. Lo, H. B. Wang, M. Dembo and Y. L. Wang, Biophys. J., 2000, 79, 144–152 CrossRef CAS PubMed.
- E. Hadjipanayi, V. Mudera and R. a. Brown, Cell Motil. Cytoskeleton, 2009, 66, 121–128 CrossRef CAS PubMed.
- P. P. Roca-Cusachs, T. T. Iskratsch and M. P. M. P. Sheetz, J. Cell Sci., 2012, 125, 3025–3038 CrossRef CAS PubMed.
- F. Beroz, L. M. Jawerth, S. Münster, D. A. Weitz, C. P. Broedersz and N. S. Wingreen, Nat. Commun., 2017, 8, 16096 CrossRef CAS PubMed.
- J. C. Hoffmann and J. L. West, Integr. Biol., 2013, 5, 817–827 RSC.
- C. Sears and R. Kaunas, J. Biomech., 2016, 49, 1347–1354 CrossRef PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- J. L. Ifkovits and J. a. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125–148 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- G. H. Underhill, A. a. Chen, D. R. Albrecht and S. N. Bhatia, Biomaterials, 2007, 28, 256–270 CrossRef CAS PubMed.
- E. H. Nguyen, M. R. Zanotelli, M. P. Schwartz and W. L. Murphy, Biomaterials, 2014, 35, 2149–2161 CrossRef CAS PubMed.
- G. P. Raeber, M. P. Lutolf and J. a. Hubbell, Biophys. J., 2005, 89, 1374–1388 CrossRef CAS PubMed.
- M. P. Schwartz, B. D. Fairbanks, R. E. Rogers, R. Rangarajan, M. H. Zaman and K. S. Anseth, Integr. Biol., 2010, 2, 32–40 RSC.
- A. M. Kloxin, C. J. Kloxin, C. N. Bowman and K. S. Anseth, Adv. Mater., 2010, 22, 3484–3494 CrossRef CAS PubMed.
- A. Tondon and R. Kaunas, PLoS One, 2014, 9, e89592 Search PubMed.
- L. Wang, Y. Li, B. Chen, S. Liu, M. Li, L. Zheng, P. Wang, T. J. Lu and F. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 15088–15097 CAS.
- Y. Li, G. Huang, M. Li, L. Wang, E. L. Elson, T. J. Lu, G. M. Genin and F. Xu, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- S.-W. Chang, B. P. Flynn, J. W. Ruberti and M. J. Buehler, Biomaterials, 2012, 33, 3852–3859 CrossRef CAS PubMed.
- M. Eastwood, V. C. Mudera, D. A. Mcgrouther and R. A. Brown, Cell Motil. Cytoskeleton, 1998, 40, 13–21 CrossRef CAS PubMed.
- T. K. Tonge, J. W. Ruberti and T. D. Nguyen, Biophys. J., 2015, 109, 2689–2700 CrossRef CAS PubMed.
- D. Vader, A. Kabla, D. Weitz and L. Mahadevan, PLoS One, 2009, 4, e5902 Search PubMed.
- M. P. Rubbens, A. Driessen-Mol, R. A. Boerboom, M. M. J. Koppert, H. C. Van Assen, B. M. Terhaar Romeny, F. P. T. Baaijens and C. V. C. Bouten, Ann. Biomed. Eng., 2009, 37, 1263–1272 CrossRef PubMed.
- Q. Wen and P. A. Janmey, Exp. Cell Res., 2013, 319, 2481–2489 CrossRef CAS PubMed.
- J. Raeber, J. A. Lutolf and G. P. Hubbell, Biomech. Model. Mechanobiol., 2008, 7, 215–225 CrossRef PubMed.
- E. K. U. Larsen, M. B. L. Mikkelsen and N. B. Larsen, Biomicrofluidics, 2014, 8, 64127 CrossRef PubMed.
- S. P. Singh, M. P. Schwartz, J. Y. Lee, B. D. Fairbanks and K. S. Anseth, Biomater. Sci., 2014, 2, 1024–1034 RSC.
- M. P. Schwartz, R. E. Rogers, S. P. Singh, J. Y. Lee, S. G. Loveland, J. T. Koepsel, E. S. Witze, S. I. Montanez-Sauri, K. E. Sung, E. Y. Tokuda, Y. Sharma, L. M. Everhart, E. H. Nguyen, M. H. Zaman, D. J. Beebe, N. G. Ahn, W. L. Murphy and K. S. Anseth, PLoS One, 2013, 8, e81689 Search PubMed.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- C.-C. Lin, C. S. Ki and H. Shih, J. Appl. Polym. Sci., 2015, 132 DOI:10.1002/app.41563.
- C. M. Denais, R. M. Gilbert, P. Isermann, A. L. Mcgregor, M. Lindert, B. Weigelin, P. M. Davidson, P. Friedl and J. Lammerding, Science, 2016, 352, 353–358 CrossRef CAS PubMed.
- J. Irianto, C. R. Pfeifer, R. R. Bennett, Y. Xia, I. L. Ivanovska, A. J. Liu, R. A. Greenberg and D. E. Discher, Mol. Biol. Cell, 2016, 27, 4011–4020 CrossRef CAS PubMed.
- C. P. Broedersz, X. Mao, T. C. Lubensky and F. C. MacKintosh, Nat. Phys., 2011, 7, 983–988 CrossRef CAS.
- K. Wolf and P. Friedl, Clin. Exp. Metastasis, 2009, 26, 289–298 CrossRef CAS PubMed.
- K. M. Schultz, K. A. Kyburz and K. S. Anseth, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3757–E3764 CrossRef CAS PubMed.
- M. Wyart, H. Liang, A. Kabla and L. Mahadevan, Phys. Rev. Lett., 2008, 101, 215501 CrossRef CAS PubMed.
- M. Sheinman, C. P. Broedersz and F. C. MacKintosh, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 21801 CrossRef CAS PubMed.
- C. P. Broedersz and F. C. Mackintosh, Rev. Mod. Phys., 2014, 86, 995–1036 CrossRef CAS.
- K. a Kyburz and K. S. Anseth, Acta Biomater., 2013, 9, 6381–6392 CrossRef CAS PubMed.
- K. M. Schultz and K. S. Anseth, Soft Matter, 2013, 9, 1570–1579 RSC.
- A. Raza and Ch.-C. Lin, Macromol. Biosci., 2013, 8, 1048–1058 CrossRef PubMed.
- T. D. Hansen, J. T. Koepsel, N. N. Le, E. H. Nguyen, S. Zorn, M. Parlato, S. G. Loveland, M. P. Schwartz and W. L. Murphy, Biomater. Sci., 2014, 2, 745–756 RSC.
- E. Y. Tokuda, J. L. Leight and K. S. Anseth, Biomaterials, 2014, 35, 4310–4318 CrossRef CAS PubMed.
- J. Foolen, V. S. Deshpande, F. M. W. Kanters and F. P. T. Baaijens, Biomaterials, 2012, 33, 7508–7518 CrossRef CAS PubMed.
- J. Raeber, J. A. Mayer and G. P. Hubbell, Biomech. Model. Mechanobiol., 2008, 7, 203–214 CrossRef PubMed.
- C. Storm, J. J. Pastore, F. MacKintosh, T. Lubensky and P. A. Jamney, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- J. P. Winer, S. Oake and P. A. Janmey, PLoS One, 2009, 4, e6382 Search PubMed.
- A. S. G. Van Oosten, M. Vahabi, A. J. Licup, A. Sharma, P. A. Galie, F. C. MacKintosh and P. A. Janmey, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- R. J. Klebe, H. Caldwell and S. Milam, Matrix, 1990, 9, 451–458 CrossRef.
- I. B. Bischofs and U. S. Schwarz, PNAS, 2003, 100, 9274–9279 CrossRef CAS PubMed.
- A. S. Adhikari, J. Chai and A. R. Dunn, J. Am. Chem. Soc., 2011, 133, 1686–1689 CrossRef CAS PubMed.
- A. S. Adhikari, E. Glassey and A. R. Dunn, J. Am. Chem. Soc., 2012, 134, 13259–13265 CrossRef CAS PubMed.
- J. W. Ruberti and N. J. Hallab, Biochem. Biophys. Res. Commun., 2005, 336, 483–489 CrossRef CAS PubMed.
- T. Majima and W. Schnabel, Makromol. Chem., 1991, 192, 2307–2315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm00018b |
| This journal is © The Royal Society of Chemistry 2018 |