DOI:
10.1039/C7SM02347B
(Paper)
Soft Matter, 2018,
14, 1375-1388
Self-diffusiophoresis induced by fluid interfaces
Received
29th November 2017
, Accepted 18th January 2018
First published on 18th January 2018
Abstract
The influence of a fluid–fluid interface on self-phoresis of chemically active, axially symmetric, spherical colloids is analyzed. Distinct from the studies of self-phoresis for colloids trapped at fluid interfaces or in the vicinity of hard walls, here we focus on the issue of self-phoresis close to a fluid–fluid interface. In order to provide physically intuitive results highlighting the role played by the interface, the analysis is carried out for the case that the symmetry axis of the colloid is normal to the interface; moreover, thermal fluctuations are not taken into account. Similarly to what has been observed near hard walls, we find that such colloids can be set into motion even if their whole surface is homogeneously active. This is due to the anisotropy along the direction normal to the interface owing to the partitioning by diffusion, among the coexisting fluid phases, of the product of the chemical reaction taking place at the colloid surface. Different from results corresponding to hard walls, in the case of a fluid interface the direction of motion, i.e., towards the interface or away from it, can be controlled by tuning the physical properties of one of the two fluid phases. This effect is analyzed qualitatively and quantitatively, both by resorting to a far-field approximation and via an exact, analytical calculation which provides the means for a critical assessment of the approximate analysis.
1 Introduction
The active motion of micro- and nano-sized particles has gained significant interest (see recent reviews such as ref. 1–4) due to a wide, associated spectrum of applications including drug-delivery systems5–7 as well as due to the ability to mimic motile biological cells such as bacteria.8 In view of these perspectives diverse approaches aiming at the understanding of active motion of micro- and nano-sized particles have been put forward; a detailed account of them can be found in ref. 2. A particularly promising approach is the case of self-phoretic particles,9–12 which attain net motion by means of chemical reactions catalyzed on parts of their surface. Indeed, if the rate of the chemical reaction on the surface of such particles is distributed inhomogeneously, local density gradients in the concentration of reactants and products lead to imbalances in the local chemical potential and pressure which eventually, by generating fluid velocity profiles, set the particle into motion. Several theoretical10,12–20 as well as experimental2,9,11,21–24 studies have characterized the performance of such systems. Recently it has been shown that the dynamics of Janus particles (i.e., catalytic particles with the catalyst distributed only across a part of their surface) is quite sensitive to the presence of boundaries, obstacles, or other means which can distort the density profiles of the reaction products and the local velocity profiles, hence inducing a modulation of the net displacement of such kind of particles. Resembling the well-documented “wall-attraction” for micro-organisms and mechanical swimmers (see, e.g., ref. 25–29), for self-phoretic particles it has been shown that, in addition to the modulation of the velocity,14,30,31 wall-bounded steady states emerge from the interplay of hydrodynamic and phoretic interactions with the wall (i.e., via the wall-induced distortions of the hydrodynamic flow and of the distribution of the chemical species).30,32–34 This can be exploited to achieve a guided motion of the active colloid.35–37 The emergence of similar effective interactions between pairs of active colloids has been studied theoretically in ref. 15 and 38–40. Recently, experimental24,41 and theoretical42–47 studies have started to tackle the issue of motion of active colloids near or trapped at a liquid-fluid interface. With respect to the single particle motion, a certain increase in the persistence length of the self-phoretic motion has been reported for particles trapped at the interface.24,41,45 Instabilities of interfaces covered by active particle, which can be considered themselves as surfactants, have been reported in ref. 44. Alternatively, the motion of active particles which induce Marangoni stresses at the interface via the chemical species or heat they release (or absorb) has been studied both in terms of the emergence of motion for a single particle42,43,46 as well as concerning the issue of the dynamics and stability of monolayers of such particles trapped at the interface.42,47 (See also earlier studies on thermocapillary motion near interfaces, e.g., in ref. 48.)
In this context, here we address self-phoresis of a catalytic particle in the vicinity of a fluid–fluid interface. First, in order to emphasize the influence of the interface, we study the case of a particle which is homogeneously catalytic. In order to keep the system as simple as possible, in the following we assume that the concentration of reactants is kept constant in space and time. In such a situation the particle releases the products of the catalytic reaction isotropically. Therefore, even though the system is kept out of equilibrium, due to the isotropic coverage of the catalyst in the bulk there is no symmetry breaking† and hence no net displacement. However, the presence of an interface breaks the homogeneity of the transport coefficients (i.e., the diffusivities of the reaction products in the two fluid phases). This leads to an inhomogeneous distribution of the concentration of the reaction products along the interface normal. Accordingly, such an inhomogeneous density profile leads to an interface-induced phoresis, the direction of which is normal to the interface, similarly to what has already been reported for the case of a hard wall.30,32 Interestingly, in the present case the sign of the resulting velocity depends not only on the surface properties of the particle, as it is the case near a hard wall, but it depends on both the contrast between the diffusivities and the distinct solvability of the catalysis products in the two fluid phases.
If the catalyst is not homogeneously distributed along the surface of the particle, a net motion will arise due to self-diffusiophoresis even in a homogeneous fluid. Accordingly, if such particles are close to a fluid–fluid interface, a competition arises between the intrinsic motility and the one induced by the interface. Since the motility induced by the interface is directed solely along its normal, in order to grasp the interplay between the self-phoresis and the interface-induced phoresis we focus on the case of an asymmetrically coated colloid (ideal Janus particle) the axis of symmetry of which is parallel to the normal of the interface.
The paper is organized as follows. In Section 2 we formulate the model describing the dynamics of active colloids close to a fluid interface. In Section 3 we study the velocity of these active colloids by using an approximate far-field expansion as well as an exact solution. While the approximate far-field expansion allows us to straightforwardly grasp the phenomenology emerging from the dynamics of active colloids close to fluid interfaces, the exact solution, by providing quantitatively reliable results, allows us to critically asses the strengths, as well as the shortcomings, of the approximate far-field approach. Finally in Section 4 we provide concluding remarks. The details of the necessary calculations beyond the ones presented in the main text are included in the Appendices A–D.
2 Model system
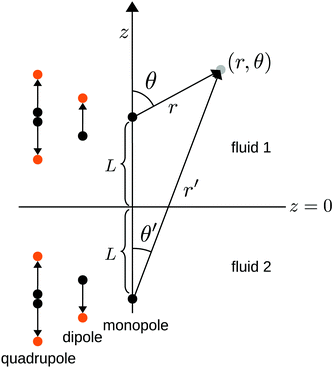
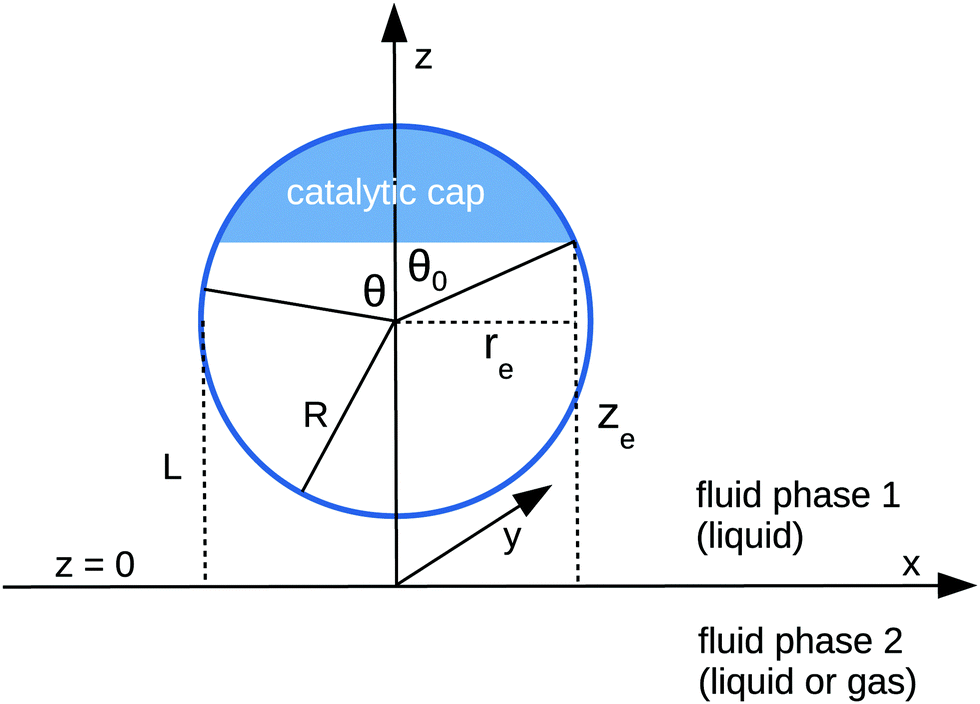
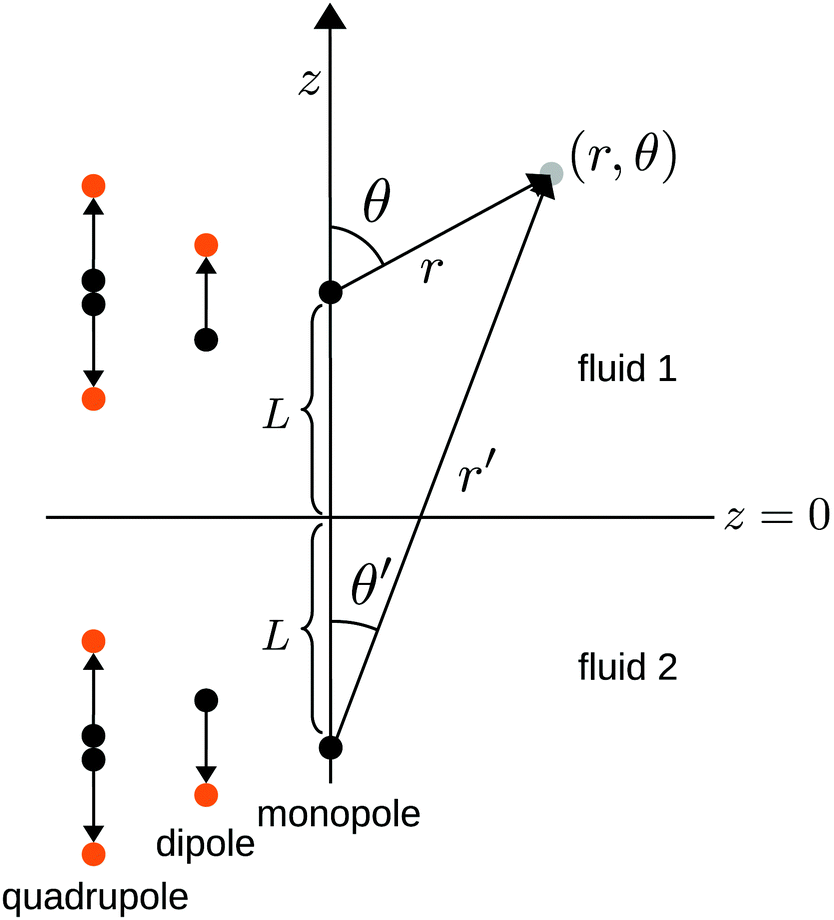
We consider the following simple model for a chemically active colloid10 (see Fig. 1 for a schematic description of the system). The spherical colloid of radius R is partially (or completely) covered by a catalyst which promotes the conversion of “fuel” molecules into product (solute) molecules. The particle is immersed in a solution, which is a fluid mixture composed of solvent 1 as well as fuel and product molecules occupying the upper half-space (z > 0). The lower half-space (z < 0) is occupied by fluid mixture composed of a solvent 2 as well as fuel and product molecules. We assume the two solvents to be phase separated, in thermodynamical equilibrium, and the interface between them, i.e., the plane z = 0, to be microscopically thin. The center of the particle is located at a distance L > R from the interface. For simplicity, we assume that both the fuel and the product molecules diffuse, albeit differently, in both fluids. Furthermore, we assume that neither of the two species called fuel and product exhibits preferential adsorption at the interface. Both solutions are considered to be Newtonian fluids. The bulk viscosities of the two fluids are denoted as μ1 and μ2, respectively, and are taken to be unaffected by the densities of the fuel (which is kept constant) and that of the product molecules. The system is thought of being in contact with reservoirs of particles which fix the bulk number densities of each molecular species. The diffusion of the fuel molecules, controlled by the diffusion constants D1 and D2 in the two fluids, respectively, is considered to be very fast compared with the reaction rate, which corresponds to the so-called reaction-limited kinetics regime.51 Under these assumptions, the chemical reaction can approximately be captured by considering the catalyst-covered part of the particle as an effective source of product molecules with a time- and position-independent rate Q of solute (i.e., product) release per area of catalyst.
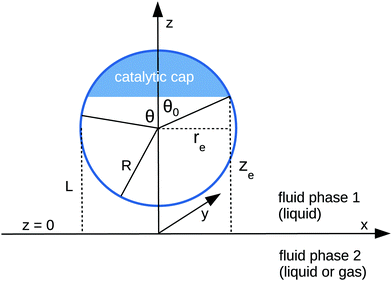 |
| | Fig. 1 Schematic description of the geometry of the system. The particle, of radius R, is positioned with its center at a distance L from the interface located at z = 0. Its axis of symmetry is parallel (“cap up”, as shown in the figure) or antiparallel (“cap down”) to the normal of the interface which points into the positive z-direction. The size of the catalytic cap is controlled by the coverage angle θ0. The base of the catalytic cap forms a circular edge of radius re at a distance ze from the interface. | |
We restrict our study to the case in which the diffusion of the solute in the two fluids is sufficiently fast such that the transport of solute by advection due to induced hydrodynamic flow is negligible compared to the one by diffusion. Accordingly, the solute number density distribution is determined solely by diffusion. Furthermore, generically in experiments with chemically active particles, the liquid media are aqueous solutions and the flows induced by self-phoresis correspond to very small Reynolds numbers. Therefore, in the following we shall describe the corresponding hydrodynamics by the Stokes equation for an incompressible Newtonian fluid.
Concerning the solute we assume that the role of the interface is simply to provide distinct diffusion coefficients and distinct solvation energies in the two fluid phases. This amounts to a partitioning effect, which is associated with the Donnan potential (see p. 75 in ref. 52). The transport by advection is negligible relative to that by diffusion (see the discussion above), so that at steady state the number density c(r) of solute is the solution of the Laplace equation
subject to the following boundary conditions (see the corresponding detailed discussion in
ref. 46):
(a) constant values in each half-space far from the particle:
| | 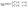 | (2) |
(b) discontinuity at the interface solely due to the different solvability in the two media (Donnan potential):
| | | λc(r)|z→0+ = c(r)|z→0−, | (3) |
with
λ determined by
(c) no transport parallel to the interface, i.e., all the solute flux leaving medium “1” enters into medium “2”:
| | | n1·(−D1∇c)|z→0+ = −n2·(−D2∇c)|z→0−, | (5) |
where
n1 and
n2 denote the outer normals of the domains
![[scr D, script letter D]](https://www.rsc.org/images/entities/char_e523.gif) 1
1 = {
z > 0} (
i.e.,
n1 = −
ez) and
2= {
z < 0} (
i.e.,
n2 =
ez), respectively, and ∇ is the gradient operator;
(d) at the catalyst covered part of the surface Σp of the particle the molecular flux of solute (i.e., the number density current along the outward normal n of the particle surface Σp) equals the “production” rate of solute by the catalytic reaction, while on the rest of the surface it vanishes (i.e., the particle is impermeable):
where
θ is the polar angle (see
Fig. 1) and
| |  | (7) |
describes the coverage of the particle by catalyst.
Due to the asymmetry introduced via the partial coverage by catalyst as well as due to the presence of the interface, the distribution of solute around the colloidal particle is spatially inhomogeneous. Since the solute molecules interact with the colloid differently than the solvent ones, this inhomogeneity induces gradients in the local osmotic pressure along the surface of the particle. As a result, flow of the fluid and motion of the particle emerge.10,53 If the range of these molecular interactions is much smaller than the size of the particle, which is a plausible assumption for most of the experimental realizations, the effects of the osmotic pressure gradient are captured via a so-called phoretic slip velocity vp(rp) of the fluid relative to the surface of the particle. The phoretic slip is related to the local gradient of the number density of solute via10,53
where ∇
‖ is the gradient along the surface of the particle,
b is the so-called phoretic mobility and
rp denotes the points on the surface of the particle. The latter encodes the effective particle–solute interaction according to
| |  | (9) |
where 1/
β =
kBT,
kB is the Boltzmann constant,
T the absolute temperature,
![[scr U, script letter U]](https://www.rsc.org/images/entities/char_e534.gif)
is the effective interaction potential between the particle and the solute relative to that between the particle and the solvent molecules, and
μ1 is the viscosity of the fluid in which the particle is suspended.
Once the slip velocity is provided one can set out to solve the Stokes equation under the proper boundary conditions at the fluid interface. Alternatively, the velocity of the particle in the lab reference frame can be obtained via the reciprocal theorem.14,54–56 The reciprocal theorem states that in the absence of volume forces any two incompressible flow fields u(r) and û(r), which are distinct solutions of the Stokes equations within the same domain , i.e., solutions subject to different boundary conditions but on the very same boundaries ∂, obey the relation
| | 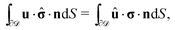 | (10) |
where
σ and
![[small sigma, Greek, circumflex]](https://www.rsc.org/images/entities/b_char_e111.gif)
denote the stress tensors corresponding to the two flow fields
u and
û, respectively, and
n is the outward normal of ∂
.
However, the present system is somewhat different, in that the phase-separated condition divides the fluid domain into two sub-domains (on each side of the interface the flow velocity along the interface normal vanishes), but the flows in the two domains 1 and 2 are connected due to the requirement of continuity of the tangential stress and of the tangential velocity at the interface. In spite of these complications, it has been shown that for an infinitely large fluid domain with a planar interface the reciprocal theorem takes the exact same form as in the case of a particle immersed in a single fluid medium,57i.e.,
| | 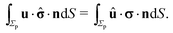 | (11) |
According to
eqn (11) the velocity (rotational or translational) of the active particle can be determined
via the stress field in a certain “dual” problem typically associated with known solutions for spatially uniform translation or rotation.
In the following we restrict the discussion to cases in which the system exhibits axial symmetry, i.e., the symmetry axis of the particle coincides with the z-direction, which in turn is the normal of the interface, and we neglect the effects of thermal fluctuations, in particular the rotational diffusion of the axis of the active colloid. Accordingly, concerning the description of the present system there is a single unknown quantity, which is the translational velocity of the particle along the direction normal to the interface. We therefore select as the dual problem the Stokes problem of the translation of a chemically inert particle (located at z = L) with velocity Û(L) along the normal of the interface, due to a force ![[F with combining circumflex]](https://www.rsc.org/images/entities/b_char_0046_0302.gif) acting on it (located at z = L) with a no-slip boundary condition at its surface, i.e.,
acting on it (located at z = L) with a no-slip boundary condition at its surface, i.e.,
Accounting for the phoretic slip, at the surface of the active particle one has
with
vp(
rp) defined by
eqn (8) and
U denoting the velocity of the active particle, which points into the
z-direction. In the usual manner,
57,58 after accounting for the fact that the active particle is force free (
i.e.,

) and that
Û and
U do not vary along the surface of the particle and therefore are independent of
rp (even though they retain a dependence on the distance
L of the center of the sphere from the interface), and that
Û and
U have a
z-component only, one arrives at
| | 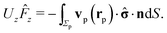 | (14) |
3 Results
In order to facilitate the use of eqn (14), it is necessary to solve the diffusion equation, thus determining the distribution of solute at the surface of the particle, and to find the stress tensor in the auxiliary problem of a spherical particle moving at zero Reynolds number towards or away from a fluid–fluid interface. Both these problems can be solved analytically in terms of series representations in bi-polar coordinates (see, e.g., ref. 38, 46, 48, 59 and 60). These solutions are concisely summarized in Appendix A. Since it is difficult to straightforwardly gain physical insight from such series representations, here we focus on an analysis in the far-field approximation (R/L ≪ 1, see Fig. 1 and the description below), which provides closed-form, easy to decipher and interpret results, as a means to survey the phenomenology. The predictions of this approximate analysis are compared with the exact results obtained from using bi-polar coordinates, which allows one to critically asses (both quantitatively and qualitatively) the reliability of this far-field approximation.
We discuss separately two cases, first the one in which the whole surface of the particle is chemically active, and second the case of a Janus colloid for which only a spherical cap is chemically active. The first case allows one to highlight those effects which are solely due to the way in which the solute partitions between the two fluid phases. The second case provides insight into the interplay between the above mechanism and the asymmetry in the distribution of the reaction sites across the surface of the particle.
3.1 Far-field approach
Within the far-field approximation, the number density c(r) and the stress tensor (r) are expressed in terms of multipole (singularity) expansions by keeping only the lowest order terms as well as the first set of images needed to account for the boundary conditions at the fluid–fluid interface. In order to keep the analysis simple, in the following we shall disregard contributions stemming from the images needed to enforce the boundary conditions on the particle surface. Rather than a priori analyzing the reliability of this approximation we shall a posteriori check it via comparison against the exact solutions. Indeed, the comparison of this approximate analysis with the exact solution shows that this truncated far-field approximation is sufficient to grasp the most important effects of the interface on the dynamics. (The comparison, however, also identifies certain qualitative discrepancies emerging from the severe truncation of far-field expansions (as noted previously61), which underlines the importance of cross-checking against analytical or numerical exact results.)
We start our analysis by decomposing the stress tensor of the auxiliary problem:
where
0 denotes the contribution of the Stokeslet (the lowest order singularity),
i.e., the external force
acting on the particle and introducing its translation with velocity
Û, while
im denotes the contribution stemming from the image system of the Stokeslet
55,62 (
i.e., the lowest order contribution of the images). Concerning the first contribution, with
Û = Û
ez it is known that
58| | 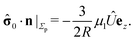 | (16) |
In the far field,
i.e., for
R ≪
L the second term
im in
eqn (15) can be expanded into a Taylor series about its value at the center of the particle
| | | im(x, y, z) = im(0, 0, L) + δim(x, y, z). | (17) |
Accordingly, the deviation, δ
im(
x,
y,
z), from
im(0, 0,
L) is subdominant,
i.e., one has
| | 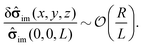 | (18) |
The first term on the rhs of
eqn (17) can be computed explicitly (see Appendix B):
| | 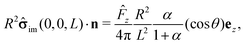 | (19) |
where
| |  | (20) |
(We remark that,
via the Stokes–Einstein relation, one has
D1,2 ∝ 1/
μ1,2; hence
α also equals the reciprocal of the ratio of the viscosities,
i.e.,
α =
μ1/
μ2.) By using
![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) z
z = −6π
μ1RÛz, employing spherical coordinates
z =
R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
cos
θ +
L,
x =
Rsin
θcos
ϕ, and
y =
R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif)
sin
θsin
ϕ, and plugging
eqn (16) and (19) into the rhs of
eqn (14) one arrives at
| | 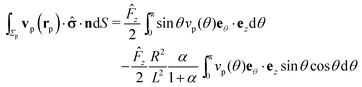 | (21) |
where
| |  | (22) |
Eqn (21) shows that the contributions to the stress tensor stemming from the images (
i.e., the last term in
eqn (21)) are sub-leading corrections to the term of leading order in
R/
L due to the Stokeslet contribution (
i.e., the first term in
eqn (21)).
3.2 Homogeneous active colloid
For a homogeneously covered active colloid the number density distribution of the solute around the particle, within the far-field expansion truncated at order ![[scr O, script letter O]](https://www.rsc.org/images/entities/char_e52e.gif) ((R/L)3), is governed by the point-source term and reads:‡
((R/L)3), is governed by the point-source term and reads:‡| |  | (23) |
By plugging the above result into eqn (8), one obtains the z-component of the slip velocity (which is needed for the rhs of eqn (21)):| | 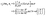 | (24) |
Finally, after substituting eqn (21) and (24) into eqn (14) and performing the integral, one obtains to leading order in R/L the following expression for the velocity of the active particle:§| |  | (25) |
where| |  | (26) |
has indeed the dimension of a velocity.
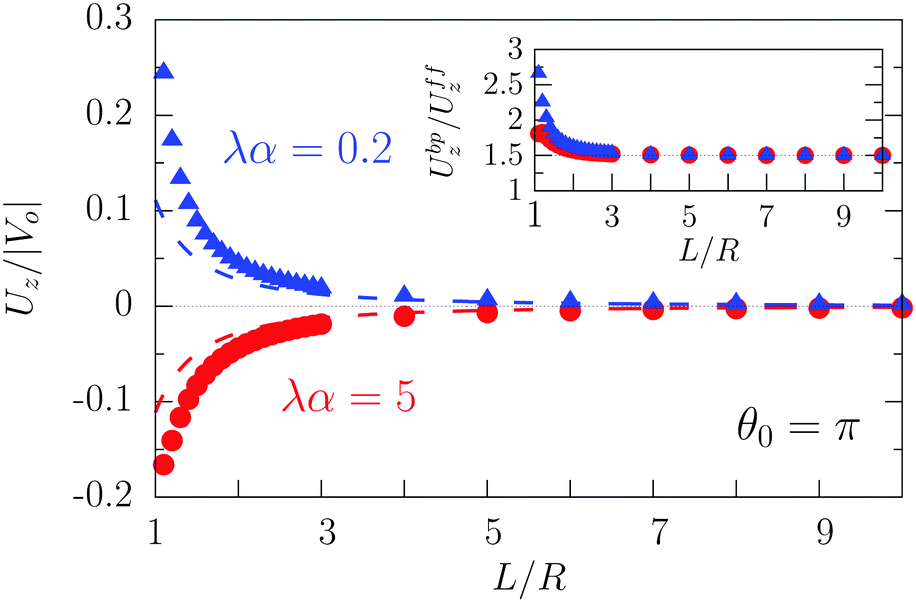
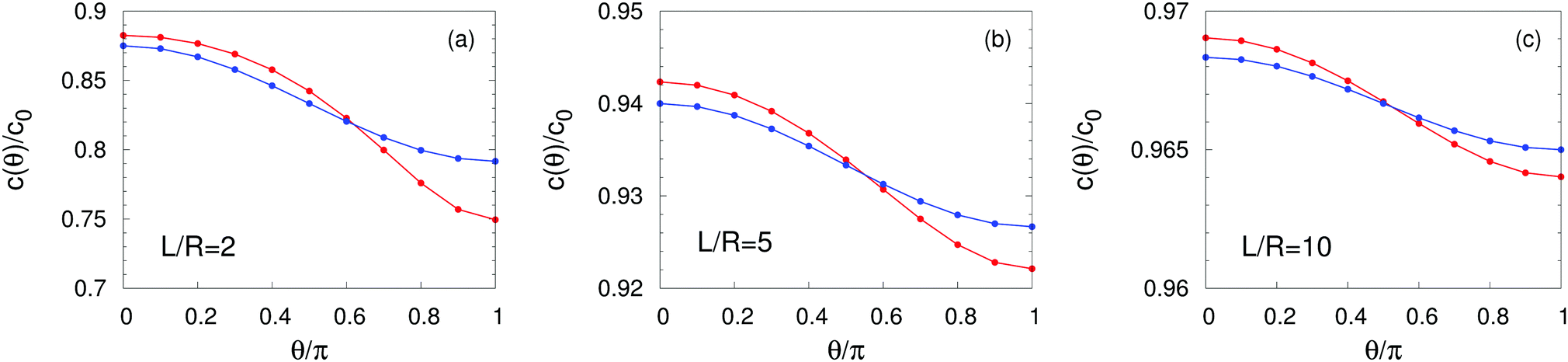
As expected from the behavior reported for similar active particles near a hard wall or near another particle,30,32,63 in the vicinity of the interface the particle exhibits motion along the direction normal to the interface. Upon leaving the interface the magnitude of the velocity of the particle decays as (R/L)2. Near the interface the velocity can reach values of the same order of magnitude as the maximum velocity Vo/4 of a Janus particle in an unbounded fluid15 (see Fig. 2). We note that in the limit α → 0 eqn (25) takes the form corresponding to a hard wall,30,32 whereas for α → ∞ it is expected that it will recover the form corresponding to a direct calculation for a fluid–gas interface.
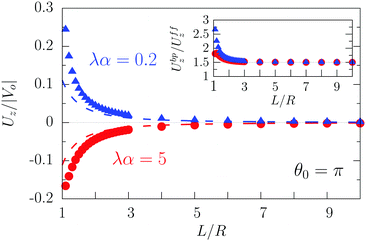 |
| | Fig. 2 Rescaled velocity Uz/|Vo| (see eqn (26)) as a function of the distance L/R of the center of the particle from the interface. The whole surface of the particle is chemically active, i.e., θ0 = π. The symbols (blue triangles for λα = 0.2 and red circles for λα = 5) show the exact results calculated by using bi-polar coordinates (see Appendix A), while the dashed lines show the results from the far-field approximation (eqn (25)), for λα = 0.2 (blue) and λα = 5 (red) (see eqn (3) and note that  ), and Vo < 0 (eqn (26)). The inset shows the ratio of the exact velocity Ubpz obtained by using bi-polar coordinates and the velocity Uffz calculated within the far-field approximation. ), and Vo < 0 (eqn (26)). The inset shows the ratio of the exact velocity Ubpz obtained by using bi-polar coordinates and the velocity Uffz calculated within the far-field approximation. | |
Apart from the parameters included in Vo (eqn (26)), the velocity depends on the ratio λ of the solvabilities (eqn (3)) and on the ratio α (eqn (20)) of the diffusion constants of the solute in the two fluid phases 1 and 2 (or equivalently, due to the Stokes–Einstein relation, on the inverse ratio of the viscosities of the two fluids). These additional dependences are particularly interesting because they imply that, in contrast to the behavior near a hard wall, also the direction of the motion depends on λα and not solely on the sign of the phoretic mobility b, which enters via Vo (eqn (26)).
Taking, for example, b < 0, which implies Vo < 0, and λ = 1 (the discussion can be straightforwardly extended to the cases in which λ ≠ 1 or b > 0), one infers from eqn (25) that for α < 1, i.e., if the particle is suspended in the less viscous fluid phase, Uz is positive and thus the particle moves away from the interface. For α > 1, i.e., if the particle is suspended in the more viscous fluid phase, Uz turns negative and thus the particle moves towards the interface. Furthermore, eqn (25) exhibits the symmetry relation  . Since replacing λα by
. Since replacing λα by  amounts to interchanging media 1 and 2, one concludes that, if one would perform an experiment in which the active particle is placed at z = L and another one with an identical particle now placed at z = −L the outcome will be the following. If in the first experiment the particle will move towards the interface, then in the second experiment it will move away from it, the speed of the motions being the same in the two cases. Vice versa, if in the first experiment the particle will move away from the interface, in the second it will move towards the interface, with the same speed of motion.
amounts to interchanging media 1 and 2, one concludes that, if one would perform an experiment in which the active particle is placed at z = L and another one with an identical particle now placed at z = −L the outcome will be the following. If in the first experiment the particle will move towards the interface, then in the second experiment it will move away from it, the speed of the motions being the same in the two cases. Vice versa, if in the first experiment the particle will move away from the interface, in the second it will move towards the interface, with the same speed of motion.
As shown in Fig. 2, the predictions of the far-field approximation (eqn (25), dashed lines) accurately capture the qualitative behavior of Uz, in particular the most important feature of the above noted change of sign of the velocity for λα ≷ 1. Fig. 2 also shows that the magnitude of the velocity changes upon the replacement λα → 1/(λα) (see the asymmetry of the blue and red symbols); thus the symmetry predicted by eqn (25) is an artifact of the truncation in the far-field analysis.
Quantitative discrepancies are noticeable, as highlighted by the inset of Fig. 2: the inset shows that, even in the limit L/R → ∞, the amplitude of the far-field result deviates from that of the exact solution by ca. 50%. As it is apparent from the main panel of Fig. 2, the far-field calculation correctly captures the asymptotic result of a vanishing velocity. The fact that the ratio of the far-field approximated velocity and the exact one happens in this case to be a constant, rather than a function of the distance from the interface, is somewhat peculiar. However, it merely translates into the difference between the approximated value and the exact one being 1/2 of the exact value. Since the latter decays to zero with increasing distance from the interface, the deviation of the approximation from the exact value also vanishes with increasing distance, as it should.
We have identified two causes of these discrepancies. First, close to the interface the mismatch between the far-field predictions and the exact solution is due to the fact that we have kept only the lowest order singularities in the far-field approximation, while in such situations higher order terms are clearly not negligible. Second, in the far-field approximation we have accounted solely for those images which are needed to enforce the boundary conditions at the fluid interface, while the changes induced by those images in the boundary conditions at the surface of the particle have been disregarded, irrespectively of the distance from the interface. In this case, this kind of issue, which demonstrates the need for critical comparisons with exact solutions, leads to discrepancies between the far-field approximation and the exact result even in the limit L → ∞ (see the inset in Fig. 2 and Appendix C).
3.3 Active Janus colloid
In this subsection we turn to the case of a Janus colloid, for which only a portion of the surface is chemically active. We focus on configurations in which the active side is facing either fully “up” or fully “down”. Within the far-field approximation, the number density profile induced by a Janus particle comprises a dipolar contribution in addition to the monopolar one considered in the previous subsection, i.e.,| | 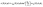 | (27) |
where the point source contribution cps(R, θ, ϕ) is defined in eqn (23). The factor sin2(θ0/2) accounts for the fact that only a portion of the surface is chemically active (Fig. 1). Straightforward calculations (see Appendix D) lead to the following expression for the dipolar contribution:¶| |  | (28) |
where (see ref. 37)| |  | (29) |
the plus sign “+” holds for catalytic caps pointing “up” while the minus sign “−” holds for catalytic caps pointing “down”. Accordingly, the contribution of the dipole to the z-component of the slip velocity is given by| |  | (30) |
combining this result with eqn (29) and (25), one arrives at| | 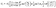 | (31) |
where ± holds for catalytic caps “down” and “up”, respectively; note the flip of sign as compared to eqn (29). For θ0 = π eqn (31) reduces to eqn (25).
The result in eqn (31) deserves further discussion. Similarly to the symmetry discussed in the context of eqn (25), a straightforward calculation shows that upon simultaneously replacing λα → 1/(λα) and “cap up” → “cap down”, Uz changes sign but its magnitude stays the same. Thus an experiment with a particle at z = L and oriented “cap up” (away from the interface) and one in which an identical particle is immersed at z = −L in the other fluid but oriented “cap-down” (towards the interface) will show in one case the particle approaching the interface and in the other moving away from the interface, in both cases with precisely the same speed. Furthermore, it can be inferred that there are cases in which the two terms within the square brackets on the rhs of eqn (31) have opposite signs. To this end we consider the situation of a “cap-up” particle, i.e., in eqn (31) for the second term the “−” sign holds so that for λα < 1 the first term is positive, while the second is negative. In this case it is straightforward to show that if
| | 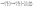 | (32) |
i.e., the cap opening
θ0 is larger than the critical value
θcr defined above, then there is a particular distance
L0, given by
| |  | (33) |
at which the velocity of the particle is vanishing. For example, according to
eqn (32) for a wall (
i.e.,
α → 0) the critical opening (
i.e., the size of the catalytic cap) is
θcr ≲ 3π/4. This state, provided it is stable (the stability depends also on the sign of
Vo), is the equivalent of the “hovering” steady state for such a chemically active particle near a wall, as reported in
ref. 32. For the latter the exact critical value is
θcr ≃ 0.83π which is somewhat larger than the former far-field approximation. Upon increasing
θ0,
θcr of this state shifts towards larger values and, as expected, diverges in the limit
θ0 → π (
i.e., for a homogeneously active particle). The symmetry exhibited by
eqn (31) and discussed above implies that similar states occur for a “cap-down” configuration for
λα > 1 under the same constraint as stated by
eqn (32).
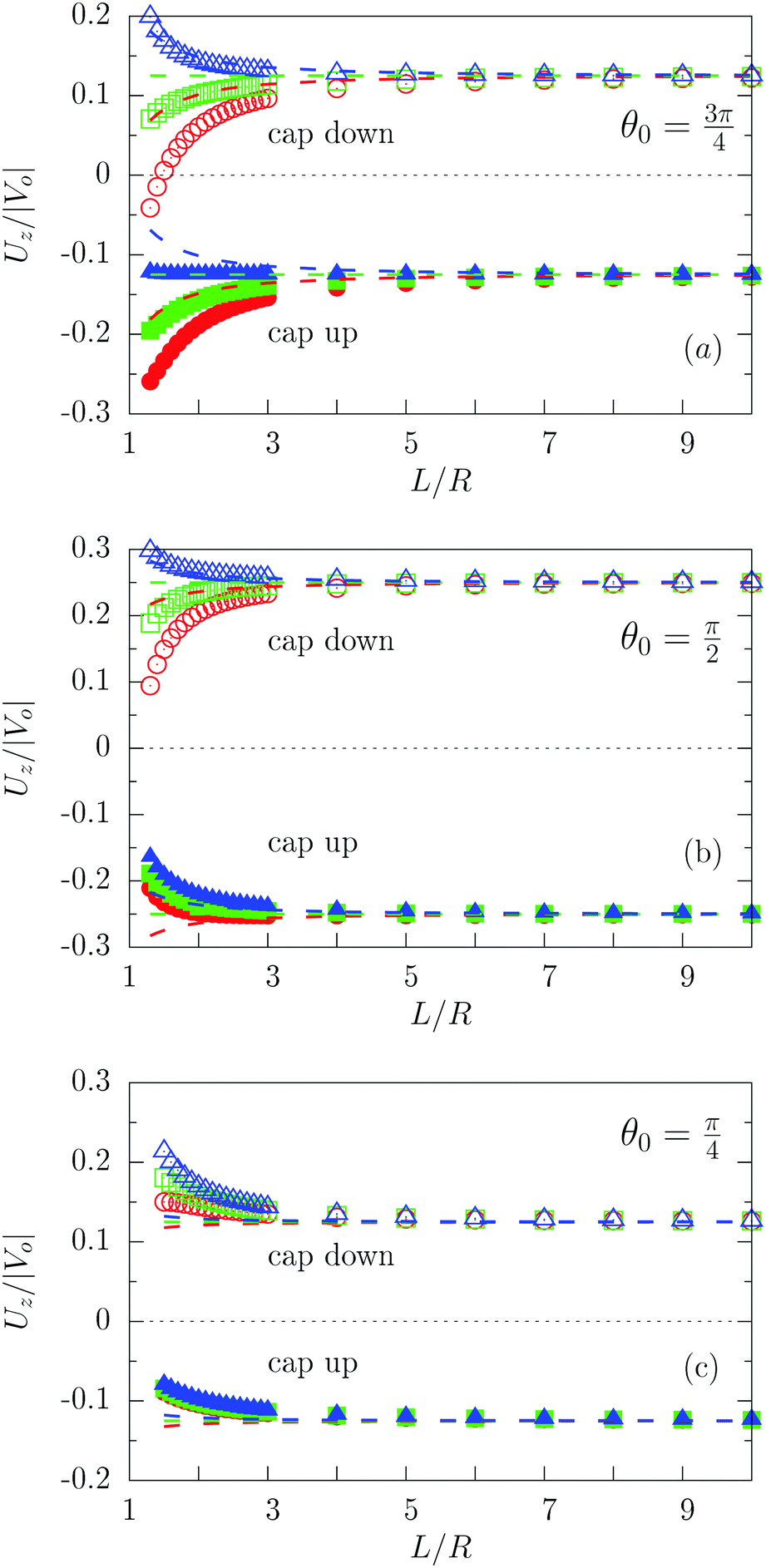
Fig. 3 shows the dependence of Uz/|Vo| for three values of the size of the catalytic cap and for Vo < 0. In particular, Fig. 3(a) indeed confirms that for sufficiently large coverages, such as θ0 = 3π/4, active particles can be trapped at a finite distance (i.e., Uz(L0) = 0 for cap down and λα = 5), similar to what has been reported for active particles close to hard walls.32 Actually, in Fig. 3(a) the cap-down configuration with zero velocity is an unstable configuration in that, if the particle deviates from this position L0, it does not return there, as can be inferred by inspection of the sign of the velocity to the left and to the right of the zero-crossing. As discussed above, such states do occur only if the coverage is sufficiently large, and therefore they are not observed in Fig. 3(b) and (c). As in the previous case of a homogeneously active particle, the far-field approximation (dashed lines in Fig. 3) misses to quantitatively capture the magnitude of the velocity if the active Janus particle is close to the interface – even though it captures the asymptotic values of Uz at large distances L. As in the case of homogeneously covered particles, the symmetry properties inferred from eqn (31) are not confirmed by the exact solution, but at a qualitative level the predicted change in sign of the velocity upon the simultaneous change λα → 1/(λα) and “cap up” → “cap down” indeed holds for large L/R. Finally, we remark that in the case λα = 1 there is no distortion of the number density profile due to the presence of the interface. Therefore in this case the variation of the velocity as function of L (green symbols in Fig. 3) is of purely hydrodynamic origin. It stems from the boundary conditions imposed on the velocity profile by the fluid–fluid interface.
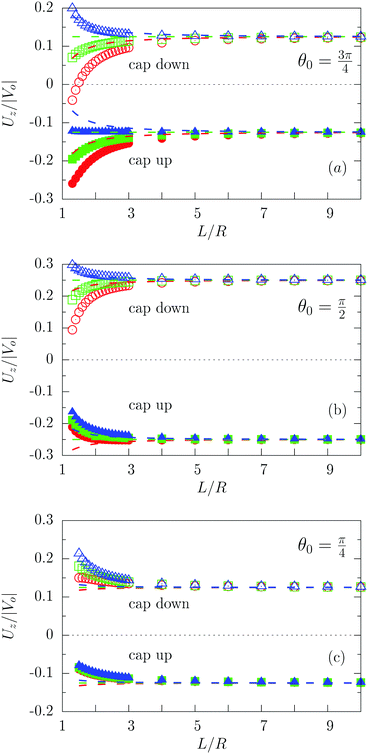 |
| | Fig. 3 Rescaled velocity Uz/|Vo| calculated by using bi-polar coordinates (full and open symbols), as a function of the distance L/R of the center of mass of the particle from the interface for θ0 = 3π/4 (panel (a)), θ0 = π/2 (panel (b)), and θ0 = π/4 (panel (c)) with the chemically active cap pointing upwards (full symbols) or downwards (open symbols) for λα = 0.2 (blue), λα = 1 (green), and λα = 5 (red). The results shown correspond to the choice Vo < 0 (b < 0). The predictions of the far-field approximation are reported as dashed lines using the same color scheme as for the results obtained by employing bi-polar coordinates. As expected, the comparison between Fig. 2 and 3 shows that Uz(L → ∞, θ0 = π) = 0 whereas Uz(L → ∞, θ0 < π) ≠ 0. | |
4 Conclusions
In order to capture the essence of the influence of a fluid–fluid interface on the self-diffusiophoresis of active particles, first we have studied the dynamics of an active colloid homogeneously covered with catalyst and being close to a fluid interface. For such a particle, in a homogeneous and unbounded fluid and in the absence of spontaneous symmetry breaking, no motion occurs. However, the interface breaks the translational symmetry in the transport coefficients of the products of the catalysis along the direction of the interface normal. This results in a velocity along the normal of the interface even for homogeneously covered particles.
In order to characterize the dynamics of catalytic active particles close to a fluid–fluid interface we have developed a truncated far-field expansion of the diffusion equation for the product molecules of the catalysis and of the Stokes equation. As well, an exact solution, in terms of bi-polar coordinates, has been constructed based on the results available in ref. 46. We have found that both the magnitude and the sign of the velocity of the particle can be controlled by tuning the ratio of the diffusivities and solvabilities of the catalysis products in the two coexisting fluid phases. In particular, both the exact solution and the far-field approximation show that the sign of the velocity is controlled by the product λα of the ratio α of the diffusion coefficients of the catalysis products in the two fluid phases, and the ratio λ of the equilibrium solvabilities. Indeed, for a positive phoretic mobility b > 0 an active particle with λα > 1 moves towards the interface, whereas it moves away for λα < 1; for b < 0, the directions are reversed. This effect is strongest close to the interface; the interface-induced velocity decays algebraically (∼L−2) with the distance L from the interface.
If the coverage of the particle is not homogeneous, there is a velocity  (eqn (31)) already in a homogeneous fluid. Therefore, if such particles are close to a fluid–fluid interface the interface induced velocity
(eqn (31)) already in a homogeneous fluid. Therefore, if such particles are close to a fluid–fluid interface the interface induced velocity  (eqn (31)) sums up with v0, leading to rich scenarios. For example, a particle with sufficiently large coverage θ0 (eqn (32)) experiences a stagnation point close to the interface (eqn (33)) where its velocity vanishes (Fig. 3(a)). If stable, these states are the equivalent of the steady-state hovering near a planar wall. Clearly, for this kind of particles adsorption at the interface is hindered due to the interface-induced component of the velocity. On the contrary, for coverages below the critical value given by eqn (32) such a tug-of-war scenario between the eigen-velocity of the particle and the interface-induced velocity does not occur and the sign of the velocity is always determined by the eigen-velocity v0.
(eqn (31)) sums up with v0, leading to rich scenarios. For example, a particle with sufficiently large coverage θ0 (eqn (32)) experiences a stagnation point close to the interface (eqn (33)) where its velocity vanishes (Fig. 3(a)). If stable, these states are the equivalent of the steady-state hovering near a planar wall. Clearly, for this kind of particles adsorption at the interface is hindered due to the interface-induced component of the velocity. On the contrary, for coverages below the critical value given by eqn (32) such a tug-of-war scenario between the eigen-velocity of the particle and the interface-induced velocity does not occur and the sign of the velocity is always determined by the eigen-velocity v0.
Conflicts of interest
There are no conflicts to declare.
Appendices
A Solutions in terms of bi-polar coordinates
Both the diffusion process (eqn (1)–(6)) and the hydrodynamics of a no-slip sphere moving at zero Reynolds number towards a planar fluid–fluid interface exhibit axial symmetry and involve boundary conditions at a spherical and at a planar surface only. Accordingly, both problems can be solved exactly in terms of bi-polar coordinates. A detailed derivation of this solution is provided in the supplementary material of ref. 46. In order to be self-contained and for reasons of clarity, here we succinctly summarize the main steps and provide the formulae required for obtaining and using this solution.
A.1 System of bi-polar coordinates.
The bi-polar coordinates (ξ,η) with −∞ < ξ < ∞ and 0 ≤ η ≤ π are defined such that the vertical coordinate z and the radial distance r from the z-axis are given by55,64| |  | (34) |
where  with ξ0 = arccosh(L/R) is chosen such that the manifold ξ = ξ0 corresponds to the spherical surface of radius R centered at z = L (which is the surface of the particle). The plane z = 0 of the interface corresponds to ξ = 0. In order to simplify the notations we introduce the quantity ω := cosη.
with ξ0 = arccosh(L/R) is chosen such that the manifold ξ = ξ0 corresponds to the spherical surface of radius R centered at z = L (which is the surface of the particle). The plane z = 0 of the interface corresponds to ξ = 0. In order to simplify the notations we introduce the quantity ω := cosη.
Here we focus on the case in which the catalytic cap, characterized by the opening angle θ0 (which is expressed in terms of the spherical coordinates attached to the particle), is oriented away from the interface, as drawn in Fig. 1. (The opposite case, i.e., the cap facing the interface, follows from a minor change in the equations determining the coefficients in the expansion of the solute number density; this change will be pointed out at the corresponding step in the derivation.) In this case, the point (ξ = ξ0, η = 0) farthest from the interface (the “north” pole) belongs to the catalytic cap, while the point (ξ = ξ0, η = π) closest to the interface (the “south” pole) belongs to the chemically inert part. The boundary between the catalytic and the inert caps (the “edge”) is a circle parallel to the plane z = 0. The points Pe = (re, ze) on the edge have the spherical coordinates re = Rsinθ0 and ze = L + Rcosθ0 (see Fig. 1); since the edge is part of the sphere ξ = ξ0, i.e., re and ze satisfy eqn (34) with ξ = ξ0, the points on the edge have the same coordinate η0 given by
| |  | (35) |
i.e., the edge is the intersection of the manifolds
ξ =
ξ0 and
η =
η0. Therefore, for the “cap up” setup the catalyst covered area corresponds to (
ξ0, 0 ≤
η ≤
η0), while for a “cap down” setup the catalyst covered area corresponds to (
ξ0,
η0 ≤
η ≤ π).
A.2 Solution of the diffusion problem.
Since all details of the corresponding calculations are provided by the openly accessible supplementary material of ref. 46, the brief outline given below for obtaining the solution of the diffusion problem and of the auxiliary stream function is considered to be sufficient.
The solution of the diffusion problem (eqn (1)–(6)) can be expressed in terms of Legendre polynomials Pn as65
| | 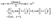 | (36) |
in fluid 1, with a similar expression but with different coefficients

,
Ân, and
![[B with combining circumflex]](https://www.rsc.org/images/entities/i_char_0042_0302.gif) n
n in fluid 2 (
ξ < 0). The prefactor
QR/
D1 has the units of a number density, so that the coefficients
An,
Bn,
Ân, and
n are dimensionless. (We note that the same prefactor
QR/
D1 is used for both
ξ > 0 and
ξ < 0.) We focus on the solution in fluid 1 (
ξ > 0) because only that one enters into the expression for the phoretic slip at the surface of the colloid.
Inserting these two series representations into the boundary conditions at infinity (eqn (2) and (4)) leads to
| |  | (37a) |
inserting them into the boundary conditions at the interface (
eqn (3) and (5)) in combination with the requirement that the density is bounded everywhere leads to
| | | Ân = n = λBn, An = λαBn, | (37b) |
with
α =
D2/
D1 (as defined in
eqn (20) in the main text). (Note that for a constant flux boundary condition on the particle surface, which is time- and position-independent over the catalyst part, the velocity of the particle turns out to be independent of the value of the constant
c∞1, see,
cf.,
eqn (46) and (48).) By combining
eqn (37) with the flux boundary condition (
eqn (6)) at the surface of the particle and by projecting the lhs and the rhs of
eqn (6) onto the Legendre polynomial
Pn(
ω), one arrives at the following set of linear equations determining the coefficients
Bn:
| | 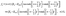 | (38) |
with the convention
B−1 = 0. With
ω0 := cos
η0 defining, as discussed above, the edge between the active and the passive parts of the surface, the coefficients
fn are given in terms of the activity function
f(
θ) (
eqn (6) in the main text):
| | 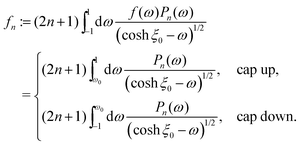 | (39) |
This infinitely large system of linear equations is solved by truncating it at a sufficiently large index
n =
N, followed by a numerical treatment. In practice, the truncation, as well as the series representation, are converging very fast as long as
L/
R ≳ 1.1. We have found that in most cases
N = 50 is sufficient for providing accurate results. This procedure is analogous to the ones used in
ref. 46 and 63.
A.3 Solution of the auxiliary hydrodynamics problem.
The auxiliary problem consists of a passive spherical particle, i.e., there is no chemical reaction, with a no slip boundary condition at its surface. The center of the particle is located at z = L and moves with velocity Û = ezÛ through fluid 1 along the direction normal to the flat fluid–fluid interface.
The corresponding solution for the velocity field û(x) of the incompressible Stokes equations can be expressed in terms of a stream function Ψaux(x) = ÛR2ψaux(x) as55
| |  | (40) |
This stream function can be represented in bi-polar coordinates:
64,65| | 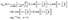 | (41) |
a similar expression, but with different coefficients
![[K with combining circumflex]](https://www.rsc.org/images/entities/i_char_004b_0302.gif) n
n,
![[L with combining circumflex]](https://www.rsc.org/images/entities/i_char_004c_0302.gif) n
n,
![[M with combining circumflex]](https://www.rsc.org/images/entities/i_char_004d_0302.gif) n
n, and
![[N with combining circumflex]](https://www.rsc.org/images/entities/i_char_004e_0302.gif) n
n, holds for
ξ < 0. In these equations
| |  | (42) |
denotes the Gegenbauer polynomial of order
n and degree −1/2.
64 The dimensionless coefficients
Kn,
Ln,
Mn, and
Nn, as well as the hatted ones, depend on
ξ0 (but not on
η0) and are determined by the boundary conditions for the velocity field. The requirement of a finite flow everywhere and the boundary conditions at infinity and at the interface lead to
| | | n = n = −n = −n, n ≥ 1, | (43a) |
| | 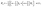 | (43b) |
and
| |  | (43c) |
where
μ1,2 denote the respective viscosities of the two fluid phases. By combining these relations with the no-slip and no-impenetrability conditions at the surface of the particle, the coefficients
Ln and
Nn are obtained as
| |  | (44a) |
and
| |  | (44b) |
where
| |  | (45a) |
| | | χ(2)n = −e−(n−1/2)ξ0 + e−(n+3/2)ξ0, | (45b) |
| | 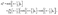 | (45c) |
| |  | (45d) |
| | 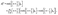 | (45e) |
and
| |  | (45f) |
With noting that
μ2/
μ1 = 1/
α, the derivation of the stream function for the auxiliary problem is complete.
A.4 Integral over the phoretic slip in bi-polar coordinates.
We start the calculation of the integral over the phoretic slip in eqn (14) by noting that: (i) in terms of the bi-polar coordinates the normal to the surface Σp of the particle is given by n = −eξ; (ii) the tangent plane to the surface of the particle is spanned by the unit vectors eη and eϕ; and (iii) since the present problems exhibit axial symmetry, the solute number density as well as the stress tensor of the auxiliary problem are independent of ϕ (see the previous subsections). Thus, the phoretic slip is given by vp := −b∇‖c(ξ0,η) = −b(hη−1|ξ0)∂ηc(ξ0,η)eη, where  denote the so-called scale (metric) factors corresponding to the η and ξ coordinates, respectively (see eqn (34) and recall the abbreviation ω = cosη). With this eqn (14) in the main text takes the form
denote the so-called scale (metric) factors corresponding to the η and ξ coordinates, respectively (see eqn (34) and recall the abbreviation ω = cosη). With this eqn (14) in the main text takes the form| |  | (46) |
where  denotes the scale factor corresponding to the ϕ coordinate.
denotes the scale factor corresponding to the ϕ coordinate.
The terms on the right hand side of eqn (46) are calculated as follows. First, is determined from the stream function Ψaux = ÛR2ψaux(x) as59,64–66
| | 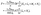 | (47) |
with
Ln and
Nn given by
eqn (44).
Second, the derivative of the number density at the surface of the particle is given by
| | 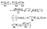 | (48) |
where

(see
eqn (36)); the coefficients
An and
Bn are determined from
eqn (38) and (39), as well as from the relation
An = (
λα)
Bn.
The contraction eη··eξ of the stress tensor at the surface of the particle (which is immersed in fluid “1”) is calculated as follows (see also ref. 38). By writing the hydrodynamic flow in the auxiliary problem as û = ûξeξ + ûηeη and by using the representation of the dyadic product ∇vaux (which one needs in order to be able to calculate the stress tensor in the auxiliary problem) in terms of the general orthogonal curvilinear coordinates provided in ref. 55 (see Appendix A-7, eqn (A-7.7) therein), one arrives at (recalling hξ = hη and eη·eξ = 0)
| | 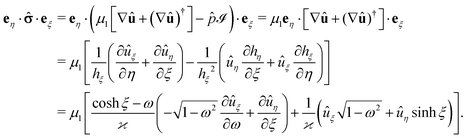 | (49) |
The flow components
uξ and
uη are obtained from the stream function
Ψaux (derived in the previous section) as (see Ch. 4-4 in
ref. 55 and
eqn (34))
| |  | (50a) |
and
| |  | (50b) |
which concludes the calculation.
The pressure component of the stress tensor| | im(x, y, z) = d(x, y, z) − p(x, y, z)![[scr I, script letter I]](https://www.rsc.org/images/entities/char_e143.gif) , , | (51) |
where is the identity matrix, renders a vanishing contribution vp(rp)··n = 0 to the integral (eqn (21)) involving the slip velocity, because the latter is orthogonal to the normal of the surface. Accordingly, one needs to compute only the contribution due to the deviatoric stress tensor| | 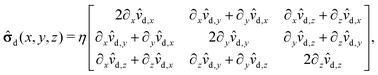 | (52) |
where each matrix element is a function of x, y, and z; ![[v with combining circumflex]](https://www.rsc.org/images/entities/b_char_0076_0302.gif) d denotes the flow field due to the image system for a Stokeslet located on the z-axis at z = L and oriented along the z-direction, i.e., normal to the fluid–fluid interface. It is given by62
d denotes the flow field due to the image system for a Stokeslet located on the z-axis at z = L and oriented along the z-direction, i.e., normal to the fluid–fluid interface. It is given by62| | 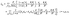 | (53) |
where α is defined in eqn (20) and| | | r = (x, y, L + z), R = (x, y, 2L + z). | (54) |
For the present problem only the value of d at the center of the particle is needed. This allows one to exploit the symmetries of the derivatives of d in order to simplify the algebra involved. In particular, one has that d is even about the x and y axis, i.e.,| | | d(x, y, z) = d(−x, −y, z), | (55) |
which implies that on the z-axis
| d,x = d,y = 0, |
| | | ∂xd = ∂yd = 0. | (56) |
Accordingly, the deviatoric stress tensor reduces to| | 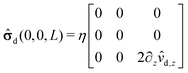 | (57) |
with 2∂z![[v with combining circumflex]](https://www.rsc.org/images/entities/i_char_0076_0302.gif) d,z evaluated at (x, y, z) = (0, 0, L). Eqn (52) yields
d,z evaluated at (x, y, z) = (0, 0, L). Eqn (52) yields| |  | (58) |
and hence| |  | (59) |
With n given by
| | | n = (sinθcosϕ, sinθsinϕ, cosθ) | (60) |
one arrives at
| | 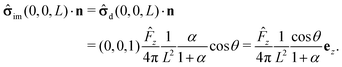 | (61) |
C Density profiles
In this appendix we report the number density profiles of the products of the catalytic reaction for homogeneously covered active particles at various distances from the interface. Fig. 4 shows the quantitative difference between the exact solution, as obtained by using the bi-polar coordinates, and the far-field approximation. In particular, we find that these differences persist even for large distances L/R ≫ 1 from the interface. Finally, in the limit L → ∞ the homogeneously covered particle is exposed to a homogeneous medium. In such a situation the density around the particle is spherically symmetric (i.e., independent of θ) and the far-field expansion, which in this case reduces to a monopole, obviously becomes exact. Accordingly, in this situation the far-field predictions coincide with those of the exact solution obtained in bi-polar coordinates.
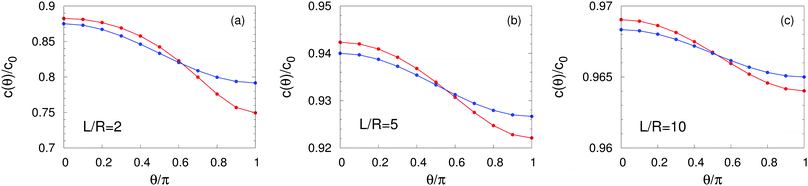 |
| | Fig. 4 Number density profiles at the surface of a homogeneously covered active particle near a fluid–fluid interface for λα = 5 and L/R = 2 (panel (a)), L/R = 5 (panel (b)), and L/R = 10 (panel (c)), normalized by the number density c0 = QR/D1 at the surface of the same particle suspended in a homogeneous unbounded fluid medium (λα = 1). The red lines correspond to the exact solution (eqn (36)) whereas the blue lines provide the far-field approximation (eqn (23)). The concentration is highest (lowest) at the north (south) pole which is distant (close) to the interface. The far-field approximation underestimates (overestimates) c(θ) on the northern (southern) hemisphere even at large values L/R (panel (c)). Upon increasing L/R → ∞ (i.e., moving the particle farther from the interface) both curves flatten and approach the value c(θ)/c0 ≡ 1, as expected for a homogeneously active sphere. | |
D Slip velocity for an arbitrary multipole contribution
We express the number density c(r), with r in fluid 1 and measured from the location of the particle, as the sum of two series (see Fig. 5 for the definition of the primed quantities):| | 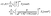 | (62) |
where| |  | (63) |
The coefficients cn are defined as| |  | (64) |
and are obtained from the solution of eqn (1), (2), and (6) in a homogeneous unbounded fluid, i.e., with μ1 = μ2, D1 = D2, λ = 1, and no interface. The coefficients cn′ are the amplitudes of the images which have to be accounted for in order to fulfill the boundary conditions at the fluid–fluid interface, i.e., eqn (3)–(5). Given the amplitude of the image for a point source, the amplitudes of the images of higher multipolar contributions can be derived straightforwardly by recalling that higher order multipoles can be obtained as sets of monopoles (see Fig. 5). Concerning the latter the boundary conditions in eqn (3) and (5) lead to| |  | (65) |
In particular, due to the reflection symmetry of the higher order images about the interface (see Fig. 5), one has cn × cn′ > 0 for n = 2i and cn × cn′ < 0 for n = 2i + 1,  , respectively. Accordingly, one finds
, respectively. Accordingly, one finds| |  | (66) |
Substituting eqn (62) into eqn (22) renders the slip velocity:| | 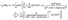 | (67) |
For the lowest orders one finds the following:
 |
| | Fig. 5 Schematic representation of the set of images occurring within the multipole expansion. The active colloid is located at z = L; its first image is located at z = −L. The red dots indicate monopoles with positive amplitudes (sources) whereas the black dots refer to monopoles with negative amplitudes (sinks). Solely for visual clarity, these higher order multipoles and their images, located at z = L and z = −L, respectively, are shown here in positions shifted to the left of the z-axis. | |
• At zeroth order,  , in the expansion only the “source” terms, which are independent of L, contribute to the slip velocity:
, in the expansion only the “source” terms, which are independent of L, contribute to the slip velocity:
| |  | (68) |
• At first order,  , in the expansion there is no contribution to the slip velocity:
, in the expansion there is no contribution to the slip velocity:
• At second order,  , in the expansion the contribution to the slip velocity reads
, in the expansion the contribution to the slip velocity reads
| | 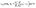 | (70) |
In order to obtain eqn (70) we have used the relation
| |  | (71) |
where
r′ is defined in
eqn (63). Upon expanding the latter one obtains
| | 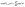 | (72) |
For a homogeneously covered particle, by using
eqn (65),
eqn (70) reduces to
eqn (24) in the main text:
| |  | (73) |
where we have substituted

.
Alternatively, for a purely dipolar contribution eqn (70) reduces to eqn (30) in the main text:
| | 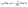 | (74) |
where we have substituted

.
Acknowledgements
Open Access funding provided by the Max Planck Society.
References
- E. Lauga and T. Powers, Rep. Prog. Phys., 2009, 72, 096601 CrossRef.
- S. Ebbens and J. Howse, Soft Matter, 2010, 6, 726–738 RSC.
- J. Elgeti, R. Winkler and G. Gompper, Rep. Prog. Phys., 2015, 78, 056601 CrossRef CAS PubMed.
- C. Bechinger, R. Di Leonardo, H. Löwen, C. Reichhardt, G. Volpe and G. Volpe, Rev. Mod. Phys., 2016, 88, 045006 CrossRef.
- S. Campuzano, B. Esteban-Fernández de Ávila, P. Yanez-Sedeno, J. Pingarron and J. Wang, Chem. Sci., 2017, 8, 6750 RSC.
- P. Diez, B. Esteban-Fernández de Ávila, D. Ramirez-Herrera, R. Villalonga and J. Wang, Nanoscale, 2017, 9, 14307 RSC.
- B. Gao, B. Esteban-Fernández de Ávila, L. Zhang and J. Wang, Adv. Drug Delivery Rev., 2017 DOI:10.1016/j.addr.2017.09.002.
-
W. C. K. Poon, Proceedings of the International School of Physics “Enrico Fermi”, Course CLXXXIV “Physics of Complex Colloids”, Amsterdam, 2013, p. 317 Search PubMed.
- W. Paxton, K. Kistler, C. Olmeda, A. Sen, S. Angelo, Y. Cao, T. Mallouk, P. Lammert and V. Crespi, J. Am. Chem. Soc., 2004, 126, 13424 CrossRef CAS PubMed.
- R. Golestanian, T. Liverpool and A. Ajdari, Phys. Rev. Lett., 2005, 94, 220801 CrossRef PubMed.
- J. Howse, R. Jones, A. Ryan, T. Gough, R. Vafabakhsh and R. Golestanian, Phys. Rev. Lett., 2007, 99, 048102 CrossRef PubMed.
- G. Rückner and R. Kapral, Phys. Rev. Lett., 2007, 98, 150603 CrossRef PubMed.
- F. Jülicher and J. Prost, Eur. Phys. J. E: Soft Matter Biol. Phys., 2009, 29, 27 CrossRef PubMed.
- M. N. Popescu, S. Dietrich and G. Oshanin, J. Chem. Phys., 2009, 130, 194702 CrossRef CAS PubMed.
- M. N. Popescu, S. Dietrich, M. Tasinkevych and J. Ralston, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 31, 351 CrossRef CAS PubMed.
- B. ten Hagen, S. van Teeffelen and H. Löwen, J. Phys.: Condens. Matter, 2011, 23, 194119 CrossRef CAS PubMed.
- B. Sabass and U. Seifert, J. Chem. Phys., 2012, 136, 064508 CrossRef PubMed.
- N. Sharifi-Mood, J. Koplik and C. Maldarelli, Phys. Fluids, 2013, 25, 012001 CrossRef.
- R. Kapral, J. Chem. Phys., 2013, 138, 202901 CrossRef PubMed.
- A. Brown, W. Poon, C. Holm and J. de Graaf, Soft Matter, 2017, 13, 1200 RSC.
- Y. Hong, D. Velegol, N. Chaturvedi and A. Sen, Phys. Chem. Chem. Phys., 2010, 12, 1423 RSC.
- S. Ebbens, M.-H. Tu, J. Howse and R. Golestanian, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 020401 CrossRef PubMed.
- T.-C. Lee, M. Alarcón-Correa, C. Miksch, K. Hahn, J. Gibbs and P. Fischer, Nano Lett., 2014, 14, 2407 CrossRef CAS PubMed.
- X. Wang, M. In, C. Blanc, M. Nobili and A. Stocco, Soft Matter, 2015, 11, 7376 RSC.
- E. Lauga, W. Di Luzio, G. Whitesides and H. Stone, Biophys. J., 2006, 90, 400 CrossRef CAS PubMed.
- A. Berke, L. Turner, H. Berg and E. Lauga, Phys. Rev. Lett., 2008, 101, 038102 CrossRef PubMed.
- D. Lopez and E. Lauga, Phys. Fluids, 2014, 26, 071902 CrossRef.
- J. Hu, A. Wysocki, R. Winkler and G. Gompper, Sci. Rep., 2015, 5, 9586 CrossRef PubMed.
- D. Pimponi, M. Chinappi, P. Gualtieri and C. M. Casciola, J. Fluid Mech., 2016, 789, 514 CrossRef CAS.
- E. Yariv, Phys. Rev. Fluids, 2016, 1, 032101(R) CrossRef.
- C. Liu, C. Zhou, W. Wang and H. P. Zhang, Phys. Rev. Lett., 2016, 117, 198001 CrossRef PubMed.
- W. E. Uspal, M. N. Popescu, S. Dietrich and M. Tasinkevych, Soft Matter, 2015, 11, 434 RSC.
- Y. Ibrahim and T. B. Liverpool, Eur. Phys. J.: Spec. Top., 2016, 225, 1843 CrossRef CAS.
- A. Mozaffari, N. Sharifi-Mood, J. Koplik and C. Maldarelli, Phys. Fluids, 2016, 28, 053107 Search PubMed.
- S. Das, A. Garg, A. Campbell, J. Howse, A. Sen, D. Velegol, R. Golestanian and S. Ebbens, Nat. Commun., 2015, 6, 8999 CrossRef CAS PubMed.
- J. Simmchen, J. Katuri, W. Uspal, M. N. Popescu, M. Tasinkevych and S. Sánchez, Nat. Commun., 2016, 7, 10598 CrossRef CAS PubMed.
- W. E. Uspal, M. N. Popescu, S. Dietrich and M. Tasinkevych, Phys. Rev. Lett., 2016, 117, 048002 CrossRef CAS PubMed.
- S. Michelin and E. Lauga, Eur. Phys. J. E: Soft Matter Biol. Phys., 2015, 38, 7 CrossRef PubMed.
- N. Sharifi-Mood, A. Mozaffari and U. Córdova-Figueroa, J. Fluid Mech., 2016, 798, 910 CrossRef.
- P. Bayati and A. Najafi, J. Chem. Phys., 2016, 144, 134901 CrossRef PubMed.
- K. Dietrich, D. Renggli, M. Zanini, G. Volpe, I. Buttinoni and L. Isa, New J. Phys., 2017, 19, 065008 CrossRef.
- H. Masoud and M. Shelley, Phys. Rev. Lett., 2014, 112, 128304 CrossRef PubMed.
- A. Würger, J. Fluid Mech., 2014, 752, 589 CrossRef.
- A. Pototsky, U. Thiele and H. Stark, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90, 030401 CrossRef PubMed.
- P. Malgaretti, M. N. Popescu and S. Dietrich, Soft Matter, 2016, 12, 4007 RSC.
- A. Dominguez, P. Malgaretti, M. N. Popescu and S. Dietrich, Phys. Rev. Lett., 2016, 116, 078301 CrossRef PubMed.
- A. Dominguez, P. Malgaretti, M. N. Popescu and S. Dietrich, Soft Matter, 2016, 12, 8398 RSC.
- A. Leshansky, A. Golovin and A. Nir, Phys. Fluids, 1997, 9, 2818 CrossRef CAS.
- S. Michelin, E. Lauga and D. Bartolo, Phys. Fluids, 2013, 25, 061701 Search PubMed.
- P. de Buyl, A. Mikhailov and R. Kapral, EPL, 2013, 103, 60009 CrossRef CAS.
- G. Oshanin, M. Popescu and S. Dietrich, J. Phys. A: Math. Theor., 2017, 50, 134001 CrossRef.
-
V. Bagotsky, Fundamentals of Electrochemistry, John Wiley & Sons, Hoboken, NJ, 2006 Search PubMed.
- J. L. Anderson, Annu. Rev. Fluid Mech., 1989, 21, 61 CrossRef.
-
H. A. Lorentz, Zittingsverslag van de Koninklijke Akademie van Wetenschappen te Amsterdam, 1896, vol. 5, p. 168 Search PubMed.
-
J. Happel and H. Brenner, Low Reynolds Number Hydrodynamics, Prentice-Hall, Englewood Cliffs, NJ, 1965 Search PubMed.
- H. K. Kuiken, J. Eng. Math., 1996, 30, 19 CrossRef.
- A. Sellier and L. Pasol, Eur. J. Mech. B, 2011, 30, 76 CrossRef.
- H. A. Stone and A. D. T. Samuel, Phys. Rev. Lett., 1996, 77, 4102 CrossRef CAS PubMed.
- S. Lee and L. Leal, J. Fluid Mech., 1980, 98, 193 CrossRef.
- D. Papavassiliou and G. Alexander, J. Fluid Mech., 2017, 813, 618 CrossRef.
- M. N. Popescu, W. E. Uspal, M. Tasinkevych and S. Dietrich, Eur. Phys. J. E: Soft Matter Biol. Phys., 2017, 40, 42 CrossRef CAS PubMed.
- K. Aderogba and J. Blake, Bull. Aust. Math. Soc., 1978, 18, 345 CrossRef.
- M. N. Popescu, M. Tasinkevych and S. Dietrich, EPL, 2011, 95, 28004 CrossRef.
- H. Brenner, Chem. Eng. Sci., 1961, 16, 242 CrossRef CAS.
- G. Jeffery, Proc. R. Soc. London, Ser. A, 1912, 87, 109 CrossRef.
- E. Bart, Chem. Eng. Sci., 1968, 23, 193 CrossRef.
Footnotes |
| † Here we assume that the advection of the products and reactants by the hydrodynamic flow is negligible compared with their diffusion so that motion of the particle due to a spontaneous symmetry breaking49,50 does not occur. |
| ‡ We remind that eqn (23) has been obtained by accounting only for those images needed to enforce the boundary conditions at the fluid interface. Additional images are needed to enforce the boundary condition on the surface of the colloidal particle.33 In order to keep the model simple we disregard the latter ones which, in principle, provide additional contributions to eqn (23). Such approximate results are sufficient to describe the qualitative behavior of active colloids close to fluid interfaces (as discussed in the main text). |
| § The velocity Uz is the instantaneous velocity an active particle will attain when its center is at distance L apart from the interface. Under the assumptions of the model (fast diffusion of solute, quasi-steady state instantaneously attained) the dynamics of the system is in the overdamped regime. Hence, the velocity of the particle does not depend on initial conditions and equals the one that would be observed by fixing an active colloid at position L (for example by an optical trap) and suddenly releasing it. |
¶ The amplitude of the image of a dipole can be obtained by recalling that a dipole is obtained from a pair of point sources of opposite magnitude located at a distance d apart (see ref. 46 for the derivation of the magnitude of the image of a point source). Accordingly, the image of a dipole with dipole moment P located at z = h is a dipole, located at z = −h, with dipole moment  (see Appendix D). (see Appendix D). |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
M. N.
Popescu
ab and
S.
Dietrich
ab
*ab,
M. N.
Popescu
ab and
S.
Dietrich
ab
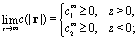
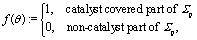
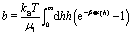
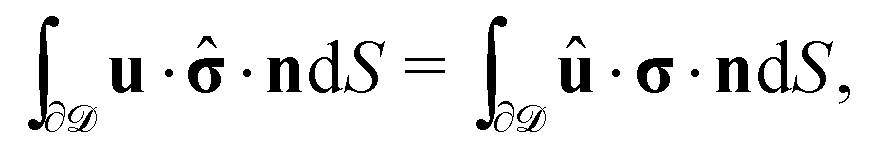
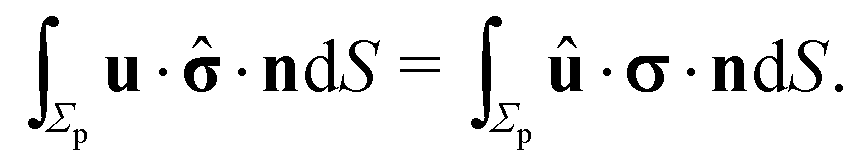
 ) and that Û and U do not vary along the surface of the particle and therefore are independent of rp (even though they retain a dependence on the distance L of the center of the sphere from the interface), and that Û and U have a z-component only, one arrives at
) and that Û and U do not vary along the surface of the particle and therefore are independent of rp (even though they retain a dependence on the distance L of the center of the sphere from the interface), and that Û and U have a z-component only, one arrives at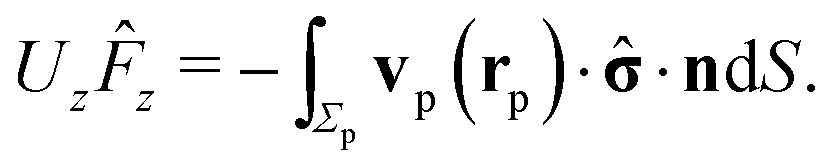
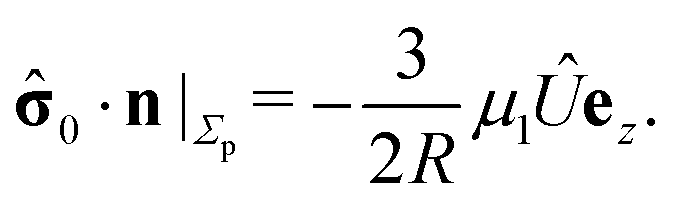
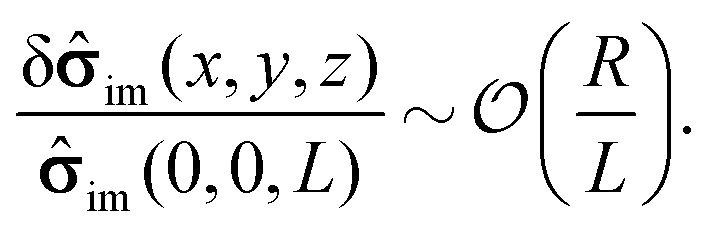
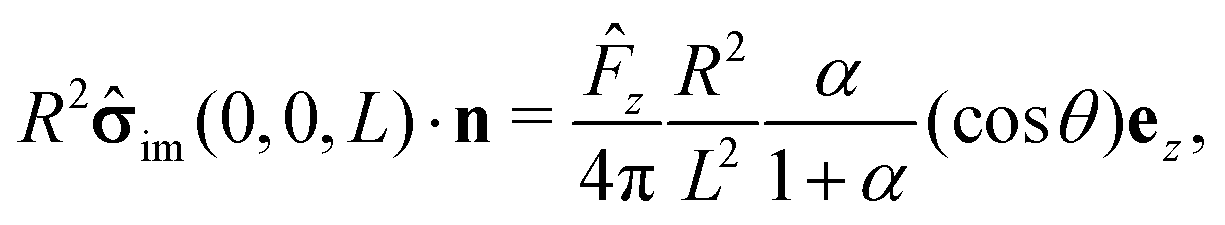
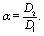
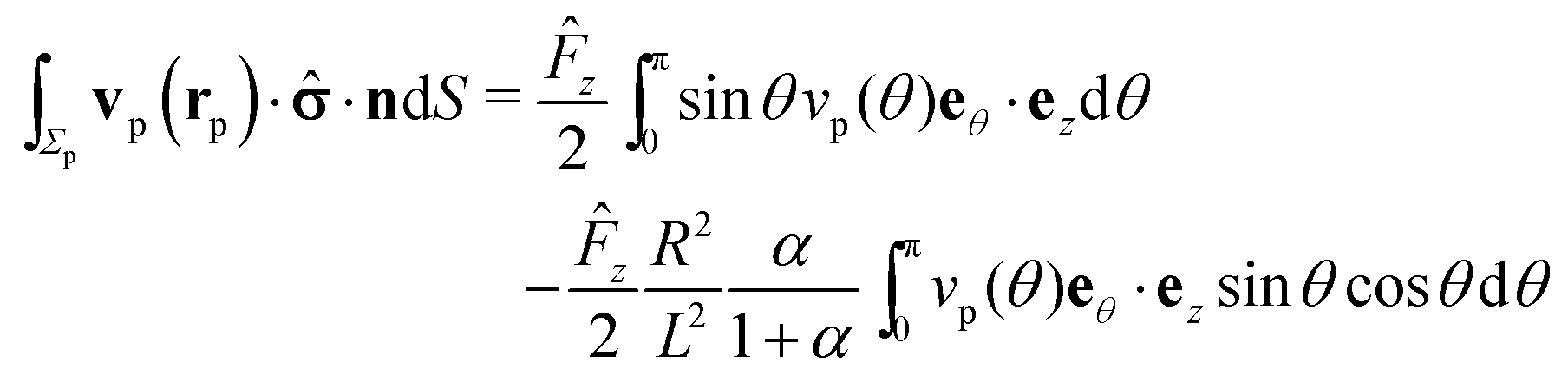
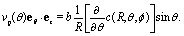
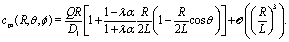
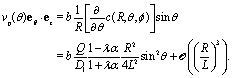
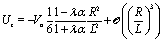
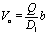
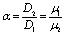 ), and Vo < 0 (eqn (26)). The inset shows the ratio of the exact velocity Ubpz obtained by using bi-polar coordinates and the velocity Uffz calculated within the far-field approximation.
), and Vo < 0 (eqn (26)). The inset shows the ratio of the exact velocity Ubpz obtained by using bi-polar coordinates and the velocity Uffz calculated within the far-field approximation.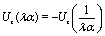 . Since replacing λα by
. Since replacing λα by 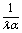 amounts to interchanging media 1 and 2, one concludes that, if one would perform an experiment in which the active particle is placed at z = L and another one with an identical particle now placed at z = −L the outcome will be the following. If in the first experiment the particle will move towards the interface, then in the second experiment it will move away from it, the speed of the motions being the same in the two cases. Vice versa, if in the first experiment the particle will move away from the interface, in the second it will move towards the interface, with the same speed of motion.
amounts to interchanging media 1 and 2, one concludes that, if one would perform an experiment in which the active particle is placed at z = L and another one with an identical particle now placed at z = −L the outcome will be the following. If in the first experiment the particle will move towards the interface, then in the second experiment it will move away from it, the speed of the motions being the same in the two cases. Vice versa, if in the first experiment the particle will move away from the interface, in the second it will move towards the interface, with the same speed of motion.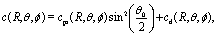
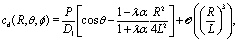

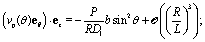
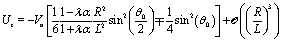
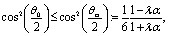
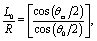
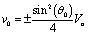 (eqn (31)) already in a homogeneous fluid. Therefore, if such particles are close to a fluid–fluid interface the interface induced velocity
(eqn (31)) already in a homogeneous fluid. Therefore, if such particles are close to a fluid–fluid interface the interface induced velocity 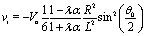 (eqn (31)) sums up with v0, leading to rich scenarios. For example, a particle with sufficiently large coverage θ0 (eqn (32)) experiences a stagnation point close to the interface (eqn (33)) where its velocity vanishes (Fig. 3(a)). If stable, these states are the equivalent of the steady-state hovering near a planar wall. Clearly, for this kind of particles adsorption at the interface is hindered due to the interface-induced component of the velocity. On the contrary, for coverages below the critical value given by eqn (32) such a tug-of-war scenario between the eigen-velocity of the particle and the interface-induced velocity does not occur and the sign of the velocity is always determined by the eigen-velocity v0.
(eqn (31)) sums up with v0, leading to rich scenarios. For example, a particle with sufficiently large coverage θ0 (eqn (32)) experiences a stagnation point close to the interface (eqn (33)) where its velocity vanishes (Fig. 3(a)). If stable, these states are the equivalent of the steady-state hovering near a planar wall. Clearly, for this kind of particles adsorption at the interface is hindered due to the interface-induced component of the velocity. On the contrary, for coverages below the critical value given by eqn (32) such a tug-of-war scenario between the eigen-velocity of the particle and the interface-induced velocity does not occur and the sign of the velocity is always determined by the eigen-velocity v0.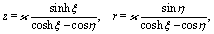
 with ξ0 = arccosh(L/R) is chosen such that the manifold ξ = ξ0 corresponds to the spherical surface of radius R centered at z = L (which is the surface of the particle). The plane z = 0 of the interface corresponds to ξ = 0. In order to simplify the notations we introduce the quantity ω := cos
with ξ0 = arccosh(L/R) is chosen such that the manifold ξ = ξ0 corresponds to the spherical surface of radius R centered at z = L (which is the surface of the particle). The plane z = 0 of the interface corresponds to ξ = 0. In order to simplify the notations we introduce the quantity ω := cos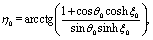
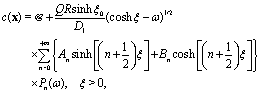
 , Ân, and
, Ân, and 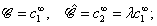
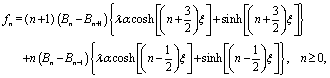
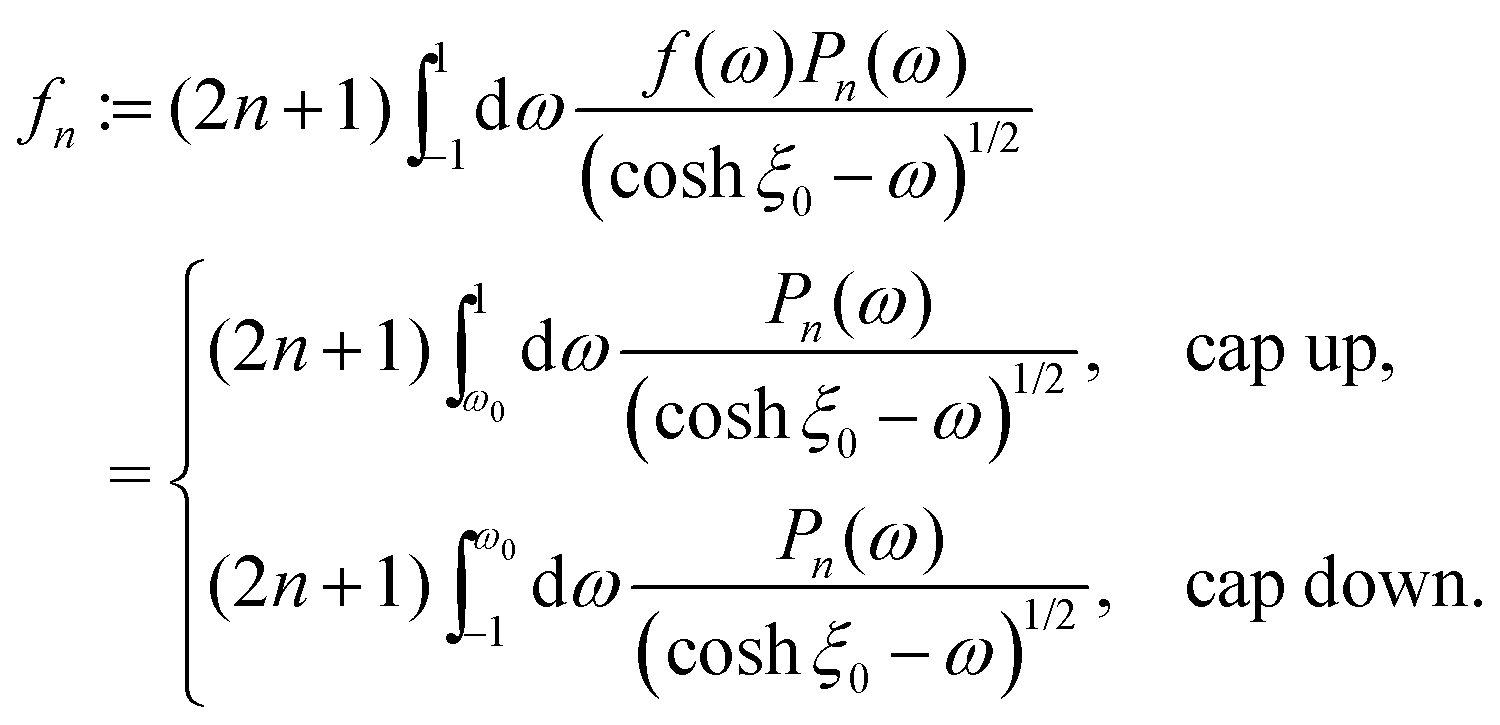
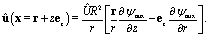
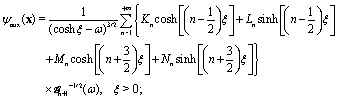
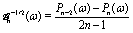
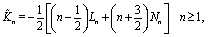
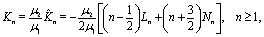
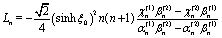
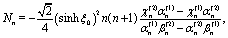
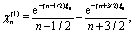
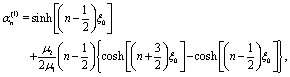
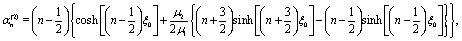
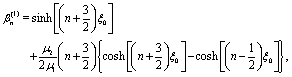
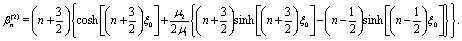
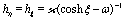 denote the so-called scale (metric) factors corresponding to the η and ξ coordinates, respectively (see eqn (34) and recall the abbreviation ω = cos
denote the so-called scale (metric) factors corresponding to the η and ξ coordinates, respectively (see eqn (34) and recall the abbreviation ω = cos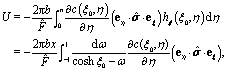
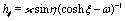 denotes the scale factor corresponding to the ϕ coordinate.
denotes the scale factor corresponding to the ϕ coordinate.
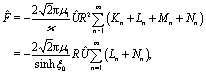
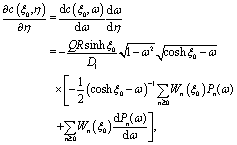
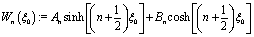 (see eqn (36)); the coefficients An and Bn are determined from eqn (38) and (39), as well as from the relation An = (λα)Bn.
(see eqn (36)); the coefficients An and Bn are determined from eqn (38) and (39), as well as from the relation An = (λα)Bn.
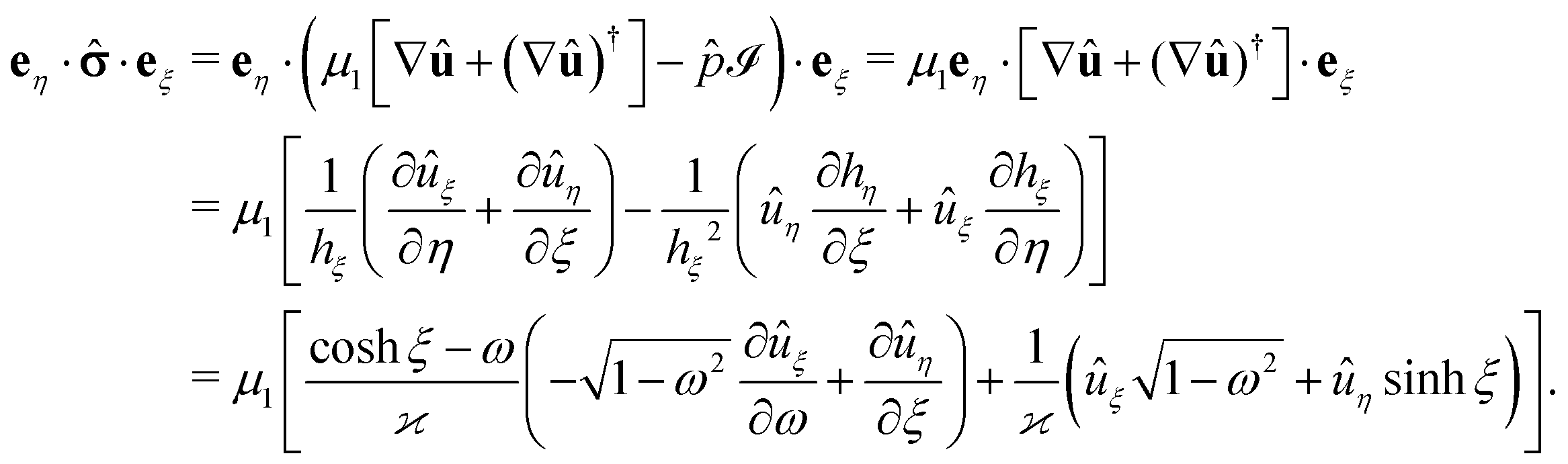
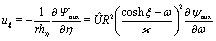
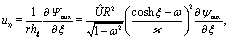
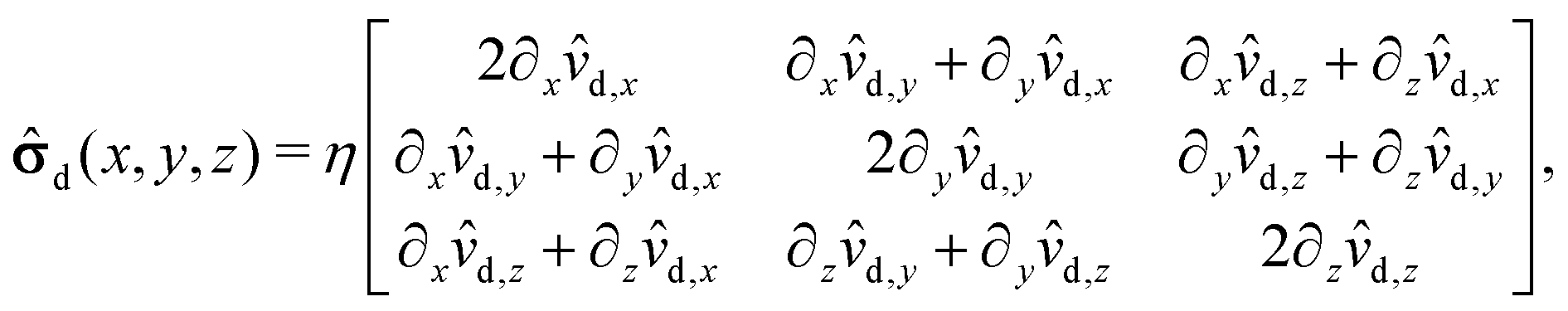
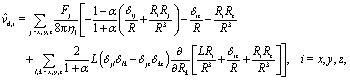
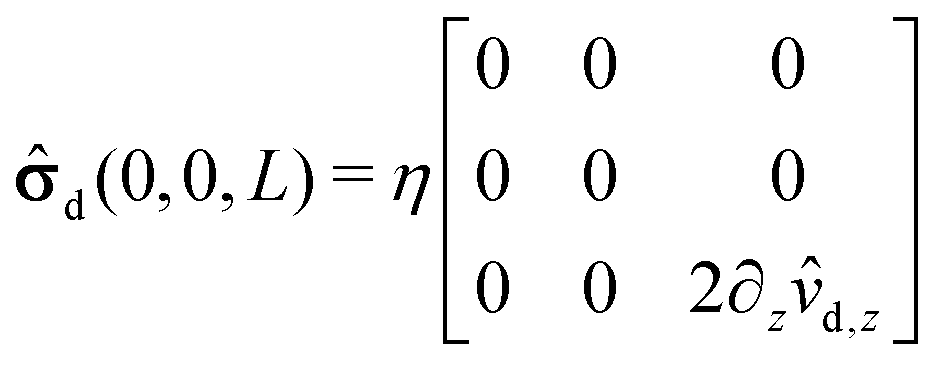
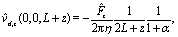
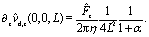
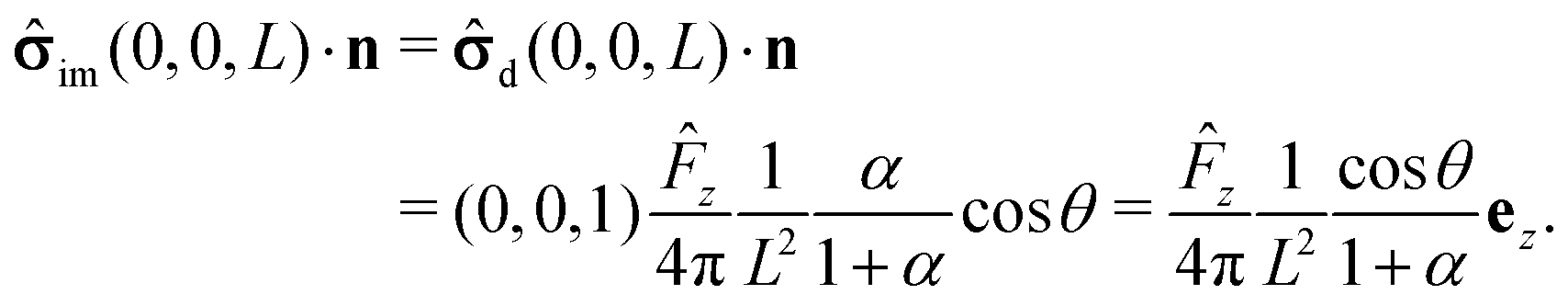
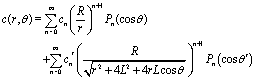
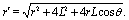
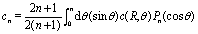
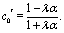
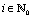 , respectively. Accordingly, one finds
, respectively. Accordingly, one finds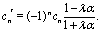
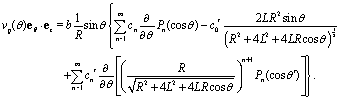
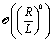 , in the expansion only the “source” terms, which are independent of L, contribute to the slip velocity:
, in the expansion only the “source” terms, which are independent of L, contribute to the slip velocity: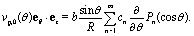
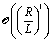 , in the expansion there is no contribution to the slip velocity:
, in the expansion there is no contribution to the slip velocity: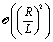 , in the expansion the contribution to the slip velocity reads
, in the expansion the contribution to the slip velocity reads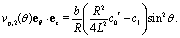
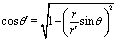
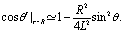
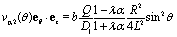
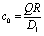 .
.
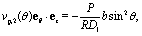
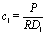 .
.
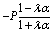 (see Appendix D).
(see Appendix D).