
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antimicrobial action of the cationic peptide, chrysophsin-3: a coarse-grained molecular dynamics study†
Andrea
Catte
a,
Mark R.
Wilson
 b,
Martin
Walker
b and
Vasily S.
Oganesyan
*a
b,
Martin
Walker
b and
Vasily S.
Oganesyan
*a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: v.oganesyan@uea.ac.uk
bDepartment of Chemistry, Durham University, University Science Laboratories, South Road, Durham, DH1 3 LE, UK
First published on 29th March 2018
Abstract
Antimicrobial peptides (AMPs) are small cationic proteins that are able to destabilize a lipid bilayer structure through one or more modes of action. In this study, we investigate the processes of peptide aggregation and pore formation in lipid bilayers and vesicles by the highly cationic AMP, Chrysophsin-3 (chrys-3), using coarse-grained molecular dynamics (CG-MD) simulations and potential of mean force calculations. We study long 50 μs simulations of chrys-3 at different concentrations, both at the surface of dipalmitoylphosphatidylcholine (DPPC) and palmitoyloleoylphosphatidylcholine (POPC) bilayers, and also interacting within the interior of the lipid membrane. We show that aggregation of peptides at the surface, leads to pronounced deformation of lipid bilayers, leading in turn to lipid protrusions for peptide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratios > 1:12. In addition, aggregation of chrys-3 peptides within the centre of a lipid bilayer leads to spontaneous formation of pores and aggregates. Both mechanisms of interaction are consistent with previously reported experimental data for chrys-3. Similar results are observed also in POPC vesicles and mixed lipid bilayers composed of the zwitterionic lipid palmitoyloleoylphosphatidylethanolamine (POPE) and the negatively charged lipid palmitoyloleoylphosphatidylglycerol (POPG). The latter are employed as models of the bacterial membrane of Escherichia coli.
:ligand ratios > 1:12. In addition, aggregation of chrys-3 peptides within the centre of a lipid bilayer leads to spontaneous formation of pores and aggregates. Both mechanisms of interaction are consistent with previously reported experimental data for chrys-3. Similar results are observed also in POPC vesicles and mixed lipid bilayers composed of the zwitterionic lipid palmitoyloleoylphosphatidylethanolamine (POPE) and the negatively charged lipid palmitoyloleoylphosphatidylglycerol (POPG). The latter are employed as models of the bacterial membrane of Escherichia coli.
Introduction
Antimicrobial peptides (AMPs) are small proteins that can be found in the majority of living organisms. They play an essential role in an organism's first-line immune response. Different AMPs have been found to be active against Gram-negative and Gram-positive bacteria, and in some cases are active against fungi, yeasts, and viruses.1,2 Due to their strong antimicrobial activity, there is currently great interest in AMPs, in the design of novel antibiotics.3–5 AMPs have also been proposed as novel therapeutic agents for cancer treatment.6–10Different mechanisms of interaction between AMPs and cell membranes are possible depending on the types of peptide and lipids present in the system.11,12 Some AMPs are believed to insert into lipid bilayers and create pores using a mechanism defined as the barrel-stave model, in which peptides orient perpendicularly to the lipid membrane surface to form cylindrical pores.13 However, many AMPs are able to generate toroidal pores, in which the pore edges consist of peptides and lipid head groups that bend continuously from the top bilayer leaflet to the bottom bilayer.14 The disruption of cell membranes operated by AMPs may also involve their adsorption to the lipid surface and the formation of lipid–peptide particles, which lead to lysis of the membrane, in a mechanism known as the carpet model.15,16 Secondary structure, charge, and hydrophobicity of AMPs all play a significant role in determining the mechanisms of action on a cell membrane. Increased α-helical content in AMPs can be correlated with their increased antibacterial activity, although molecular flexibility is also known to play a role in enhancing the ability of AMPs to kill bacteria.17
While erythrocyte plasma membranes are composed of zwitterionic lipids, Gram-positive and Gram-negative bacterial membranes (as well as fungal cell membranes) are characterized by comparably large amounts of negatively charged lipids, in addition to zwitterionic lipids.18 Hence, cationic AMPs are known to interact strongly with these membranes.19 Chrysophsin-3 (chrys-3) is a recently discovered, highly cationic 20-amino acid AMP, derived from the gills of the red sea bream, Chyrsophrys major.20 It has an unusual C-terminal domain (RRRH sequence) that contributes to a net positive charge of + 5 (fractional charge of 3.2 at pH 7), is amphipathic21 and assumes a α-helical structure when in contact with a biological membrane. While the positive charge is concentrated near the C-terminus, the rest of the peptide is significantly hydrophobic, as measured by both the Urry and Eisenberg scales. A consequence of this structure is that chrys-3 has the possibility of lipid bilayer insertion as a single molecule or as an aggregate.20,22 As expected, the removal of the C-terminal sequence RRRH increases the peptide's affinity to neutral and cholesterol-rich membranes as confirmed by 2H, 15N and 31P solid-state NMR.23 Moreover, this truncated form of chrys-3, which has a reduced α-helical content, is less effective in the formation of pores and less harmful to eukaryotic cells.22,23
AMPs derived from fish have become increasingly popular due to their prominent role as pharmaceuticals with important medical applications.24 Chrys-3 has recently been shown to kill the sporulated, germinated and vegetative stages of Bacillus anthracis and to penetrate and kill the spores without full germination.25 Chrysophsin-1 (chrys-1), an isoform of chrys-3 with 25 amino acids and the same C-terminal RRRH sequence, is a promising protecting agent in oral disease treatment because of its ability to kill the cariogenic pathogen Streptococcus mutans.26 Moreover, chrys-1 has also shown antitumoral activity by both inhibiting the growth of, and killing, the cancer-derived cell line HT1080 in RAW264.7 cells.27 Antimicrobial surfaces with immobilized chrys-1 peptides show an antibacterial activity capable of killing around 82% of E. coli bacteria.28 Recently, the GXXXXG motif of chrys-1 has been studied by replacing glycine residues with alanine, valine and proline residues, showing that proline-substituted chrys-1 peptides exhibited significantly reduced cytotoxicity towards mammalian cells.29 Interestingly, in the development of self-decontaminating coatings, the antimicrobial activity of chrys-1 and chrys-3 has also been shown to be severally reduced (chrys-1) or abolished (chrys-3) by the removal of the C-terminal RRRH.30
Recently, Wang et al. have employed a quartz crystal microbalance with dissipation monitoring (QCM-D) to study the interactions of chrys-3 and other AMPs with a membrane model containing egg PC in a solid supported lipid bilayer (SLB).21 The main lipid component of egg PC is the zwitterionic lipid palmitoyloleoylphosphatidylcholine (POPC). The authors have observed that chrys-3 peptides show three modes of interaction with the lipid membrane, namely, membrane insertion, pore formation and surface adsorption.
Molecular dynamics (MD) simulations provide a powerful technique for studying the action of AMPs on membranes. All-atom (AA) MD simulations of magainin-H2 have shown that its interaction with dipalmitoylphosphatidylcholine (DPPC) lipid membranes involves both adsorption on the lipid bilayer surface and the formation of disordered toroidal pores, in agreement with NMR experiments.31 Sengupta et al. reported AA MD simulations of melittin peptides interacting with DPPC lipid bilayers showing the formation of disordered toroidal pores above a critical peptide to lipid ratio.32 The authors also found that, in the absence of counterions, pores form more rapidly leading to the rupture of the lipid membrane. AA MD simulations of melittin in water, in methanol and in a dimyristoylphosphatidylcholine (DMPC) lipid bilayer have shown the partial unfolding of the peptide in solution.33 In a recent study Irudayam et al. have observed that the binding of melittin to a POPC lipid bilayer involves the folding of the peptide to adopt a “U-shaped” conformation, which facilitates the formation of pores.34 A combination of coarse-grained (CG) and AA MD simulations has also been used to show the formation of disordered pores and peptide aggregates by alamethicin peptides interacting with DMPC lipid bilayers.35 This diversity of alamethicin structures within a lipid bilayer is in agreement with solid-state NMR experiments. CG MD simulations were also employed to study the interaction of maculatin 1.1 with DPPC lipid bilayers and a POPC liposome at different peptide:lipid (P:L) molar ratios.36 At low P:L molar ratios maculatin 1.1 interacted mainly with the surface of lipid membranes, while at higher P:L molar ratios it formed aggregates containing a minimum of four peptides in lipid bilayers. In addition to the formation of peptide aggregates, maculatin 1.1 induced major structural changes in the POPC vesicle, implying that changes in the membrane curvature might play a key role in cell lysis. Rzepiela et al. employed a similar approach to study the interaction of magainin-H2 with DPPC lipid bilayers observing that the disordered nature of the toroidal pore is conserved during both CG and AA MD simulations.37 Recently, Yoo et al. have observed through CG MD simulations the formation of lipid protrusions in POPC lipid bilayers and vesicles interacting with ionic liquids (ILs).38 CG MD simulations of pores formed by two different AMPs in DPPC lipid membranes have also highlighted that magainin-H2 forms large sized disordered toroidal pores while melittin assembles in small pores with peptides adopting a “U-shaped” conformation.39
In the current study, we perform long CG MD simulations to uncover the mechanisms of interaction of the highly cationic chrys-3 AMP with DPPC, POPC and POPE:POPG lipid bilayers and also with POPC vesicles. We simulate peptide aggregation at the membrane surface and within bilayers; analysing the structure of pores that form. We show that both peptide aggregates and peptide disordered toroidal pores form in lipid bilayers and in vesicles. We find also that at, sufficiently high concentrations, chrys-3 peptides in water will aggregate at the surface of a bilayer (or vesicle), causing disruption to the bilayer structure through formation of lipid protrusions. Both mechanisms appear to be important in the anti-microbial action of this highly cationic AMP.
This paper is organised as follows. Firstly, MD computational protocols are described. Secondly, results are presented showing aggregation of peptides at the bilayer surface and in the centre of the membrane, for both lipid bilayers and vesicles. Thirdly, we analyse the structures of the pores that form (noting the preferential interaction of chrys-3 with anionic lipids); and carry out potential of mean force calculations to characterise the free energy changes associated with the peptide moving from water to the surface adsorbed state and into a peptide pore. Finally, conclusions are presented.
Computational
The simulations in this work are carried out at a coarse-grained molecular level, using the MARTINI force field40 (as discussed in detail below). MARTINI uses a mapping of four heavy atoms to one CG interaction site and has been parametrized with the aim of reproducing thermodynamic properties (transfer free energies between different environments). Full details of each of the simulation components are given below.Chrys-3 peptide
The monomer of chrys-3 was built with the protein builder of VMD version 1.9.141 using the amino acid sequence FIGLLISAGKAIHDLIRRRH. The peptide has six positively charged amino acids, which are represented by one lysine, two histidines and three arginines, and one negatively charged amino acid, which is represented by one aspartic acid residue. The native form of the peptide has its C-terminus amidated and is modelled with a net charge of +5. Based on conclusions from previous experimental studies in trifluoroethanol and model membranes,20,23 the secondary structure assigned for the starting configuration of chrys-3 was almost completely α-helical but with the charged N- and C-termini in a random coil conformation. We note in passing that while we expect MARTINI to be able to model aggregation behaviour well (because of its original parametrisation in terms of transfer free energies), we do not expect MARTINI to be able to model any changes in peptide secondary structure arising from changes in the dielectric environment. Hence, we are effectively using the same secondary structure throughout the simulations.DPPC, POPC and POPE:POPG lipid bilayers and POPC vesicles with chrys-3 peptides
Table 1 provides a summary of each of the CG MD simulations carried out with chrys-3 peptides in lipid bilayers and vesicles. Thirteen separate systems were studied in all, using either surface or transmembrane peptides. The lipid composition of the POPE:POPG lipid bilayer systems (systems 7–10) were designed to be similar to that found in the bacterial membrane of Escherichia coli, namely 90% POPE:10% POPG and 80% POPE:20% POPG in outer and inner leaflets, respectively42,43 (see Table 2). CG DPPC, POPC and POPE:POPG lipid bilayers were generated with the CHARMM-GUI membrane builder.44–47
| System | Lipids | Peptides | P:L |
Time (μs) |
|---|---|---|---|---|
| s: surface chrys-3 peptides; tm: transmembrane chrys-3 peptides. | ||||
| 1. DPPC | 1200 | 40s | 1:30 |
30 |
| 2. DPPC | 1200 | 50tm | 1:21 |
50 |
| 3. DPPC | 1200 | 100s | 1:12 |
50 |
| 4. POPC | 1200 | 40s | 1:30 |
45 |
| 5. POPC | 1058 | 50tm | 1:21 |
50 |
| 6. POPC | 1200 | 100s | 1:12 |
30 |
| 7. POPE:POPG | 1209 | 25s | 1:48 |
50 |
| 8. POPE:POPG | 1209 | 75s | 1:16 |
50 |
| 9. POPE:POPG | 1209 | 100s | 1:12 |
50 |
| 10. POPE:POPG | 1057 | 50tm | 1:21 |
50 |
| 11. POPC ves0 | 2422 | 81s | 1:30 |
20 |
| 12. POPC ves1 | 2422 | 205s | 1:12 |
44 |
| 13. POPC ves2 | 2139 | 100tm | 1:21 |
50 |
In our initial CG-MD simulation work, we used the MARTINI force field with single CG beads to represent water molecules. By convention for MARTINI, approximately 5% of CG beads are replaced by antifreeze water particles48 to prevent the crystallization of water.49 We also carried out calculations with the newer polarizable MARTINI water model.50 Aggregation behaviour for chrys-3 in both models was found to be very similar. Results presented below, for systems 1–10 in Table 1, use the polarizable water model. Owing to very large system sizes, simulations of the vesicle systems (systems 11–13 in Table 1) are carried out only using the conventional none-polarizable MARTINI water model.
The standard cut-offs for the MARTINI force field were used for non-bonded interactions, the Lennard-Jones potential was shifted to zero between 0.9 and 1.2 nm, and the Coulomb potential was shifted to zero between 0 and 1.2 nm with a relative dielectric constant of 2.5 for polarizable water (15 for none-polarizable water). The time step used was 20 fs, and the neighbour list was updated every 10 steps.49 Initially, CG DPPC, POPC and POPE:POPG lipid bilayers containing 1200 lipids were subjected to 5 μs CG-MD simulations at 310 K and 1 atm using the CG MARTINI force field for lipids48 and Gromacs (version 4.5.5) simulation suite,51 prior to production runs (see below). In all simulations, any close molecular contacts were initially removed by energy minimization, followed by pre-equilibration with the Berendsen thermostat and barostat. Thereafter, equilibration and production runs were carried out in the constant-NpT ensemble at 310 K, representing physiological temperature and successfully employed previously in the MARTINI model,40 and 1 atm using a Nosé–Hoover thermostat52 and a Parrinello–Rahman barostat,53 respectively.
All CG lipid systems were solvated with at least 30 water molecules per lipid and ionized with a suitable amount of Na+ and Cl− ions to reach a physiological ionic strength of 150 mM. In the systems containing polarizable CG water the total number of beads, including water and ions, was approximately 50000 for every lipid bilayer.
Suitable starting structures that included chrys-3 molecules were generated by the following procedure involving all-atom models back-mapped from the coarse-grained lipid models: (i) all-atom models of DPPC, POPC and POPE:POPG lipid bilayers were generated from the final structures of the 5 μs CG runs using the back-mapping tool of the MARTINI force field, which involves the use of a library of mapping definitions encoding the geometric reconstruction, energy minimization and MD simulations based relaxation.54 (ii) Fine-grained structures of chrys-3 peptides were inserted into the all-atom simulations and close contacts were removed. (iii) These starting systems were coarse-grained using the CGTools Plugin version 0.1 of VMD.55,56 Two main starting models were generated using different P:L molar ratios: (a) chrys-3 peptides were inserted close to the lipid surface with random parallel or antiparallel orientations relative to each other (as shown in Fig. 1a); (b) chrys-3 peptides were inserted into the lipid membrane at transmembrane positions with the same randomly distribution of parallel and anti-parallel orientations (Fig. 1b and c).
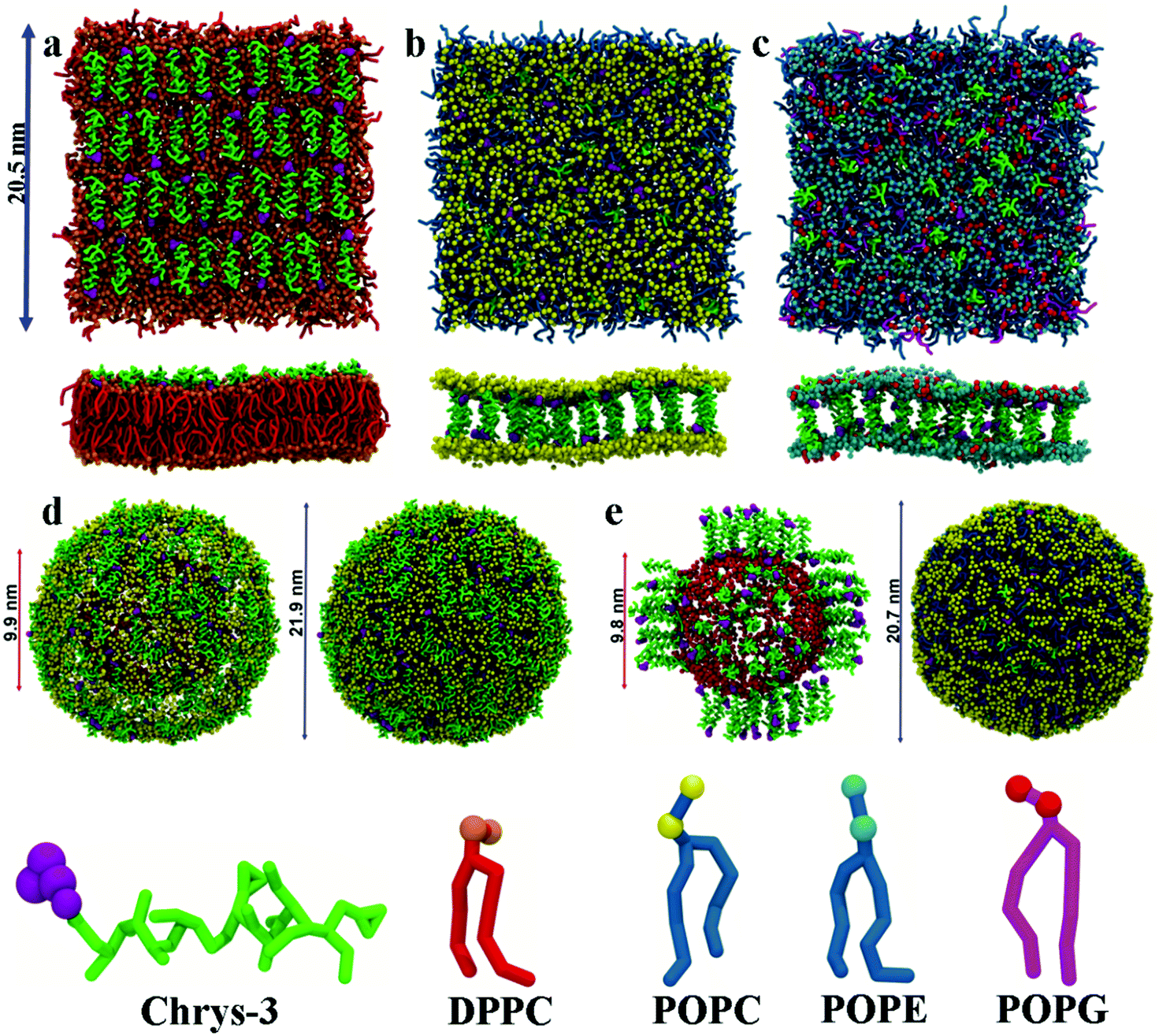 | ||
| Fig. 1 Top and side views of different CG starting structures of lipid bilayers and vesicles with chrys-3: (a) DPPC lipid bilayer with surface peptides; (b) POPC lipid bilayer with transmembrane peptides; (c) POPE:POPG lipid bilayer with transmembrane peptides; (d) two views of POPC vesicle with surface peptides; and (e) two views of POPC vesicle with transmembrane peptides. In both (d) and (e) the inner and outer leaflets diameters of the vesicles are shown. Phenylalanine and the rest of chrys-3 amino acid residues are shown in purple and green, respectively. DPPC, POPC, POPE and POPG polar head groups are shown in orange, yellow, cyan and red, respectively. DPPC, POPC, POPE and POPG hydrophobic tail groups are shown in red, skyblue and magenta, respectively. POPC phosphate beads of the inner leaflet are shown in red. Water and ions are not shown. | ||
The final CG models of DPPC, POPC and POPE:POPG lipid bilayers containing different peptide:lipid molar ratios were subjected to at least 50 μs of CG MD simulations at 310 K and 1 atm using the CG MARTINI force field for lipids48 and proteins57 and Gromacs version 4.5.551 on 32 processors of a 302 node cluster.
CG POPC vesicles containing 2422 lipids were generated via CG MD simulation of a POPC lipid bilayer.58 CG chrys-3 peptides were docked to the surface of the vesicle to generate systems with different P:L molar ratios (Fig. 1d). Insertion of transmembrane chrys-3 peptides into the vesicles was achieved by initially fine graining the CG POPC vesicle, inserting peptides, and deleting any lipid within 0.3 Å of any inserted peptide atom. This resulted in a final P:L molar ratio of 1:21 (Fig. 1e). This starting system was coarse-grained using the CGTools Plugin version 0.1 of VMD.55,56
All CG lipid vesicles with surface and transmembrane chrys-3 peptides were solvated with a large amount of regular MARTINI CG water (at least 300 water molecules per lipid in order to avoid the interaction between periodic images of the vesicle) and ionized with a suitable amount of Na+ and Cl− ions to reach a physiological ionic strength of 150 mM. The largest CG POPC vesicle reached a total number of approximately of 230000 beads (∼1135000 atoms), including waters and ions.
Potential of mean force calculations for chrys-3 peptide with POPC bilayers
In order to calculate approximate Gibbs's free energy of insertions for a chrys-3 peptide into a POPC lipid bilayer, the potential of mean force (PMF) for the insertion of a chrys-3 peptide was calculated using umbrella sampling. Here, two systems were studied. In the first, a single peptide was initially equilibrated within the aqueous component of a solvated POPC bilayer to provide a starting geometry. In the second, a starting configuration was obtained from the endpoint of a 50 μs CG-MD simulation of a lipid bilayer containing 271 POPC lipids and 5 peptides forming a pore. The pull code of Gromacs was used to generate umbrella sampling windows using a force constant of 2200 kJ mol−1 nm−2. For the first system, pulls were carried out with the pulling distance measured relative to the centre of the membrane (defined by the mean position of the terminal aliphatic beads for each lipid chain). For the second system, a pull was started from an initial structure in which the pulling distance was defined as the inter-helical distance between the peptide that was subjected to the pulling force and a reference peptide from the pore.59 In the latter, the initial reference distance was 1.6 nm. Initially, 29 windows with a spacing of 0.1 nm were created to cover the range from 1.6 to 4.5 nm. Twenty-five additional windows were also generated using a force constant of 500 kJ mol−1 nm−2 and spacing values of 0.1, 0.05 and 0.025 nm to improve the sampling at inter-helical distances in which the transitions from transmembrane orientation to surface-adsorbed and from surface-adsorbed to solution state take place. A pulling rate of 0.0001 nm ps−1 was used for both force constants. In the additional windows, we used an umbrella potential with a force constant of 1000 kJ mol−1 nm−2.60 Each window was equilibrated for 2 μs and subjected to a 20 μs production run at 310 K and 1 atm. The first 2.5 μs of each production run were not used for analysis. The weighted histogram analysis method (WHAM) was used to unbias the umbrella window potentials.61Analysis of simulation runs was performed using the utilities of Gromacs version 4.5.5, with additional TCL scripts written for VMD version 1.9.1,41 over the last 40% of each simulation trajectory. Images of all simulated structures and movies were generated with VMD version 1.9.1.
Results and discussion
Aggregation of chrys-3 peptides on the surface of lipid membranes
Initial, CG MD simulations were performed for peptides positioned close to the membrane surface. In all simulations, regardless of lipid composition or peptide concentration, we observe adsorption of peptides on the membrane surface with subsequent formation of peptide surface aggregates, as shown in Fig. 2. In none of these simulations were we able to observe spontaneous insertion of peptides into bilayers, even for extremely long simulation times exceeding 50 μs. The results indicate that the surface-bound state of the peptide is very stable within the MARTINI parameterisation.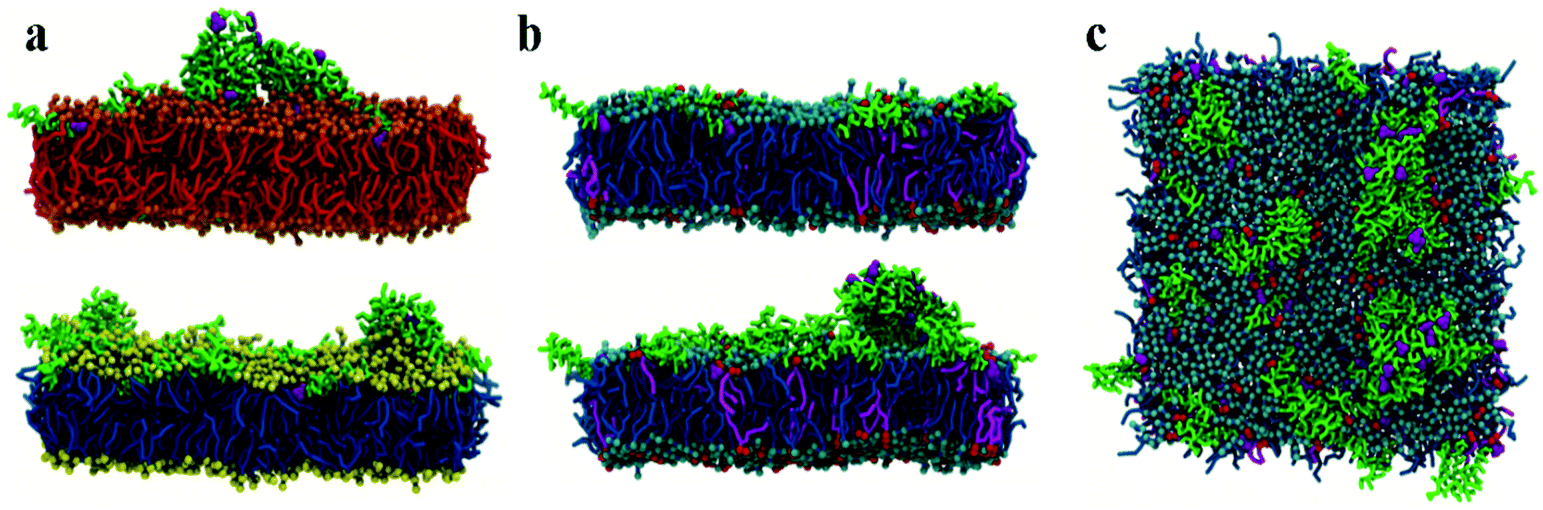 | ||
| Fig. 2 Side views of CG lipid bilayers with surface chrys-3 peptides: (a) DPPC (top) and POPC (bottom) simulated for 30 μs and 45 μs, respectively; (b) 50 μs structures of POPE:POPG with 25 (top) and 75 (bottom) peptides. (c) Top view of the POPE:POPG lipid bilayer with 75 peptides. | ||
In POPE:POPG lipid bilayers similar results are observed together with a clustering of negatively charged POPG molecules around the positively charged surface-adsorbed chrys-3 peptides (see Fig. 2b and c where the head groups of POPG are represented by red spheres). It is worth noting that when the P:L molar ratio reaches a value of 1:12 the formation of differently sized lipid protrusions are observed in every simulated lipid bilayer (Fig. 3a–c). In this respect, chrys-3 peptides act rather like detergents extracting lipid molecules from the membrane, as previously observed experimentally for other antimicrobial peptides.23,62–65 The formation of these lipid protrusions has been monitored also by plotting x and y dimensions of the box, and the product of the x and y dimensions of the box versus the time of simulation (see Fig. S1, ESI†). These plots clearly show that lipid protrusions are formed within the first 20 μs of simulation for POPC and POPE:POPG lipid bilayers, while it takes about 27.5 μs of simulation before a plateau is reached in each subplot of Fig. S1 (ESI†) for the DPPC lipid bilayer.
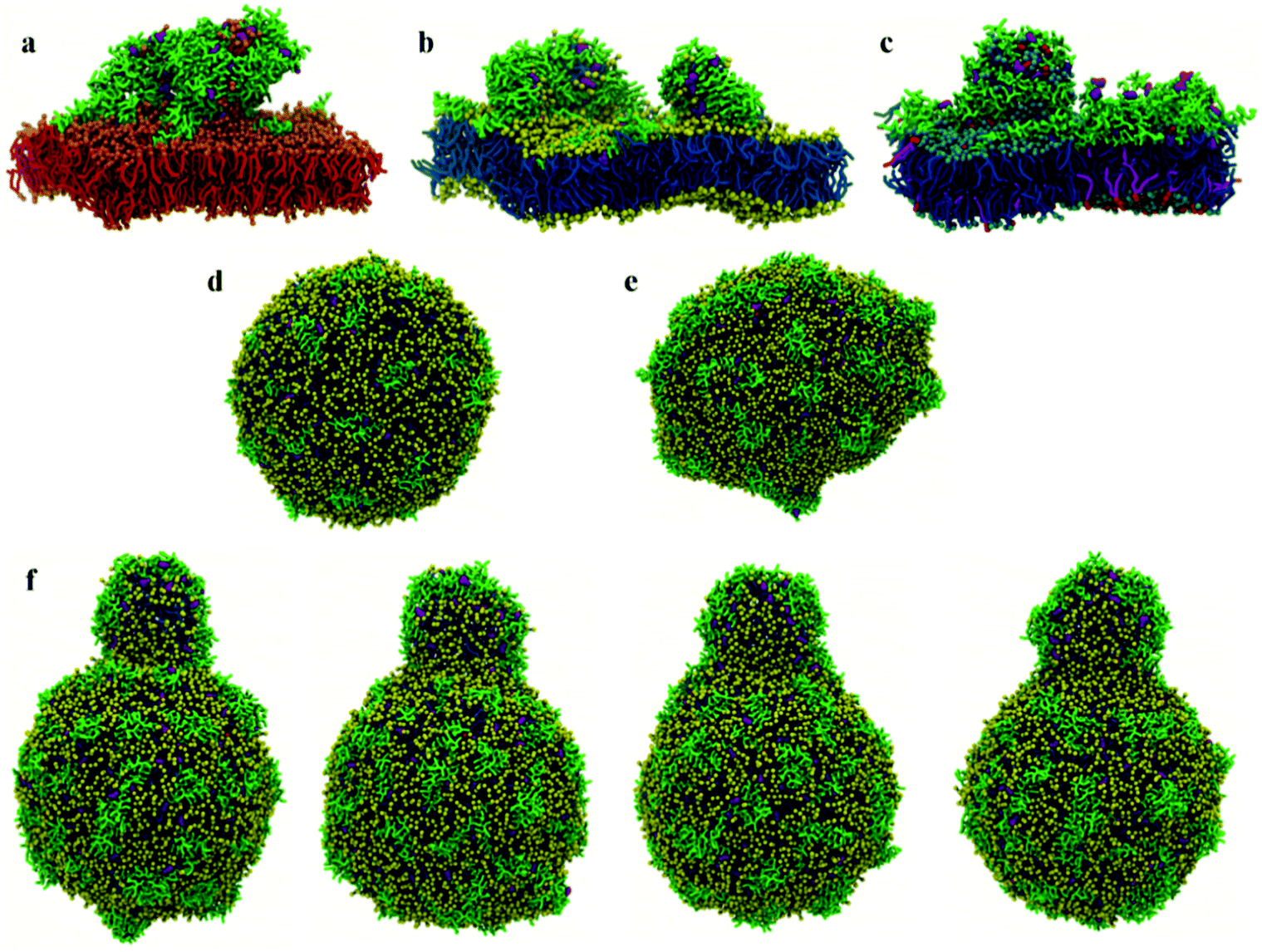 | ||
| Fig. 3 50 μs structures of CG (a) DPPC, (b) POPC and (c) POPE:POPG lipid bilayers with surface chrys-3 peptides (P:L = 1:12) showing the formation of different sized lipid protrusions. CG POPC vesicles with surface chrys-3 peptides: (d) 20 μs structure (P:L = 1:30); (e) 11 μs structure (P:L = 1:12). Note the deformation of the vesicle in (e) at the highest P:L molar ratio; (f) 20 μs, 24 μs, 34 μs and 44 μs structures (P:L = 1:12) show the formation and the growth of a lipid protrusion (from left to right). | ||
At low P:L molar ratios, the interaction of surface chrys-3 peptides with POPC vesicles leads to similar results to the bilayers, with strong surface-adsorption and some clustering of peptides (Fig. 3d). An increase in the P:L molar ratio induces a significant deformation of the POPC vesicle (Fig. 3e), which appears to be an initial staging point prior to the rupture of the vesicle. This is in good agreement with mechanisms seen in previous computational studies of other AMPs.36 Interestingly, in the current system, a significant lipid protrusion is observed after 20 μs of simulation (Fig. 3f) showing the detergent-like behaviour of chrys-3 in contact with lipids. Similar behaviour has been reported previously for apolipoprotein A-I (apoA-I) and other amphipathic peptides interacting with lipid bilayers and vesicles.58 The growth in the size of the lipid protrusion, as the simulation time increases (Fig. 3f), suggests that this behaviour could lead to the formation of lipid particles similar to those observed experimentally.21
Aggregation of chrys-3 peptides within lipid membranes
Starting from initial transmembrane arrangements of peptides, simulations demonstrate peptide aggregation within the transmembrane region of the lipid bilayer. A range of aggregate sizes is seen, as discussed in detail below.At a low P:L molar ratio of 1:45, regardless of their relative starting orientation or lipid composition, peptides adsorb at the bilayer surface within the first 40 ns of a simulation (see Fig. S2, ESI†) and remain there for the remainder of the simulation. However, at a P:L molar ratio of approximately 1:21 the formation of disordered toroidal pores and transmembrane peptide aggregates is observed. Here, a disordered toroidal pore is obtained when there is a local reorganisation of the lipid membrane, with leaflets curving to have some lipid headgroups entangled with peptides; while a peptide aggregate is constituted by a cluster of transmembrane peptides (which may or may not be aligned) without any lipid headgroups between them. Here, during the first 40 ns of the simulation, the lateral diffusion of peptides leads to the formation of small aggregates. Typical behaviour is illustrated by the POPC system in Fig. 4a. Within the POPC bilayer, small peptide clusters form, then grow in size, and during the first 5 μs form pores of different sizes and also a larger aggregate. During the next 45 μs of simulation time, only minor structural rearrangements of pores and the larger aggregate are observed, indicating the stability of these peptide structures (Fig. 4b). The timescale required for the formation of small pores containing 3–4 peptides varies between 100 and 200 ns.32 Interestingly, a single pore containing 3 peptides is not particularly stable with two peptides becoming surface adsorbed after about 500 ns. For the POPC system, there are also 9 peptides that undergo a fast transition from the transmembrane state to the head-group localized state and remain in that state for the remainder of the 50 μs simulation. The complete formation of larger disordered toroidal pores containing 9 and 13 peptides is associated with longer times of 1.7 μs and >25 μs, respectively. For instance, one of the two larger pores is formed by merging two smaller ones containing 7 and 6 peptides, respectively. The fusion of the two pores starts at about 23.5 μs and is completed within 27 μs of simulation time. Once the formation of the pores and the transmembrane aggregate are completed, they remain stable through the remaining time of the simulation. In order to estimate the sizes of the formed pores we have fine grained the system using the last frame of the 50 μs structure according to the reverse coarse graining procedure of VMD described in ref. 66 and applied the HOLE program.67 The estimated average radii were 1.08, 1.96 and 2.30 Angstroms for the pores containing 4, 9 and 13 peptides, respectively. For the original MARTINI model, as discussed previously in ref. 35, pores tend to be dry, i.e. water is not transported. However, for the polarizable MARTINI model used here, we do see evidence for water diffusing through the pores. We illustrate this with the snapshot in Fig. S9 (ESI†). In addition, for the pore with 9 peptides we have estimated the water permeability coefficient which was P = 8.4 × 10−17 cm3 s−1. The calculation of this coefficient was performed according to the procedure described in the literature68–70 and was based on the averaging over the last 40% of the 50 μs trajectory.
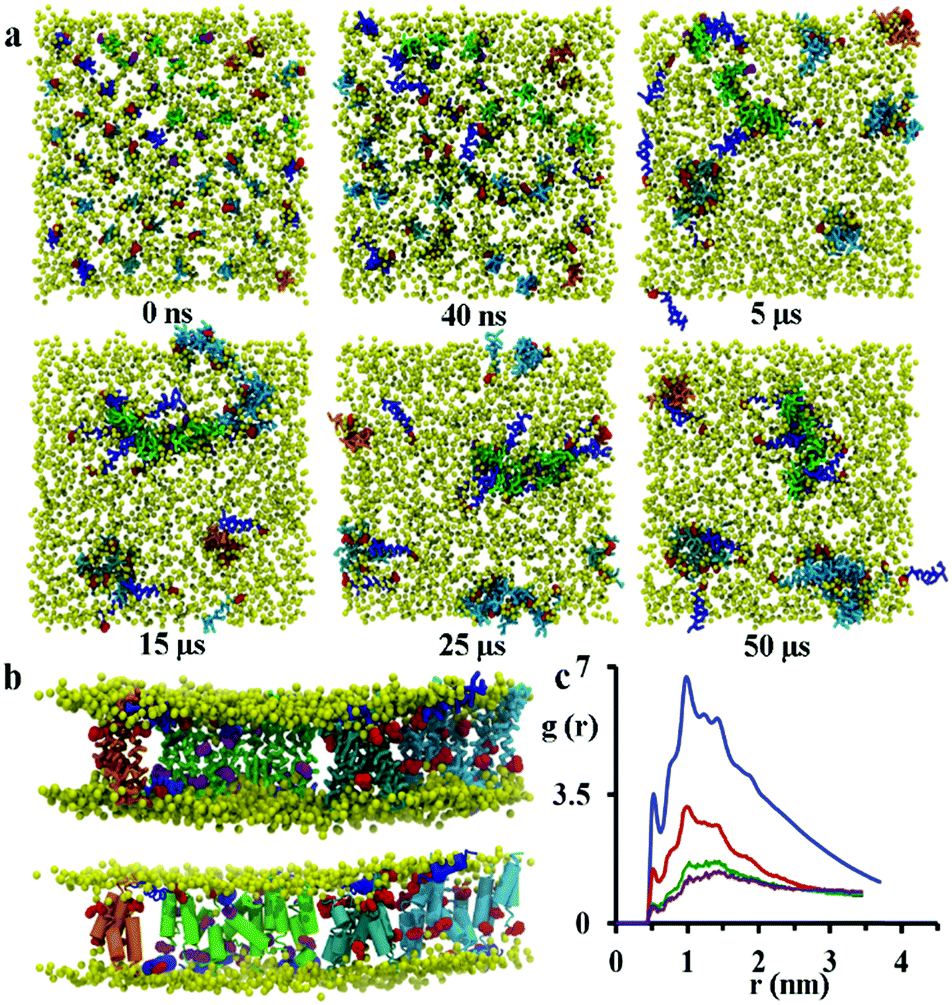 | ||
| Fig. 4 (a) Top views of different snapshots of the 50 μs CG MD simulation of a POPC lipid bilayer with transmembrane chrys-3 peptides. (b) Side view of the 50 μs structure (top: coarse grained, bottom: fine grained) showing pores with 4 (orange), 9 (cyan) and 13 (skyblue) peptides and an aggregate with 13 peptides (green). Peptides adsorbed on the membrane surface are shown with phenylalanine and the rest of chrys-3 residues in red and blue, respectively. Phenylalanine residues of peptides belonging to the pores and the aggregate are shown in red and purple, respectively. (c) Radial distribution functions (RDFs) of peptide backbone beads after 50 ns (purple), 100 ns (green), 1 μs (red) and over the last 20 μs (blue) of the simulation. | ||
The observation of the formation of peptides pores and an aggregate are in agreement with experimental QCM-D results reported recently by Wang et al.21 The close packing of peptides on aggregation was monitored by analysing the radial distribution function (RDF) of peptide backbone beads (shown in Fig. 4c). The peptides have initial distances of 2.7, 3.2 and 3.8 nm from the starting configuration, but the presence of peaks at 0.52, 0.78, 1.0, 1.24 and 1.44 nm that increase in intensity over the course of the simulation indicate the formation of peptide aggregates.
Results for DPPC lipid bilayers are very similar to those seen (above) for POPC (see Fig. S3, ESI†). In addition, Fig. 5 shows results for a POPE:POPG system, designed as a model for the bacterial membrane of E. coli. Here, transmembrane chrys-3 peptides again form pores and aggregates similar to those observed in POPC and DPPC lipid bilayers (Fig. 5 top). Interestingly, for this system, pores containing 4, 5 and 7 peptides and an aggregate composed of 5 peptides are observed, suggesting that the formation of smaller sized pores and aggregates are favoured in POPE:POPG lipid bilayers (Fig. 5 top). A similar formation of peptide pores and aggregates is confirmed by the CG MD simulation of transmembrane chrys-3 peptides in a POPC vesicle (Fig. 5 bottom), with similar pore and aggregate sizes as seen in POPC bilayers.
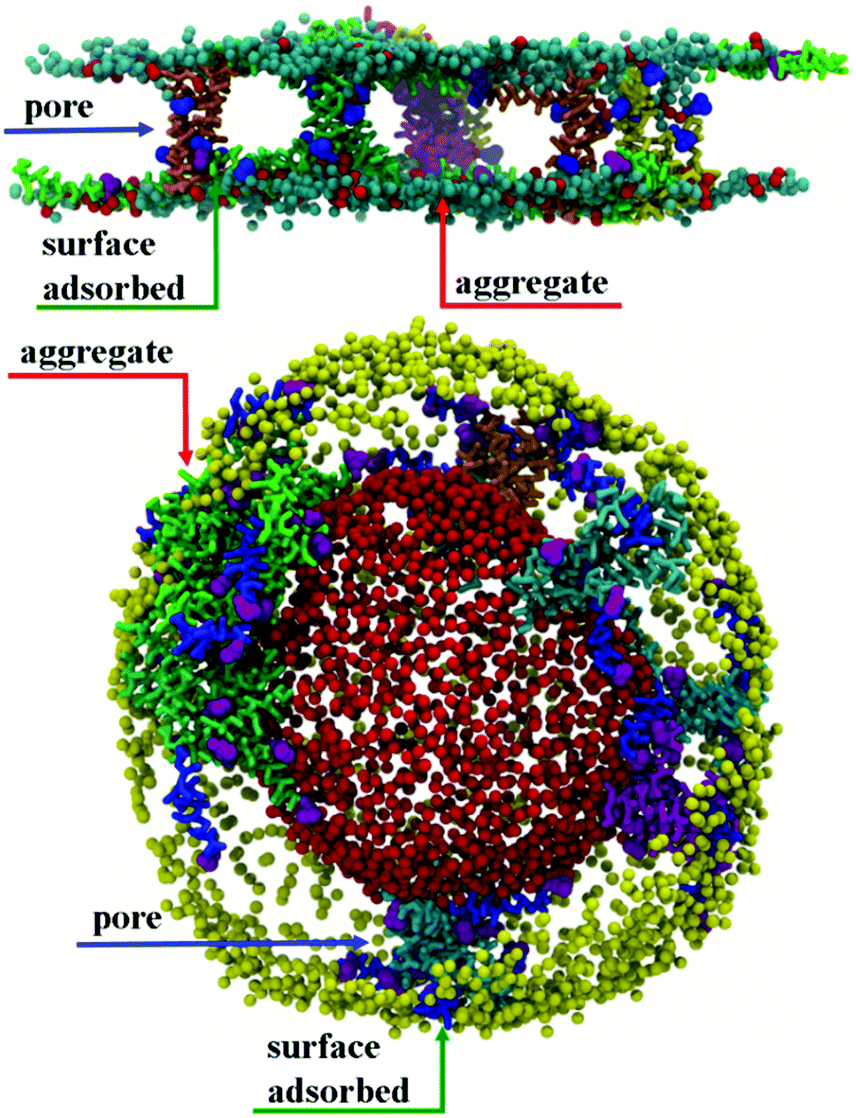 | ||
| Fig. 5 (top) Side view of the 50 μs structure of a CG POPE:POPG lipid bilayer with transmembrane chrys-3 peptides showing pores with 4 (orange), 5 (yellow) and 7 (seagreen) peptides and an aggregate with 5 peptides (magenta). Peptides adsorbed on the membrane surface are shown with phenylalanine and the rest of chrys-3 residues in purple and green, respectively. Phenylalanine residues of peptides belonging to the pores and the aggregate are shown in blue and purple, respectively; (bottom) section of the 50 μs structure of a CG POPC vesicle with transmembrane chrys-3 peptides showing pores with 4 (orange), 6 (magenta), 7 (skyblue), 9 (cyan) and 11 (seagreen) peptides and an aggregate with 32 peptides (green). Peptides adsorbed on the membrane surface are shown with phenylalanine and the rest of chrys-3 residues in purple and blue, respectively. Phenylalanine residues of peptides belonging to the pores and the aggregate are shown in purple. Lipids acyl chains, water and ions are not shown for clarity. | ||
Peptide–lipid interactions
The effect of the interaction of peptides with lipid molecules can be monitored by measuring the order parameters of bonds along the sn-1 and sn-2 acyl chains of lipid molecules. The second-rank order parameter, P2 = 〈1/2(3cos2θ − 1)〉, in which θ is the angle between the direction of the bond and the bilayer normal, has been calculated for consecutive bonds starting from the glycerol backbone beads. Here, we differentiate between two groups of lipid molecules, one within the immediate environment of chrys-3 and the second representing the rest of the lipids. We define peptide-interacting lipids as those lipid molecules that have at least one CG bead within a distance of 0.52 nm (the first peak of the RDFs shown in Fig. 4c) from any peptide bead. The lipids outside a cutoff of 0.52 nm are defined as “bulk lipids”. The calculated order parameters for both groups of lipids are shown in Fig. 6a from left to right for pure POPC, and for POPE and POPG lipids in the POPE:POPG bilayer, respectively. The results among all three types of phospholipids clearly show that the order parameters for both sn-1 and sn-2 acyl chains are significantly lower for peptide-interacting lipids compared to bulk lipids. The latter exhibit values comparable to those found in pure lipid membranes.40 Similar results are seen for an analysis of the DPPC simulations (Fig. S4, ESI†).
 | ||
| Fig. 6 Analysis of order parameter distributions and bilayer thicknesses from simulations 5 and 10 of Table 1, for POPC and POPE:POPG mixtures, respectively. (a) Sn-1 (blue) and sn-2 (red) lipid acyl chain order parameters of bulk lipids are compared with sn-1 (green) and sn-2 (purple) order parameters of peptide-interacting lipids. Peptide-interacting lipids are defined as those lipids with at least one bead within 0.52 nm of any peptide bead. (b) Time average order parameter for the acyl chains of lipids, plotted in the x, y plane of the bilayer. In both (a) and (b) results for POPC, POPE and POPG are shown from left to right, respectively; (c) lipid bilayer thickness around chrys-3 peptides in POPC (left) and POPE:POPG (right), plotted in the x, y plane of the bilayer. (d) Comparison between order parameters of POPE and POPG peptide lipids. | ||
The decreased ordering of lipids in the vicinity of peptides can also be appreciated from 2D plots representing the average order parameter of lipid acyl chains, shown in Fig. 6b, from left to right for pure POPC lipids and POPE and POPG lipids in the POPE:POPG bilayer, respectively. These plots show the distribution of the order parameter for different lipids across the membrane plane, showing the lower lipid acyl chains order parameters that occur in the regions of peptide pores and aggregates. Also, the formation of disordered toroidal pores of different sizes as well as peptide aggregates affects other bilayer structural characteristics such as the layer thickness (Fig. 6c) and the area per lipid (Table 3).
| Lipid | Area per lipid (nm2)a | |
|---|---|---|
| Outer leaflet | Inner leaflet | |
| a The area per lipid is averaged over the final 40% of each trajectory and the errors are standard deviations. | ||
| DPPC | 0.608 ± 0.004 | 0.608 ± 0.004 |
| DPPC + chrys-3 | 0.563 ± 0.005 | 0.587 ± 0.006 |
| POPC | 0.653 ± 0.002 | 0.651 ± 0.002 |
| POPC + chrys-3 | 0.620 ± 0.005 | 0.625 ± 0.005 |
| POPE | 0.612 ± 0.003 | 0.617 ± 0.004 |
| POPE + chrys-3 | 0.588 ± 0.006 | 0.600 ± 0.007 |
| POPG | 0.607 ± 0.018 | 0.615 ± 0.012 |
| POPG + chrys-3 | 0.450 ± 0.020 | 0.465 ± 0.015 |
The observed thinning of the POPC lipid membrane in the proximity of chrys-3 peptides is in good agreement with a previous computational study of WALP23 peptide59 in a DOPC lipid bilayer. We observe that the decrease in the values of the order parameters for POPG molecules is significantly stronger compared to both POPE and POPC molecules. This can be seen from the 2D plots in Fig. 6b, and also from the comparison between POPE and POPG order parameters of peptide-interacting lipids given in Fig. 6d. From Fig. 6d it is also seen that the decrease in order is more pronounced in the region of the acyl chains that is closest to the bilayer surface.
The average area per lipid of POPC, POPE and POPG molecules is reduced in the presence of chrys-3 peptides (Table 3) with the POPG lipids again experiencing the strongest effect, confirming that the interaction of cationic chrys-3 peptides with the anionic POPG lipids is stronger than that with the neutral POPC and POPE lipids.
These computational findings are broadly in agreement with previous experimental results reported for different AMPs in lipid bilayers containing POPG molecules,71–74 indicating the crucial role played by favourable interactions, between positively charged chrys-3 peptides and negatively charged POPG lipids, in terms of disruption to bilayer structure.
Fig. 7a presents partial density profiles calculated for chrys-3 peptides and POPC acyl chains. The partial density of chrys-3 peptides is distributed between the POPC glycerol backbone (surface adsorbed) and the POPC acyl chains (transmembrane orientation). The latter points towards the interaction of transmembrane chrys-3 peptides with POPC acyl chains at the centre of the lipid bilayer. By comparison, in the POPE:POPG lipid bilayer (Fig. 7b) the shift of the partial density of chrys-3 peptides towards the lipids’ acyl chains is less pronounced.
 | ||
| Fig. 7 Partial density profiles from 50 μs CG MD simulations of POPC (a) and POPE:POPG (b) lipid bilayers containing 50 chrys-3 peptides. POPC and POPE phosphate and glycerol densities are shown in blue and red, respectively. POPC and POPE palmitoyl and oleoyl chains densities are shown in dark purple and orange, respectively. POPG phosphate and glycerol densities are shown in magenta and purple, respectively. POPG palmitoyl and oleoyl chains densities are shown in cyan and skyblue, respectively. Chrys-3 peptide density is shown in green. | ||
The interaction between chrys-3 and lipid molecules has been monitored also by means of pair RDFs. Here, we calculate RDFs between peptide backbone beads and the beads representing phosphate groups (Fig. 8a) and terminal beads of lipid acyl chains (Fig. 8b). The highest intensity peak at ∼0.5 nm, in Fig. 8a is associated with close packing of peptides with lipid phosphate groups (see, for example, the snapshot in Fig. 8d showing a typical peptide–lipid interaction). Both RDF plots show that the interaction of chrys-3 peptides with POPG molecules is much stronger (i.e. more localized) than with POPE and POPC lipids, due to the strong electrostatic interaction of positively charged peptides with the overall negatively charged head group of POPG. The presence of two additional peaks with lower intensities at 0.8 and 1 nm supports the strength of this interaction. The RDFs arising from peptide backbone beads interacting with the terminal hydrophobic beads of the palmitoyl and oleoyl chains indicate that this interaction is not localized (Fig. 8b). Fig. 8b, confirms also the strong preference for chrys-3 peptides to exhibit closer interactions with POPG in preference to either of the zwitterionic lipids, POPC and POPE.
 | ||
| Fig. 8 RDFs of peptide backbone beads interacting with (a) phosphate beads and (b) terminal beads of palmitoyl (dashed lines) and oleoyl (straight lines). (c) Average number of contacts of amino acid residues of chrys-3 peptides with lipids per peptide-interacting lipid (within a contact cut-off of 0.52 nm). (d) Transmembrane chrys-3 peptide (yellow) interacting with two POPC molecules (skyblue). Backbone beads of chrys-3 are shown in purple. Choline, phosphate, terminal palmitoyl and oleoyl chains beads of POPC are shown in blue, orange, red and green, respectively. The numbers represent characteristic distances in nm. | ||
Additionally, we performed analysis of the interaction of the lipids with individual amino acid residues of chrys-3, averaged over the last 40% of the simulation, by measuring the average number of contacts between them. A contact is counted when at least one bead of the amino acid residue is within 0.52 nm of any bead of the lipid molecule. Contacts are normalised for each amino acid since the number of peptide-interacting lipids is different in the POPC and POPE:POPG systems. The results are presented in Fig. 8c. They show that for all three phospholipids there is a preferential interaction with hydrophobic residues of chrys-3 such as phenylalanine (PHE) and to a slightly lesser extent leucine (LEU) and isoleucine (ILE). These findings are in good agreement with experimental results by Wimley and White.75 However, we see a significant number of contacts with a wide range of amino acids, including, alanine (ALA), serine (SER) and glycine (GLY), and even hydrophilic residues such as histidine (HIS). Asparagine (ASP) though has a low number of contacts with lipid molecules. As expected, the number of contacts, indicate that both hydrophobic and hydrophilic amino acid residues of chrys-3 have stronger interactions with POPG compared to POPC and POPE. This conclusion is further supported by a relatively large percentage of POPG lipids (57 ± 3%) that are in close contact with at least one chrys-3 molecule, compared to POPE (26 ± 1%) and POPC (24 ± 1%) (see Fig. S5, ESI†) and also by Voronoi diagrams comparing the distribution of lipids in the absence and presence of chrys-3 peptides (Fig. S6, ESI†).
Free energy of insertion of a chrys-3 peptide into the POPC lipid bilayer
In order to determine the free energy involved in the transition from the aqueous solution, via the surface adsorbed state, to the transmembrane state of the peptide, we calculated the free energy profile (potential of mean force (PMF)) for the insertion of a chrys-3 peptide into the POPC lipid bilayer within the coarse-grained MARTINI model.Here, we carry out the calculation for two cases, corresponding to pulling (out of the bilayer) (i) an isolated chrys-3 peptide located in the membrane, and (ii) a selected chrys-3 peptide belonging to a small disordered toroidal pore (Fig. S7, ESI†). The PMFs for these processes, shown in Fig. 9, were calculated using the WHAM method employing 63 and 45 different windows, for cases (i) and (ii), respectively. Case (ii) is illustrated in Fig. S8 (ESI†). In both cases the reference free energy of the peptide assumed to be zero. High resolution in energy states in the PMF plot have been achieved by extensive sampling using relatively long (17.5 μs) trajectories of CG MD simulation for each window.60
 | ||
| Fig. 9 PMF of insertion of a chrys-3 peptide into POPC lipid bilayers. The chrys-3 peptide shown in blue is subjected to different pulling forces in order to sample the different states of the peptide. Top: The pulling distance, r, is measured for a single chrys-3 molecule relative to the centre of the membrane. Bottom: The pulling distance (inter-helical distance) is calculated between the centres of masses of the blue and cyan (reference) peptides (see text) for a peptide pore within a POPC membrane. | ||
From Fig. 9, the PMF for the single chrys-3 molecule shows two energy minima corresponding to the surface-bound and transmembrane states (Fig. S8, ESI†) identified above. These are separated by a sufficiently high energy barrier of ca. 20 kJ mol−1 to make it difficult for peptides to spontaneously jump between these two states on the 50 μs time-scale. The surface-bound state for the peptide is lower in energy by ≈85 kJ mol−1 compared to the peptide in aqueous solution, indicating a very strong driving force for surface adsorption of peptides.
Formation of a peptide pore however dramatically changes the PMF, leading to a three potential wells, the lowest corresponding to the transmembrane state where the chrys-3 molecule is fully bound to other pore-forming peptides. A potential well is also seen between the surface adsorbed and transmembrane states, where the peptide partially interacts with both the surface and other pore forming peptides. The free energy difference for transition from the fully solvated peptide in its preferred transmembrane state in the pore (i.e. in association with other peptides within the transmembrane lipid environment) is ca. −110 kJ mol−1, though a large component of this is the free energy change from solution to the surface adsorbed state (−80 kJ mol−1). In future, it would be interesting to compare these data with an atomistic simulation of this process, similar to the one reported by Rzepiela et al.37 though this will be extremely computationally demanding for the current system.
Conclusions
We have performed CG-MD simulations in order to investigate the interaction of chrys-3, a highly cationic peptide, with 4 positively charged amino acids at the C-terminus, with lipid bilayers and vesicles. By employing long, 50 μs, CG-MD simulations we observe both the formation of peptide aggregates and peptide disordered toroidal pores in lipid bilayers and vesicles. We see also surface-adsorption of peptides, followed by aggregation and abstraction of lipids from bilayers and vesicles, leading to lipid protrusions. It has previously been hypothesised,15,16 that these mechanisms may be important in anti-microbial action. Both mechanisms are consistent also with experimental QCM-D studies of the behaviour of chrys-3 peptide and both are likely to play crucial roles in cationic AMP-mediated cell lysis.We have observed similar behaviour for chrys-3 with different types of lipid bilayers and with a model vesicle system. In particular, we note the ability of chrys-3 to interact with, and disrupt, a POPE:POPG lipid bilayer, which acts as a model for a Gram-negative bacterial membrane. For this system, it is observed that the average size of pores and aggregates formed are smaller than the ones in pure POPC and DPPC lipid membranes. We observe also preferential interactions of chrys-3 with the anionic lipid, POPG, in comparison to the zwitterionic POPC and POPE lipids, suggesting that electrostatics interaction play a significant role in the antimicrobial activity of cationic AMPs.64,65
PMF calculations show that the initial adsorption, and subsequent insertion, of chrys-3 into a POPC lipid bilayer are associated with favourable free energy changes. At high concentration of peptide within the bilayer, the PMF for insertion is significantly perturbed, leading to the formation of three potential wells, the deepest of which corresponds to the transmembrane state where the peptide forms part of a membrane pore.
The formation of multiple pores by chrys-3 can be considered as one of the main steps leading to the disruption of a bacterial lipid membrane. In addition, the prediction that chrys-3 induces lipid protrusions in membranes might have important implications for its antimicrobial function. In summary, our results provide detailed structural and dynamical insights into chrys-3:lipid systems, paving the way for future studies of how changes in AMP structure can influence cell lysis.
Author contributions
A. C. performed the main part of the research. M. W performed some of the PMF calculations; A. C., M. R. W., M. W. and V. S. O. analysed the data and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. S. O. gratefully acknowledges the financial support of this project by EPSRC (grant EP/L001322/1). A. C. and V. S. O. thank University of East Anglia (UEA) for access to the High-Performance Computer Cluster (Grace) supported by the Research and Specialist Computing Support service. M. R. W. gratefully acknowledges the financial support of EPSRC (grant EP/L00111X/1). M. R. W. and M. W. thank Durham University for the use of its High-performance computer system (Hamilton).References
- A. M. Carmona-Ribeiro and L. Dias de Melo Carrasco, Int. J. Mol. Sci., 2014, 15, 18040–18083 CrossRef CAS PubMed.
- B. F. Kraemer, R. A. Campbell, H. Schwertz, M. J. Cody, Z. Franks, N. D. Tolley, W. H. A. Kahr, S. Lindemann, P. Seizer, C. C. Yost, G. A. Zimmerman and A. S. Weyrich, PLoS Pathog., 2011, 7, e1002355 CAS.
- A. A. Bahar and D. Ren, Pharmaceuticals, 2013, 6, 1543–1575 CrossRef PubMed.
- W. Aoki and M. Ueda, Pharmaceuticals, 2013, 6, 1055–1081 CrossRef PubMed.
- G. Wang, B. Mishra, K. Lau, T. Lushnikova, R. Golla and X. Wang, Pharmaceuticals, 2015, 8, 123–150 CrossRef CAS PubMed.
- F. Pinheiro da Silva and M. C. C. Machado, Peptides, 2012, 36, 308–314 CrossRef CAS PubMed.
- D. Gaspar, A. S. Veiga and M. A. R. B. Castanho, Front. Microbiol., 2013, 4, 1–16 Search PubMed.
- A. J. Moore, D. A. Devine and M. C. Bibby, Pept. Res., 1994, 7, 265–269 CAS.
- D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta, 2008, 1778, 357–375 CrossRef CAS PubMed.
- C.-Y. Huang, H.-Y. Huang, M. D. Forrest, Y.-R. Pan, W.-J. Wu and H.-M. Chen, PLoS One, 2014, 9, e109174 Search PubMed.
- H. Jenssen, P. Hamill and R. E. W. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS PubMed.
- Y. Shai, Biopolymers, 2002, 66, 236–248 CrossRef CAS PubMed.
- Y. Shai, Biochim. Biophys. Acta, 1999, 1462, 55–70 CrossRef CAS.
- L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Biophys. J., 2003, 81, 1475–1485 CrossRef.
- H. Steiner, D. Andreu and R. B. Merrifield, Biochim. Biophys. Acta, 1988, 939, 260–266 CrossRef CAS.
- E. Gazit, A. Boman, H. G. Boman and Y. Shai, Biochemistry, 1995, 34, 11479–11488 CrossRef CAS PubMed.
- L. Liu, Y. Fang and J. Wu, Biochim. Biophys. Acta, 2013, 1828, 2479–2486 CrossRef CAS PubMed.
- S. E. Blondelle, K. Lohner and M. Aguilar, Biochim. Biophys. Acta, 1999, 1462, 89–108 CrossRef CAS.
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS PubMed.
- N. Iijima, N. Tanimoto, Y. Emoto, Y. Morita, K. Uematsu, T. Murakami and T. Nakai, Eur. J. Biochem., 2003, 270, 675–686 CrossRef CAS PubMed.
- K. F. Wang, R. Nagarajan and T. A. Camesano, Biophys. Chem., 2015, 196, 53–67 CrossRef CAS PubMed.
- K. F. Wang, R. Nagarajan, C. M. Mello and T. A. Camesano, J. Phys. Chem. B, 2011, 115, 15228–15235 CrossRef CAS PubMed.
- A. J. Mason, P. Bertani, G. Moulay, A. Marquette, B. Perrone, A. F. Drake, A. Kichler and B. Bechinger, Biochemistry, 2007, 46, 15175–15187 CrossRef CAS PubMed.
- J. A. Masso-Silva and G. Diamond, Pharmaceuticals, 2014, 7, 265–310 CrossRef PubMed.
- P. A. Pinzon-Arango, R. Nagarajan and T. A. Camesano, J. Phys. Chem. B, 2013, 117, 6364–6372 CrossRef CAS PubMed.
- W. Wang, R. Tao, Z. Tong, Y. Ding, R. Kuang, S. Zhai, J. Liu and L. Ni, Peptides, 2012, 33, 212–219 CrossRef CAS PubMed.
- J. C. Hsu, L. C. Lin, J. T. Tzen and J. Y. Chen, Peptides, 2011, 32, 900–910 CrossRef CAS PubMed.
- I. E. Ivanov, A. E. Morrison, J. E. Cobb, C. A. Fahey and T. A. Camesano, ACS Appl. Mater. Interfaces, 2012, 4, 5891–5897 CAS.
- A. K. Tripathi, T. Kumari, M. K. Harioudh, P. K. Yadav, M. Kathuria, P. K. Shukla, K. Mitra and J. K. Ghosh, Sci. Rep., 2017, 7, 3384 CrossRef PubMed.
- P. A. Fulmer, J. G. Lundin and J. H. Wynne, ACS Appl. Mater. Interfaces, 2010, 2, 1266–1270 CAS.
- H. Leontiadou, A. E. Mark and S. J. Marrink, J. Am. Chem. Soc., 2006, 128, 12156–12161 CrossRef CAS PubMed.
- D. Sengupta, H. Leontiadou, A. E. Mark and S. J. Marrink, Biochim. Biophys. Acta, 2008, 1778, 2308–2317 CrossRef CAS.
- A. Glättli, I. Chandrasekhar and W. F. van Gunsteren, Eur. Biophys. J., 2006, 35, 255–267 CrossRef PubMed.
- S. J. Irudayam and M. L. Berkowitz, Biochim. Biophys. Acta, 2012, 1818, 2975–2981 CrossRef CAS PubMed.
- L. Thøgersen, B. Schiøtt, T. Vosegaard, N. C. Nielsen and E. Tajkhorshid, Biophys. J., 2008, 95, 4337–4347 CrossRef PubMed.
- P. J. Bond, D. L. Parton, J. F. Clark and M. S. P. Sansom, Biophys. J., 2008, 95, 3802–3815 CrossRef CAS PubMed.
- A. J. Rzepiela, D. Sengupta, N. Goga and S. J. Marrink, Faraday Discuss., 2010, 144, 431–443 RSC.
- B. Yoo, Y. Zhu and E. J. Maginn, Langmuir, 2016, 32, 5403–5411 CrossRef CAS PubMed.
- K. P. Santo and M. L. Berkowitz, J. Phys. Chem. B, 2012, 116, 3021–3030 CrossRef CAS PubMed.
- S. J. Marrink, A. H. de Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. Epand, P. B. Savage and R. M. Epand, Biochim. Biophys. Acta, 2007, 1768, 2500–2509 CrossRef CAS PubMed.
- T. J. Piggot, D. A. Holdbrook and S. Khalid, J. Phys. Chem. B, 2011, 115, 13381–13388 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. MacKerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed.
- S. Jo, J. B. Lim, J. B. Klauda and W. Im, Biophys. J., 2009, 97, 50–58 CrossRef CAS PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- S. Baoukina, E. Mendez-Villuendas, W. F. D. Bennett and D. P. Tieleman, Faraday Discuss., 2013, 161, 63–75 RSC.
- S. O. Yesylevskyy, L. V. Schafer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, e1000810 Search PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- M. Parrinello and A. J. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. A. Wassenaar, K. Pluhackova, R. Böckmann, S. J. Marrink and D. P. Tieleman, J. Chem. Theory Comput., 2014, 10, 676–690 CrossRef CAS PubMed.
- A. Y. Shih, A. Arkhipov, P. L. Freddolino and K. Schulten, J. Phys. Chem. B, 2006, 110, 3674–3684 CrossRef CAS PubMed.
- A. Y. Shih, P. L. Freddolino, A. Arkhipov and K. Schulten, J. Struct. Biol., 2007, 157, 579–592 CrossRef CAS PubMed.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS PubMed.
- J. P. Segrest, M. K. Jones, A. Catte, M. Manchekar, G. Datta, L. Zhang, R. Zhang, L. Li, J. C. Patterson, M. N. Palgunachari, J. F. Oram and G. Ren, Structure, 2015, 23, 1214–1226 CrossRef CAS PubMed.
- N. Castillo, L. Monticelli, J. Barnoud and D. P. Tieleman, Chem. Phys. Lipids, 2013, 169, 95–105 CrossRef CAS PubMed.
- D. Sengupta and S. J. Marrink, Phys. Chem. Chem. Phys., 2010, 12, 12987–12996 RSC.
- B. Roux, Comput. Phys. Commun., 1995, 91, 275–282 CrossRef CAS.
- B. Bechinger, Biochim. Biophys. Acta, 2005, 1712, 101–108 CrossRef CAS PubMed.
- B. Bechinger and K. Lohner, Biochim. Biophys. Acta, 2006, 1758, 1529–1539 CrossRef CAS PubMed.
- B. Bechinger, Curr. Opin. Colloid Interface Sci., 2009, 14, 349–355 CrossRef CAS.
- B. Bechinger and E. S. Salnikov, Chem. Phys. Lipids, 2012, 165, 282–301 CrossRef CAS PubMed.
- A. Y. Shih, P. L. Freddolino, S. G. Sligar and K. Schulten, Nano Lett., 2007, 7, 1692–1696 CrossRef CAS PubMed.
- O. S. Smart, J. G. Neduvelil, X. Wang, B. A. Wallace and M. S. P. Sansom, J. Mol. Graphics Modell., 1996, 14, 354–360 CrossRef CAS.
- W. F. D. Bennett, N. Sapay and D. P. Tieleman, Biophys. J., 2014, 106, 210–219 CrossRef CAS PubMed.
- H. Leontiadou, A. E. Mark and S. J. Marrink, J. Am. Chem. Soc., 2006, 128, 12156–12161 CrossRef CAS PubMed.
- D. L. Parton, E. V. Akhmatskaya and M. S. P. Sansom, J. Phys. Chem. B, 2012, 116, 8485–8493 CrossRef CAS PubMed.
- A. J. Mason, I. N. H. Chotimah, P. Bertani and B. Bechinger, Mol. Membr. Biol., 2006, 23, 185–194 CrossRef CAS PubMed.
- T. C. B. Vogt and B. Bechinger, J. Biol. Chem., 1999, 274, 29115–29121 CrossRef CAS PubMed.
- M. R. Wenk and J. Seelig, Biochemistry, 1998, 37, 3909–3916 CrossRef CAS PubMed.
- T. Wieprecht, M. Beyermann and J. Seelig, Biochemistry, 1999, 38, 377–378 CrossRef PubMed.
- W. C. Wimley and S. H. White, Nat. Struct. Biol., 1996, 3, 842–848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sm02152f |
| This journal is © The Royal Society of Chemistry 2018 |