
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into the nature of semi-soft elasticity and “mechanical-Fréedericksz transitions” in liquid crystal elastomers†‡
Devesh
Mistry
 *a,
Philip B.
Morgan
b,
John H.
Clamp
c and
Helen F.
Gleeson
a
*a,
Philip B.
Morgan
b,
John H.
Clamp
c and
Helen F.
Gleeson
a
aSchool of Physics and Astronomy, University of Leeds, Leeds, LS2 9JT, UK
bEurolens Research, University of Manchester, Manchester, M13 9PL, UK
cUltraVision CLPL, Commerce Way, Leighton Buzzard, LU7 4RW, UK. E-mail: pydam@leeds.ac.uk
First published on 25th January 2018
Abstract
The mechanical properties of an all-acrylate liquid crystal elastomer (LCE) with a glass transition of 14 ± 1 °C are reported. The highly nonlinear load curve has a characteristic shape associated with semi-soft elasticity (SSE). Conversely, measurements of the director orientation throughout tensile loading instead indicate a “mechanical-Fréedericksz” transition (MFT). Values of the step length anisotropy, r, are independently calculated from the theories of SSE (r = 3.2 ± 0.4), MFT (9.3 < r < 30.0) and thermally-induced length change (r = 3.8 ± 0.5). From simultaneously recorded polarising microscopy textures, the consequences of the above discrepancies are considered. Further, a mechanically-induced negative order parameter is observed. Results show the tensile load curve shape cannot solely be used to determine the underlying physics. Consequently, the LCE properties cannot be fully described by theories of SSE or MFTs alone. This suggests that the theory of LCEs is not yet complete. The conclusions suggest that both the LC order parameter and r must be functions of the mechanical deformation.
Introduction
Liquid crystal elastomers (LCEs) are amongst today's most interesting materials in soft matter science. Their coupling of liquid crystallinity to elastomeric networks has led to several technologically relevant functionalities such as thermal, light or chemical induced shape changes.1–3 Over the years many researchers have proposed various innovative applications of these functionalities such as soft actuators and robotics, tuneable irises and photo-driven motors.4–10Monodomain nematic LCEs in particular exhibit a plethora of mechanical and opto-mechanical phenomena when stressed perpendicular to the liquid crystal (LC) director, the average orientation of the LC molecules, including semi-soft elasticity (SSE) and the more elusive “mechanical-Fréedericksz transitions” (MFTs).1,11–14 SSE is the name given to a deformed LCE which demonstrates a tensile load curve like that illustrated in Fig. 1. The plateau-like region II of the tensile load curve has a reduced elastic modulus compared to the first and third regions and costs relatively little energy for the system to traverse. Across the plateau, the director gradually rotates from perpendicular to the stress axis at λ1, to lie parallel to the stress axis at λ2.11,15–17 MFT is an alternative deformation mode which has only been previously reported by Mitchell, Roberts and co-workers.14,18,19 In this process the director rotates sharply at a critical strain (as opposed to gradually over a strain range) to lie parallel to the stress axis at a critical extension.14,18,19
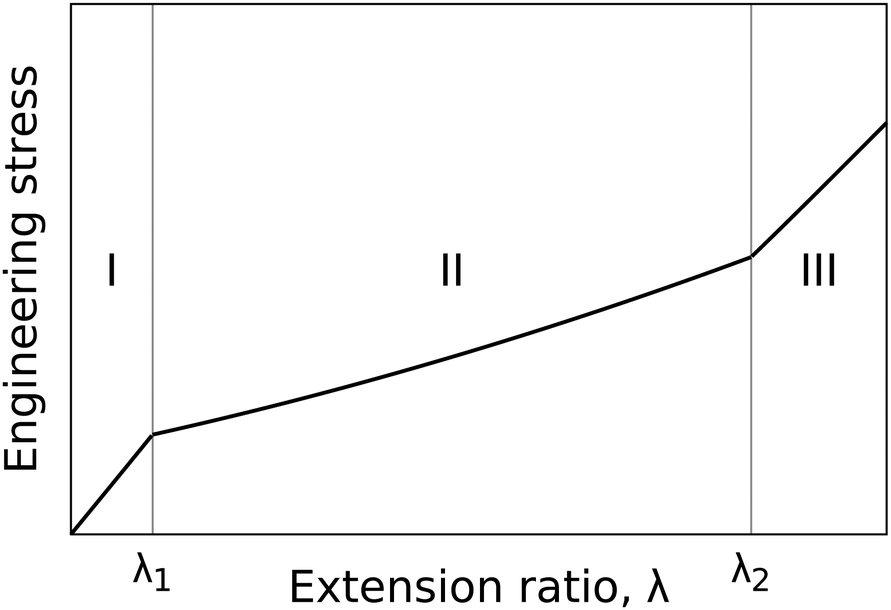 | ||
| Fig. 1 The semi-soft elastic load curve as for a monodomain nematic LCE stressed perpendicular to the director. Curve was generated using equations of SSE theory taken from Warner and Terentjev.11 | ||
One of the unknowns of LCEs is why a particular LCE will only display one of these above properties. Further, on an even more fundamental level, it is unknown to what extent the two phenomena, SSE and MFT, are related. By comparing the theory of SSE by Verwey and Warner to the theory of MFTs by Bladon, Terentjev and Warner it is certainly clear that that the processes, as currently described by theory, cannot occur simultaneously as they predict incompatible director rotation behaviours with respect to strain (as described above).11,20–23
In this work we use bespoke opto-mechanical equipment to simultaneously track the orientation of the LC director throughout tensile testing and paradoxically observe a tensile load curve which appears to conform to SSE and a director re-orientation behaviour which corresponds to a MFT. This remarkable simultaneous observation of behaviours from seemingly incompatible theories opens the possibility that the underlying physics of SSE and MFTs are more closely linked than previously thought. To explore this possibility, we calculate and discuss values for the step length anisotropy of the polymer network, r, that are independently deduced by assuming the validity of each of the two deformation modes as well as from thermally induced length changes of the LCE. Our observations also show that as the LCE is deformed perpendicular to the director, it passes through a state of negative LC order parameter.
The open questions which still surround the physical properties of LCEs and the debate about the theory behind them are perhaps typical of an exciting, complex soft matter system.24–28 The approach taken in this paper to simultaneously record the mechanical properties, direction and magnitude of liquid crystal order provides a simple but robust method to unambiguously record the complete response of LCEs under mechanical testing. We believe the results of this work demonstrate the importance of simultaneous and complete characterisation of a LCE's response to mechanical fields.
Experimental
Sample preparation
The LCE used in this work was made by adapting the LCE first described by Urayama et al. in 2005 which, briefly, is produced as follows.29 6-(4-Cyano-biphenyl-4′-yloxy)hexyl acrylate (A6OCB) is a monofunctional reactive mesogen which forms liquid crystal (LC) polymer chains. 4′-Hexyloxybiphenyl (6OCB) is a non-reactive mesogen used to broaden the nematic phase range of the monomer mixture prior to polymerisation. 1,6-Hexanediol di-acrylate (HDDA) is a non-mesogenic crosslinking group used to crosslink the polymerised chains of A6OCB together. Finally Irgacure 784 is a visible light photoinitiator used to trigger polymerisation. The structures of A6OCB and 6OCB are given in Fig. 2 and the molar percentage of each component used is given in the ‘monomer mixture’ column of Table 1. After polymerisation, the 6OCB, photoinitiator and any unreacted components are removed by washing the film in dichloromethane. The resultant material has a composition given by the ‘final LCE’ column of Table 1 and has a reported glass transition temperature (Tg) of approximately 50 °C.30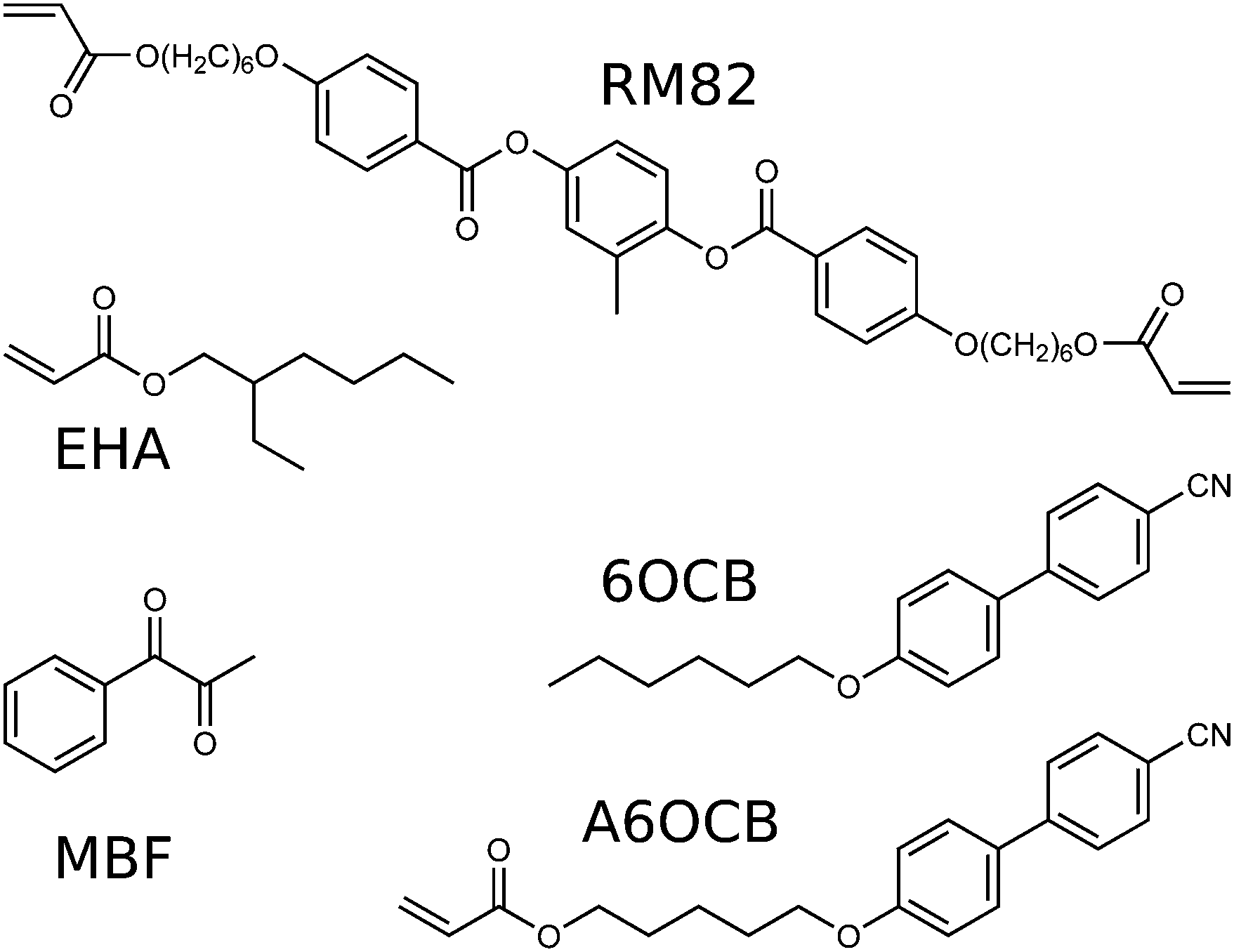 | ||
| Fig. 2 Structures of chemicals used here. The LCE produced by Urayama (2005) also used A6OCB and 6OCB.29 | ||
| % by mol. of each component in the LCE | ||
|---|---|---|
| Component | Monomer mixture | Final LCE |
| A60CB | 46.25 | 87 |
| 6OCB | 46.25 | |
| HDDA | 7.0 | 13 |
| Irgacure 784 | 0.5 | |
The Urayama material can be adapted in two ways to reduce Tg to below the ambient room temperature while maintaining a nematic phase prior to crosslinking (important in producing monodomain samples). Firstly the monomer 2-ethylhexyl acrylate (EHA) is introduced to increase the flexibility of the polymer backbone. Secondly, the non-mesogenic crosslinker, (HDDA), can be replaced with the bifunctional reactive mesogen, 1,4-bis-[4-(6-acryloyloxyhex-yloxy)benzoyloxy]-2-methylbenzene (RM82), to maintain a nematic phase in the monomer mixture prior to polymerisation (this also affects Tg). For this work we chose the UV-photoinitiator methyl benzoylformate (MBF). The structures of EHA, RM82 and MBF are given in Fig. 2.
The LCEs were polymerised inside LC devices of nominal dimensions of 60 mm × 20 mm × 100 μm made with one glass and one 100 μm thick Melinex® (DuPont Teijin Films) substrate. Melinex® was chosen for one of the cell substrates as its flexibility means it was easily peeled away from the LCE following polymerisation. Prior to device assembly, the internal surfaces were prepared with a uniaxially rubbed alignment layer of poly(vinyl alcohol). Full details of the cell fabrication method can be found in the ESI‡ alongside a diagram of the cells used (Fig. S1).
A6OCB, 6OCB and RM82 were purchased from Synthon Chemical GmbH, and EHA and MBF from Sigma Aldrich. Monodomain LCEs were prepared according to the composition shown in the ‘monomer mixture’ column of Table 2; the procedure followed that of Urayama (2005).29 A complete description of the process can be found in the ESI.‡ The final composition of the LCE is shown in the ‘final LCE’ column of Table 2.
| % by mol. of each component in the LCE | ||
|---|---|---|
| Component | Monomer mixture | Final LCE |
| A60CB | 14.6 ± 0.2 | 34.4 ± 0.3 |
| 6OCB | 55.9 ± 0.4 | |
| RM82 | 7.1 ± 0.1 | 16.6 ± 0.1 |
| EHA | 20.9 ± 0.2 | 49.0 ± 0.2 |
| MBF | 1.56 ± 0.01 | |
Experimental instruments
A TA Instruments Q20 DSC was used to determine the phase behaviour of the monomer mixture prior to polymerisation and of the final LCE. Sample masses between 4.9 and 8.8 mg were used. For each test at each heating rate the samples were thrice cycled across the temperature range to ensure consistency of results. For determination of the Tg of the final LCE, the samples were cycled between −20 and 150 °C at heating rates of 20, 10 and 5 K min−1. Thermal stability of the final LCE at high temperatures was investigated by evaluating the value of Tg for a separate sample cycled first between −40 and 120 °C and then between −40 and 330 °C at 20 °C min−1. TA Universal Analysis 2000 software was used to analyse the DSC curves for Tg of the final LCE. From the three values at 5, 10 and 20 °C min−1, Tg was determined by extrapolating the inflection point to a heating rate of 0 °C min−1.Opto-mechanical tests were performed using bespoke equipment created for simultaneous tensile load and polarising microscopy experiments. This allowed us in a single experiment to: measure the tensile load curves of LCEs under test; determine the orientation of the director across the samples; and qualitatively assess the degree of liquid crystalline order. Crucially, this ensures all the observables are recorded with the LCE deformed under exactly the same conditions of temperature, geometry and strain rate and thus removes the possibility that conflicting physical observations could be due to different test conditions.
The enclosing chamber of the apparatus was maintained at 23 °C by external control. Strips of monodomain LCE were cut to nominal dimensions of 2 mm wide and 25 mm long and at angles of 2 ± 1° and 88 ± 1° to the director (the aim was to have samples with their long axes either effectively parallel or perpendicular to the director). The unstressed sample thicknesses were measured using a micrometer with 1 μm accuracy. The samples were then loaded into the clamps of opposing actuators inside the chamber which were used to impose strains. One of the actuators included a calibrated 5N load cell mounted in series, allowing the stress applied to the samples at each strain step to be measured. Glass windows in the chamber allowed sample observation using a 4.2Mpx camera and lens system with a field of view of 16 × 16 mm in the plane of focus. During analysis, sample dimensions (including the initial width) were measured in pixels from photographs and calibrated to using a standard of known length, accurate to ±5 μm. The precision of the calibration means that the accuracy in determining the sample dimensions was limited solely the uncertainty in pixel measurements. Details of the entire opto-mechanical testing equipment can be found in the ESI‡ under Fig. S2.
Each sample was mechanically tested by sequentially imposing extension steps of 5% of the LCE strip's initial length, until failure. At each step, the sample was allowed to stress relax for 120 s before any measurements were taken. This is important as meaningful measurements of the director angle require the sample to be sufficiently close to equilibrium. After sample relaxation, the load cell reading was logged and a photograph taken under white light illumination along with 36 photographs taken using a crossed polariser arrangement with the polariser and analyser rotated by 10° between each photograph. As the polarisers had to be rotated manually, the overall time between successive extensions was typically 8 minutes.
Stress–extension curves were calculated using the load cell readings and the photographs taken under white light illumination. A Python particle tracking package, trackpy, was used to analyse the photographs taken with white light by monitoring the position of tracer particles embedded within the LCE strips.31 Figures demonstrating the effectiveness of the tracking can be found in ESI,‡ Fig. S3. From such measurements, the localised extension ratio, λ = L/L0, was readily deduced.
The transmitted light intensity was measured at each of the 10° rotation steps from the photographs taken under crossed polarisers using average intensities for 100 × 100 pixel regions of interest at the centre of film deformations. The transmitted intensity for a birefringent material rotated between crossed polarisers is expected to fit to the function
 | (1) |
There are four minima in eqn (1) for a 360° rotation. Therefore there are two possible orientations of the director separated by 90°, (since 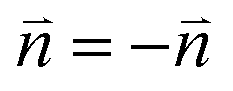 the minima 180° apart are degenerate). As we cut the samples with the true director orientation known, we can deduce the position of the relevant minima to track the director orientation.
the minima 180° apart are degenerate). As we cut the samples with the true director orientation known, we can deduce the position of the relevant minima to track the director orientation.
Temperature-dependent changes in the sample length and birefringence were measured using a Leica DM2700 P polarising microscope equipped with a Berek compensator and a Linkam THMS600 hot stage. A Nikon D7100 DSLR camera recorded images of the sample for analysis using the software ImageJ (National Institutes of Health, USA).32,33 Using the compensator, the sample retardance, Γ = Δn × d (where Δn is the birefringence and d is the sample thickness), was measured.34 Dividing the retardance by sample thickness gave the sample birefringence which in turn is related to the LC order parameter. Prior to thermal testing, the sample thickness was measured using a micrometer with 1 μm accuracy. The thickness at each temperature step was deduced by dividing the initial thickness by the fractional changes in sample length and width (measured in ImageJ), assuming a constant sample volume.11
Theory
Of the several theoretical models of LCEs, the theories of SSE by Verwey and Warner and MTFs by Bladon, Terentjev and Warner have been most effectively used to account for the widest variety of observed phenomena.20–23 For this reason we use these theories here and test them against the results for our material. A complete description of the theories used in this paper can be found in ref. 11. Here we briefly outline the steps relevant to the physical observables used in this work.In nematic LCEs, the LC anisotropy causes a macroscopic alignment of the individual polymer chains resulting in an anisotropic polymer conformation with a uniaxial ellipsoidal shape. Therefore the polymer conformation has unequal radii of gyration parallel (R‖) and perpendicular (R⊥) to the symmetry axis of the polymer conformation.
An effective step length tensor, 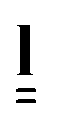 , can also be used to describe the anisotropy of the polymer conformation. For a prolate polymer conformation, with principal axis parallel to the z axis of the reference frame,
, can also be used to describe the anisotropy of the polymer conformation. For a prolate polymer conformation, with principal axis parallel to the z axis of the reference frame, 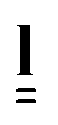 is given by
is given by
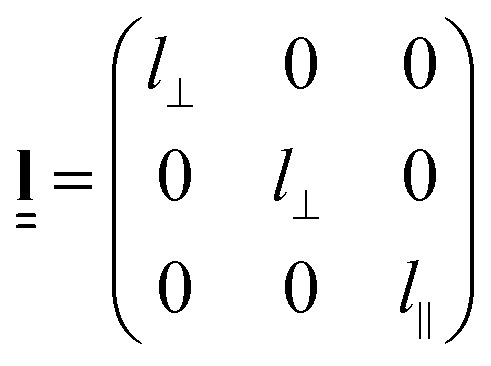 | (2) |
The unique physical properties of LCEs are all intrinsically linked to the fact that r is greater than unity. The magnitude of r also determines the ‘magnitude’ of the physical response of the unique LCE properties. For instance, the shape of tensile load curves, the length of the (semi-) soft elastic plateau, and the contraction ratio on heating, are all dependent on r. As such, r is possibly the most important parameter of LCEs and its accurate determination is key to their understanding and practical use. We next outline the methods that are used in this work to determine the value of r.
Semi-soft elasticity
As shown in Fig. 1, when an LCE is stressed perpendicular to the director, a highly non-linear load curve with three distinct regions is observed.1 According to the Verwey and Warner theory, in region I the stress–strain relationship is identical to that of an isotropic rubber. Above the first critical strain, λ1, the behaviour changes as the polymer conformation begins to rotate toward the stress axis. In region II, the gradient of load curve reduces significantly compared to that in the first region. In region III, once the director has completely rotated to lie parallel to the stress axis, the gradient increases once again and the stress–strain relationship takes on a modified version of the isotropic rubber response. If the first critical extension ratio occurs at λ1, then the second critical extension ratio will occur at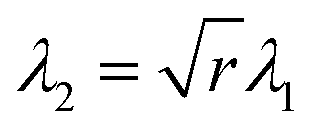 .11 Hence
.11 Hence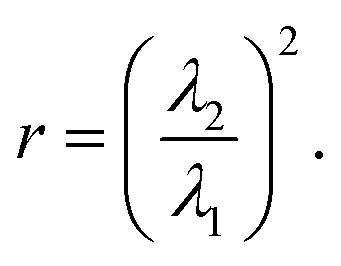 | (3) |
“Mechanical-Fréedericksz transition”
In this case the director rotation does not have a smooth behaviour but instead sharply rotates at a critical extension ratio, λc. By applying the theory of Bladon, Warner and Terentjev, upper and lower limits for the ratio r can be calculated using λc.11,22 Briefly, the free energy density can be calculated for deformations, λ, (1) before the strain, where the step length tensor remains perpendicular to the deformation axis and (2) after the threshold strain where the step length tensor has been rotated to lie parallel with the deformation axis. By comparing these, the director rotation is calculated to occur at:11 | (4) |
| λc = r1/3 ∴r = λc3. | (5) |
Thermally induced length changes
On heating from the nematic to the isotropic phase, an LCE with a prolate polymer conformation contracts along its director from its initial length L0 to its final length L = L0/λm.11 In the isotropic phase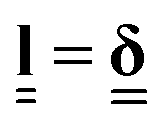 , the Kronecker delta, and hence r = 1. By minimising the free energy density, given by the “trace formula”, with respect to the deformation λm, one finds,
, the Kronecker delta, and hence r = 1. By minimising the free energy density, given by the “trace formula”, with respect to the deformation λm, one finds,| r = λm3. | (6) |
Results
In order to better discuss the results of mechanical tests in context with one another, we first present the data and then move to a detailed discussion.Differential scanning calorimetry (DSC)
DSC curves are shown in the ESI‡ Fig. S5. DSC tests of the LCE show that Tg = 14.0 ± 1.0 °C. Thrice cycling the sample at 20 °C min−1 between −40 and 120 °C gave a consistent inflection point at 19.0 °C. However, heating to 330 °C resulted in a slight decrease in the inflection point to 14.5 °C with a further decrease to 11.8 °C after a second cycle to 330 °C. The potential impact of this change is discussed later. No transition from the nematic to isotropic state was seen over the temperature range studied for this material.Quality of monodomain alignment
The two photographs in Fig. 3 show the 88° sample viewed via crossed polarisers. In the top and bottom images the polarisers are at 45° and 0° respectively to the director. The high quality of monodomain alignment is demonstrated by the high contrast in transmitted light between the two images, together with the obvious uniformity of the sample's appearance in both photographs. | ||
| Fig. 3 Polarising microscopy images of the 88° sample with the director (almost aligned with the short axis) at (a) 45° and (b) 0° to the polarisers. | ||
Tensile loading testing
Fig. 4 shows the load curves for the LCE stressed at 2° and 88° to the director. The significantly different shape of the two load curves clearly demonstrates the mechanical anisotropy of the LCE. The initial moduli and extension-at-failure were measured to be 23.1 ± 0.2 MPa and 1.21 respectively for the 2° sample and 4.0 ± 0.2 MPa and 2.34 for the 88° sample. The inset of Fig. 4a shows, perhaps surprisingly, that the 2° load curve is somewhat non-linear. Over this relatively small extension ratio we expected to observe a linear load curve. This was because the stress was applied at close to parallel to the long axis of the polymer conformation and so the non-linear effects of polymer conformation rotation should be minimal. Therefore the material was expected to have an isotropic response. We are left to conclude that 2° offset from perfectly parallel orientation is sufficient to cause the small nonlinearity observed.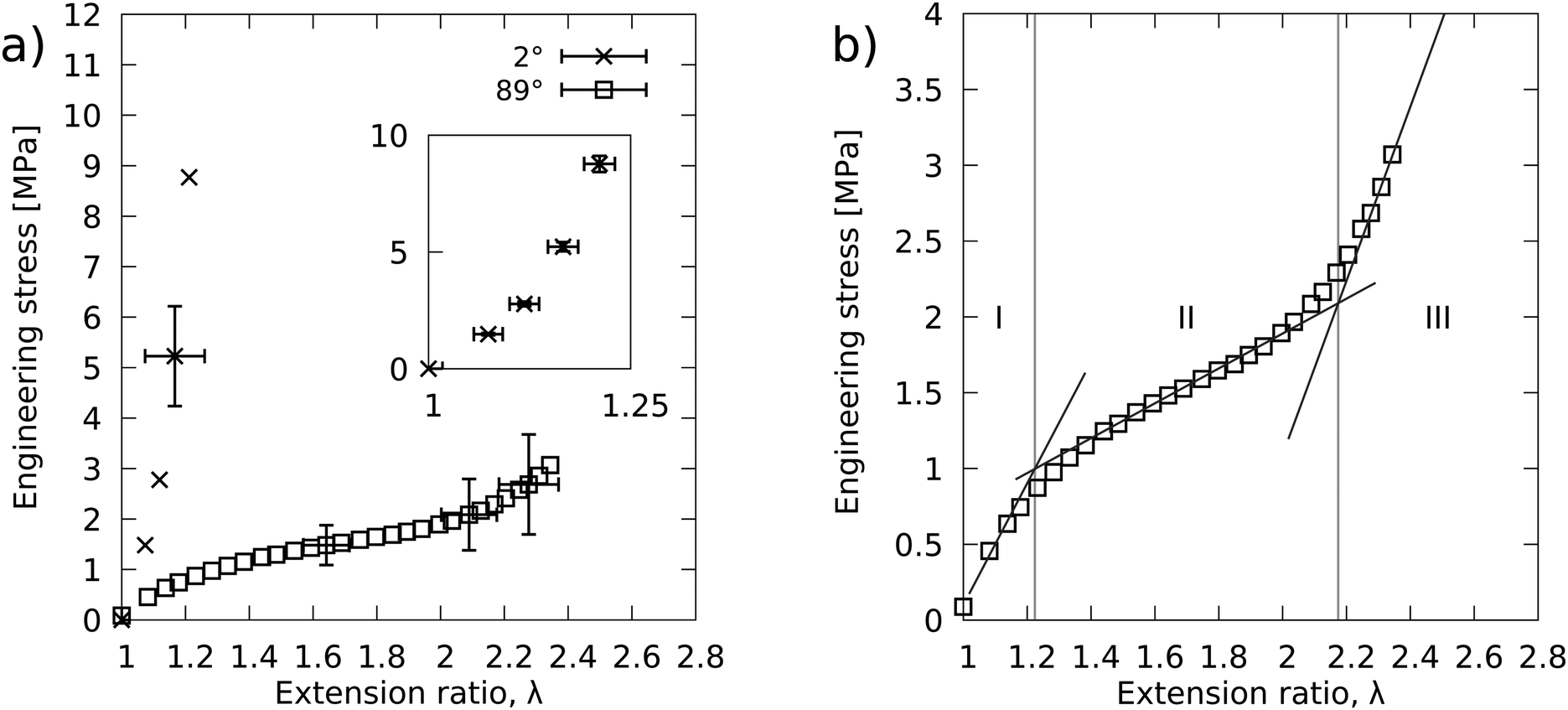 | ||
| Fig. 4 (a) Tensile load data for the LCE samples stressed at 88° and 2° to the director. In the main figure the representative error bars have been enlarged by a factor of 5 for clarity. The inset reproduces the data for the 2° sample to highlight the non-linearity of the load curve. Error bars in the inset are unscaled. (b) 88° curve is replotted to show the three distinct regions of the load curves. The lines fit to each region can be used with SSE theory to deduce the anisotropy of the effective step length tensor. | ||
The 88° load curve has been replotted in Fig. 4b to clearly show its highly non-linear shape. The three distinct regions of moduli of 4.0, 1.2 and 5.7 MPa gives the load curve the typical shape of SSE.11 The intercepts of the fitted lines gives the two critical extension ratios, λ1 = 1.22 ± 0.03 and λ2 = 2.17 ± 0.10. Using eqn (3) we find a step length anisotropy of, r = 3.2 ± 0.4.
Optical tracking of the director
Fig. 5 shows the results of fitting eqn (1) for the director angle at each strain step from Fig. 4 for both the 2° and 88° samples. The sample initially at 88° maintains a relatively constant director angle until λ ∼ 1.9 at which point it begins to rotate before sharply rotating at λc = 2.1 to an orientation almost parallel to the stress axis. Such behaviour is indicative of the “mechanical Fréedericksz” behaviour reported by Mitchell, Roberts and co-workers.14,18,19 Inserting the critical value of λc = 2.1 into eqn (4) and (5) gives another set of possible values for the upper and lower bounds on the value of the step length anisotropy: 9.3 < r < 30.0. It should be noted that eqn (4) gives two roots for the value of r, 0.7 and 30.0. The solution of r = 0.7 is discarded as the LCE contracts parallel to the director on heating (shown later in Fig. 7) and so the polymer conformation must be prolate and hence r > 1. | ||
| Fig. 5 Director angle at each strain step measured simultaneously with data from Fig. 4. For clarity, representative error bars have been enlarged by a factor of 5. The critical strains found from the 88° load curve are shown by the vertical lines. | ||
Qualitative determination of the nematic order
Fig. 6a shows polarising microscopy images of the 88° sample during mechanical testing. The sequence shows a significant change in the birefringence colours as the imposed extension was increased. For the first of the photographs (λ = 1.00), it is known that the sample was 99 μm thick with a birefringence of 0.12 and hence a retardance of ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880 nm (approximately 20th order). The increasing saturation of birefringence colours as the sample is strained indicates that the retardance is decreasing (seen by considering the colour sequence on a Michel-Levy chart). Indeed the images taken at λ = 2.00 and 2.09 have first order birefringence colours implying retardances of 0 < Γ < 600 nm.
880 nm (approximately 20th order). The increasing saturation of birefringence colours as the sample is strained indicates that the retardance is decreasing (seen by considering the colour sequence on a Michel-Levy chart). Indeed the images taken at λ = 2.00 and 2.09 have first order birefringence colours implying retardances of 0 < Γ < 600 nm.
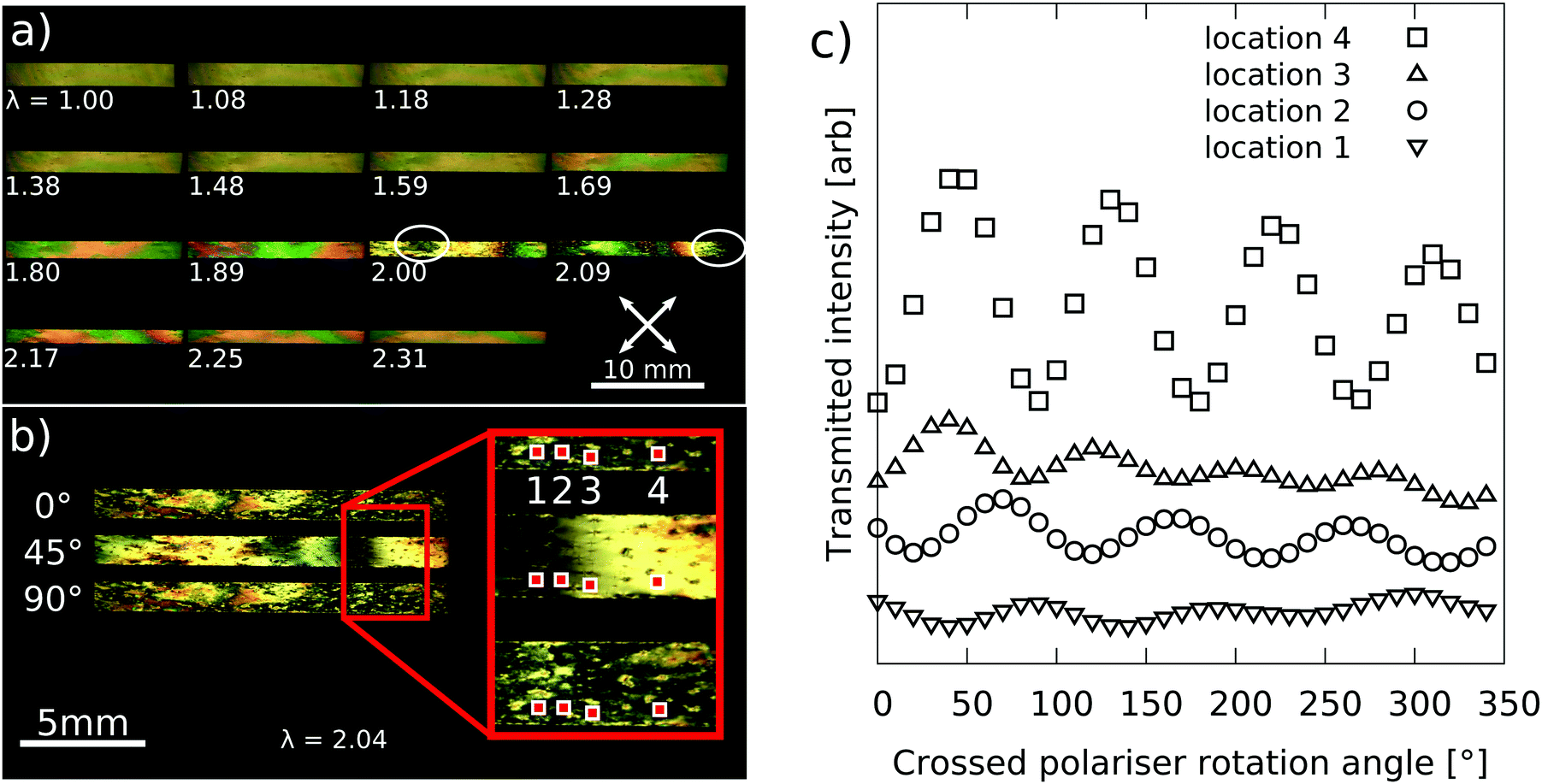 | ||
| Fig. 6 (a) Polarising microscopy images of the 88° sample at various extension steps across the deformation. The circled regions in images of λ = 2.00 and 2.09 highlight black regions of near-zero retardance. (b) Polarising microscopy photos for the sample at λ = 2.04 with the polariser at 0°, 45° and 90° to the LC director. The transmission as a function of polariser rotation angle is plotted in (c) for the four shaded 30 × 30 pixels locations in the magnified portion of (b). For clarity the baseline of each curve in (c) has been shifted to separate the curves. | ||
Fig. 6b show the sample at λ = 2.04 with the polariser at angles of 0°, 45° and 90° to the director. Each photograph has a common region which appears black, indicative of zero retardance, i.e. an effectively isotropic region. Fig. 6c plots the transmitted intensity in the four locations marked in Fig. 6b as the crossed polarisers were rotated. For clarity the baselines of the curves have been shifted to vertically separate the curves. The transmission plots show a decreasing amplitude of intensity variation near the dark regions of the sample. This observation is consistent with a near-zero retardance in the ‘black’ regions of the sample.
As the extension is increased beyond λc, we see from the birefringence colours of Fig. 6a that the retardance, and hence LC order parameter and step length anisotropy, increase.
Thermal testing
Fig. 7 shows the fractional length change (relative to room temperature) and birefringence of the elastomer as it is heated from 18 °C to 330 °C along with polarising microscopy images of the sample at 18 °C and 330 °C. Both the LCE fractional length change and the birefringence show effectively linear relationships with temperature and decrease by 1.13% K−1 and 3.5 × 10−2 K−1 respectively. Before a LCE sample reaches the isotropic phase, a sharp reduction in the LC order parameter, visible through a sharp reduction in birefringence, would be expected.5,15,36 However, our DSC results showed that unsurprisingly, some thermal degradation had begun by 330 °C. As it makes no sense to heat the sample higher than 330 °C we do not observe the nematic to isotropic transition or the associated sharp decline in Δn to zero. We therefore take 330 °C as a lower limit for the temperature at which the LCE becomes isotropic. An upper limit can be determined as 361 °C, the point at which the extrapolated birefringence in Fig. 7a reaches zero. For the purposes of calculating r, we therefore take the nematic to isotropic transition temperature (TNI) as the mid-point between 330 and 361 °C and use an uncertainty of ±15 °C to reflect this range, hence TNI = 345 ± 15 °C. Thermal degradation of the LCE in this experiment would also cause further errors in this temperature. However, its effects were taken as acceptably small for the following reasons. Firstly, in the thermal stability tests performed using DSC, the shift in the glass transition inflection point by 7.2 °C occurred only after the sample had been maintained at over 200 °C (an approximate temperature above which one may expect thermal changes to begin) for 45 minutes. By comparison, the sample tested for the data shown in Fig. 7 was tested on a microscopy hotstage and was maintained at over 200 °C for less than 15 minutes. Any thermal degradation effects are therefore small by comparison. Secondly, the data for both curves shown in Fig. 7a show no apparent change in gradient at any point and therefore it is unlikely that any significant changes are occurring in the material on the timescale of the experiments. Bearing the above in mind, we can safely assume that the thermal effects would have resulted in a maximum additional error of 10 °C for TNI. Combining the two uncertainties gives a final value of TNI = 345 ± 20 °C.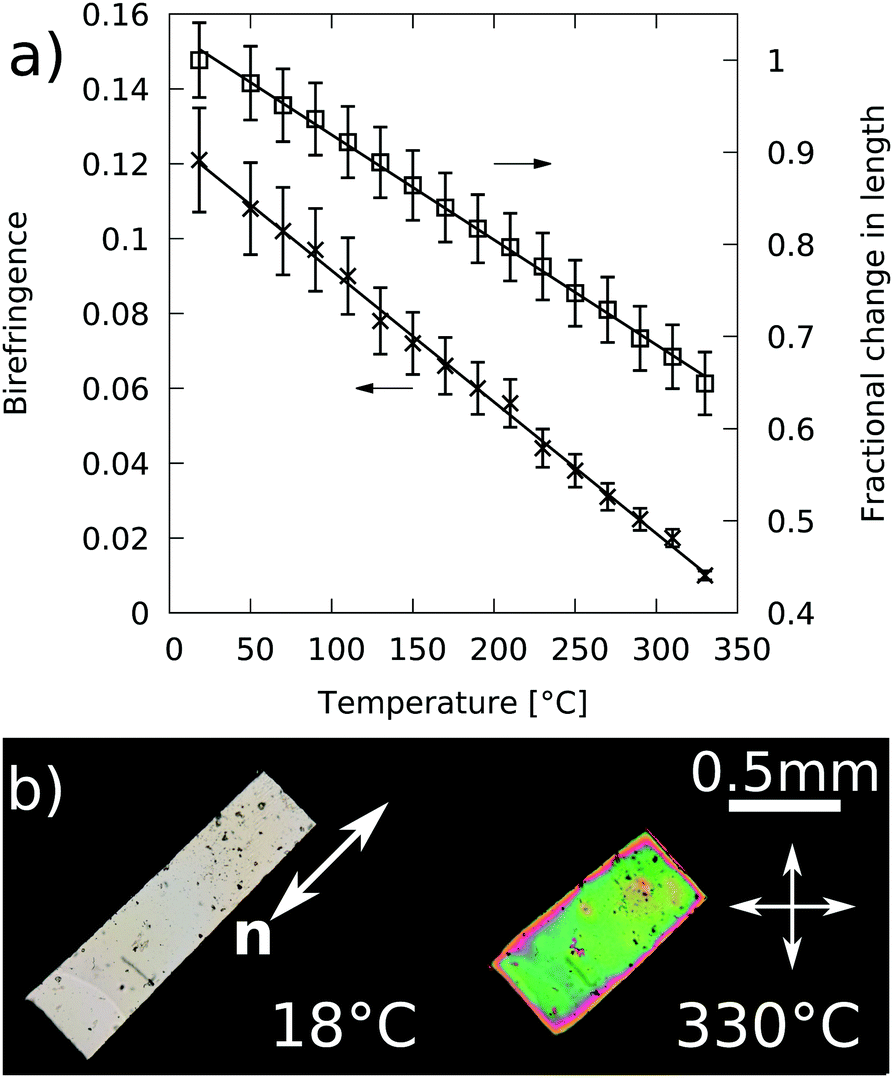 | ||
| Fig. 7 (a) Fractional length change and birefringence of a sample as it is heated from 18 °C to 330 °C. For clarity, errors have been enlarged by a factor of 5. (b) Corresponding polarising microscopy images of the sample at 18 °C and 330 °C. The director, n, has been marked on the photograph of the sample at 18 °C and remained in the same orientation at 330 °C. | ||
Since the birefringence and the step length anisotropy, r, are both related to the LC order parameter we can predict that if Δn = 0 at 345 ± 20 °C, then similarly r = 1 at that temperature. A linear fit to the fractional change in sample length extrapolated to 345 °C gives the fractional change between 18 °C and 345 °C, L18/LISO = λm = 1.56 ± 0.06. By using eqn (6) and fully propagating the errors, we arrive at a final value of r = λm3 = 3.8 ± 0.5.
Discussion
At a first glance, the results presented above have produced two similar values for r and one significantly larger with the total range of values spanning an order of magnitude. Previous reports by several researchers have found relatively good agreement between several methods for calculating r. For instance Clarke et al. found agreement between values of r calculated from thermal tests, the position of the SSE plateau and neutron scattering data for a range of polysiloxane-based LCEs.35 Further, Mitchell, Roberts and co-workers found agreement in the values of r calculated from MFT theory and wide angle X-ray scattering (WAXS) experiments.14,18 However, here we have found significant disagreement, the reasons for which are explored in the following discussion.First let us consider Fig. 4b and 5 together. The load curve in Fig. 4b has the characteristics of SSE. According to the SSE theory, such a load curve requires a gradual and continuous director rotation between λ1 and λ2.11 However, as we see from Fig. 5, the director rotation for our LCE is consistent with a mechanical Fréedericksz transition occurring close to the boundary between regions II and III of the load curve. We know of no other reports of the load curve for an LCE displaying a MFT. The similarity in the appearance of the load curve to a semi-soft elastic load curve is highly significant as that characteristic load curve shape is taken as evidence of semi-soft elasticity.11,36 We therefore suggest that the shape of the load curve cannot solely be used to determine either the mode of deformation or key parameters of LCEs such as r. Instead the director orientation throughout a mechanical experiment must also be monitored.
Since the form of the director orientation curve (Fig. 5) suggests the deformation mode of this LCE is a MFT and not SSE, the value of r = 3.2 ± 0.4 calculated from eqn (3) must be discarded. This is despite its similarity to the value of r = 3.8 ± 0.5 deduced from thermal tests.
In their experiments Mitchell et al. found a critical extension λc = 1.13. Applying eqn (4) and (5) to their data gives in a narrow range of values of r, 1.4 < r < 1.6 which is in agreement with the value they found from neutron scattering experiments.14 However, in our work the comparatively large critical extension ratio of λc = 2.1, coupled with the form of eqn (4) and (5) means a large range of values for r has been calculated which does little to provide a precise or accurate value for r. From this result it is therefore difficult for us to comment on the validity of this model when applied to the present LCE.
The observations in Fig. 6a and b of regions of near-zero retardance when the sample is extended close to λc is particularly interesting. As the sample began with a thickness of 99 μm the thickness must have remained on order of tens of microns thick at all stages of the extension. Therefore, the observation of a near zero retardance around λc corresponds to LCE going through a state of a near-zero birefringence and hence a near-zero LC ordering within the image plane. Observations by Finkelmann et al. (2001) can in part account for this behaviour. In their work they showed that if director rotation is prohibited when a LCE strained perpendicular to the director, then both the liquid crystal order and step length tensors will have to some degree become biaxial.37 In our samples, the mechanical introduction of biaxiality has resulted in an effective lowering of the order parameter within the image plane and hence is responsible for the observation of a retardance which decreases to zero. Given the intimate link between LC order and the shape of the polymer conformation, the state of zero LC ordering within the image plane similarly translates to a near-circular polymer conformation within the image plane (as illustrated in Fig. 8). While we note that the above results provide no insight into the degree LC ordering in the plane perpendicular to the stress axis, it is reasonable to expect that throughout the deformation the sample will have become thinner through the sample extension. As a result the polymer conformation will have contracted in the direction perpendicular to the image plane. By the time λ ∼ λc, where “black regions” are observed in Fig. 6a and 6b, the polymer conformation had become oblate.
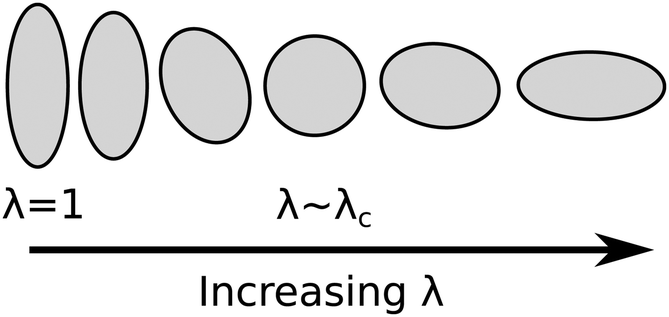 | ||
| Fig. 8 Diagram of the evolution of the polymer conformation in the plane of the sample initially oriented at 88°. | ||
The above observations must mean that at λc the sample has a negative LC order parameter for the following reason: at λc, there is zero correlation and hence zero ordering between the LC molecules within the image plane (hence observation of zero retardance). Despite this, the thinning of the polymer conformation perpendicular to the image plane with strain will have confined the long axes of the LC molecules to the image plane. The symmetry of this arrangement of LC molecules corresponds to a negative LC order parameter.38 This state of ordering is physically unachievable for the majority of LC systems and is therefore rarely reported.
The implications of the above discussions taken together with the director orientation data from Fig. 5 indicates that neither theories of SSE or MFTs are completely appropriate for describing the deformation of the present LCE. Both theories assume a constant LC order parameter and hence constant value for r. As a result, eqn (3)–(5) cannot be reliably applied to the deformation of this LCE and our calculated values of 9.3 < r < 30.0 must also be disregarded. The evident deformation of the polymer conformation does however demonstrate that the deformation process is not as discontinuous as Fig. 5 would suggest but instead behaves as shown in Fig. 8.
The question of a changing order parameter during the mechanical deformation of an LCE was explored by Mitchell, Roberts and co-workers. In one paper they saw a decrease in the LC order parameter, but concluded that the sample was spatially inhomogeneous near λc and the measurement of a reduced order parameter was an artefact of their WAXS measurements averaging over domains of different director orientation. When a pinhole was used to measure the order parameter over a smaller area, the decrease in the order parameter was only 10%.18 In two other papers they reported neutron scattering and WAXS data which showed an unambiguous decrease in the nematic order parameter and therefore a decrease in the polymer conformation anisotropy between λ = 0 and λ = λc.14,19 However, in both cases the calculation of r using eqn (4) was sufficiently close to values obtained from direct measurements of r from neutron scattering data that the authors concluded that the MFT theory accurately described their results. From our observations we would suggest that the deformation of the polymer conformation has a lower energy cost compared to the sharp director rotation of an MFT and so the director remains relatively constant until the cost of deforming the polymer conformation increases to larger than the energy required for director rotation. As such the changing order must be taken into account.
The above discussion leads to us discounting values of r calculated for this material from opto-mechanical testing. We are therefore left with r = 3.8 ± 0.5 as the most reliable value. We can perhaps say that this value is reasonable since the physical process involved and theory behind it is comparatively simple compared to that describing the mechanical behaviour of LCEs.
Table 3 compares our value of r = 3.8 ± 0.5 to values measured for other LCEs either reported or calculated by us using eqn (5) or (6) from data contained within existing literature. The last column of Table 3 gives details of how the mesogenic groups of each LCE are incorporated into the polymer backbone (see Fig. 9 for schematic representations).39 The material described in this paper consists of solely acrylate monomers and the LC-backbone coupling is entirely side-chain end on. From Table 3 we see that the value of r = 3.8 ± 0.5 for our material is comparable to the upper range of values seen for similar materials (first three rows of Table 3). Of particular interest is the value of r = 2.0 calculated from data from Tamashima et al. (2016) as this is for the LCE described by Urayama et al. (2005) which contains the same primary monofunctional mesogenic group, A6OCB.29,40 Comparatively, values for r from side-on, side chain and main chain LCEs are far greater (≳25) as would be expected given their stronger LC-backbone coupling.
| Ref. | Effective step length anisotropy, r | LC-backbone coupling type (see Fig. 9) | Additional information |
|---|---|---|---|
| Our LCE | 3.8 ± 0.5 | Side-chain, end on | From thermal testing and eqn (6) |
| Mitchell (1993)14 | 1.3 and 1.4 | Side-chain, end on | Values respectively from neutron scattering and mechanical test using eqn (5) |
| Tamashima (2016)40 | ∼2.0 | Side-chain, end on | From thermal testing and eqn (6) |
| Brömmel (2012)39 | <2.25 | Side-chain, end on | Book chapter. Values based on SANS from ref. 41 and 42 |
| Brömmel (2012)39 | <25 | Side-chain, side on | Book chapter. Values based on SANS from ref. 41 and 42 |
| Tajbakhsh (2001)43 | ∼43 | Majority main-chain, some side chain | From thermal testing and eqn (6) |
| D’Allest (1988)44 | ≤60 | Main-chain | Results from SANS on a main chain LC polymer |
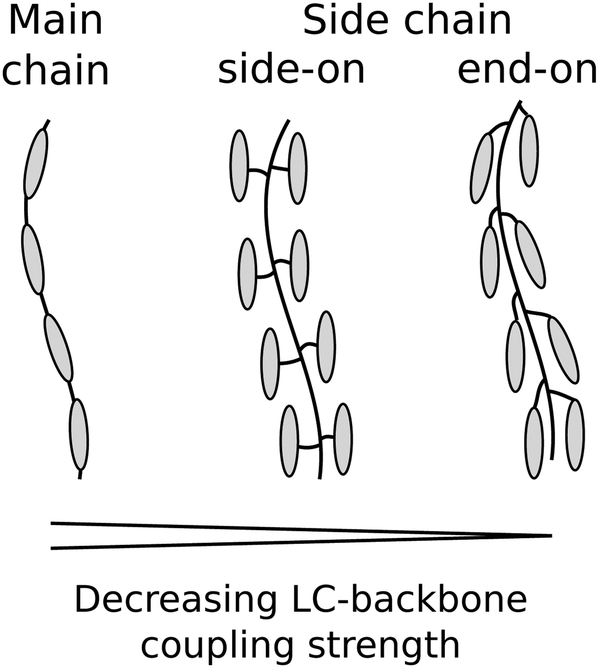 | ||
| Fig. 9 LC-backbone coupling types. | ||
Conclusions
The key finding of our work is that a characteristic load curve shape such as is shown in Fig. 4 is not necessarily indicative of semi-soft elasticity as currently accepted by the majority of the field. While the theories of Warner, Terentjev and co-workers have described many observations of LCEs with great success, our data suggest that the story is not yet complete. Indeed our results from simultaneous mechanical and opto-mechanical tests have shown for the first time behaviours which are incompatible with both theories of SSE and MFTs, though these currently offer the best theoretical description of LCEs.Our results appear to show that when an LCE is stressed perpendicular to the director, the polymer conformation is deformed from an elliptical to a circular shape within the plane of the sample. Further, by studying polarising microscopy textures we have deduced that during this process, it possible to mechanically induce a negative LC order parameter. We have also demonstrated that the deformation of the polymer conformation and LC ordering causes a plateau-like region of reduced modulus. Previously, such behaviour has been solely attributed to the rotation of the polymer conformation which maintains a constant magnitude of anisotropy throughout the LCE deformation, described by SSE theory. A theoretical understanding of why the material here shows a SSE-like load curve but has a MFT-like director re-orientation behaviour may answer the question of why some materials show SSE and others show MFT-like behaviour. We propose that such a theory should include a mechanical dependency of the LC order parameter and step length anisotropy.
Our results also highlight the importance of measuring both the LC order and tensile properties simultaneously when characterising an LCE. A single test would have led to the incorrect assumption that the behaviour conformed to one or other mode of deformation and calculated potentially vastly inaccurate values of r. It is likely to also be important that the measurements are done on a single sample, thus ensuring results that can be directly compared.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Mistry thanks UltraVision CLPL and the EPSRC for a CASE PhD studentship and the Royal Commission for the Exhibition of 1851 for an Industrial Fellowship. The authors thank T Haynes and P Thornton for building of equipment.Notes and references
- J. Küpfer and H. Finkelmann, Makromol. Chem., Rapid Commun., 1991, 12, 717–726 CrossRef.
- Y. Yu and T. Ikeda, Angew. Chem., Int. Ed., 2006, 45, 5416–5418 CrossRef CAS PubMed.
- J. M. Boothby, H. Kim and T. H. Ware, Sens. Actuators, B, 2017, 240, 511–518 CrossRef CAS.
- H. Wermter and H. Finkelmann, e-Polym., 2001, 1, 111–123 Search PubMed.
- D. L. Thomsen, P. Keller, J. Naciri, R. Pink, H. Jeon, D. Shenoy and B. R. Ratna, Macromolecules, 2001, 34, 5868–5875 CrossRef CAS.
- H. Zeng, O. M. Wani, P. Wasylczyk and A. Priimagi, Macromol. Rapid Commun., 2017, 1700224 Search PubMed.
- H. Shahsavan, S. M. Salili, A. Jákli and B. Zhao, Adv. Mater., 2015, 27, 6828–6833 CrossRef CAS PubMed.
- S. Schuhladen, F. Preller, R. Rix, S. Petsch, R. Zentel and H. Zappe, Adv. Mater., 2014, 26, 7247–7251 CrossRef CAS PubMed.
- H. Zeng, O. M. Wani, P. Wasylczyk, R. Kaczmarek and A. Priimagi, Adv. Mater., 2017, 29, 1701814 CrossRef PubMed.
- M. Yamada, M. Kondo, J. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- M. Warner and E. M. Terentjev, Liquid Crystal Elastomers, Clarendon Press, 2013 Search PubMed.
- S. Clarke and E. M. Terentjev, Phys. Rev. Lett., 1998, 81, 4436–4439 CrossRef CAS.
- I. Kundler and H. Finkelmann, Macromol. Rapid Commun., 1995, 16, 679–686 CrossRef CAS.
- G. R. Mitchell, F. J. Davis and W. Guo, Phys. Rev. Lett., 1993, 71, 2947–2950 CrossRef CAS PubMed.
- J. Küpfer and H. Finkelmann, Macromol. Chem. Phys., 1994, 195, 1353–1367 CrossRef.
- R. V. Talroze, E. R. Zubarev, S. A. Kuptsov, A. S. Merekalov, T. I. Yuranova, N. A. Plate’ and H. Finkelmann, React. Funct. Polym., 1999, 41, 1–11 CrossRef CAS.
- H. Higaki, T. Takigawa and K. Urayama, Macromolecules, 2013, 46, 5223–5231 CrossRef CAS.
- P. M. S. Roberts, G. R. Mitchell and F. J. Davis, J. Phys. II, 1997, 7, 1337–1351 CrossRef CAS.
- P. M. S. Roberts, G. R. Mitchell, F. J. Davis and J. A. Pople, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1997, 299, 181–186 CrossRef CAS.
- G. C. Verwey and M. Warner, Macromolecules, 1997, 30, 4189–4195 CrossRef CAS.
- G. C. Verwey and M. Warner, Macromolecules, 1997, 30, 4196–4204 CrossRef CAS.
- P. Bladon, E. M. Terentjev and M. Warner, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1993, 47, R3838–R3840 CrossRef CAS.
- P. Bladon, E. Terentjev and M. Warner, J. Phys. II, 1994, 75–91 CrossRef CAS.
- J. Weilepp and H. R. Brand, Europhys. Lett., 1996, 34, 495 CrossRef CAS.
- E. M. Terentjev and M. Warner, Europhys. Lett., 1997, 37, 495 CrossRef CAS.
- J. Weilepp and H. R. Brand, Europhys. Lett., 1997, 37, 499 CrossRef CAS.
- E. Fried and S. Sellers, J. Appl. Phys., 2006, 100, 043521 CrossRef.
- H. R. Brand, H. Pleiner and P. Martinoty, Soft Matter, 2006, 2, 182–189 RSC.
- K. Urayama, Y. O. Arai and T. Takigawa, Macromolecules, 2005, 38, 3469–3474 CrossRef CAS.
- K. Urayama, R. Mashita, I. Kobayashi and T. Takigawa, Macromolecules, 2007, 40, 7665–7670 CrossRef CAS.
- D. Allan, T. Caswell, N. Keim and C. Van Der Wel, trackpy: Trackpy v0.3.2. Zenodo. http://doi.org/10.5281/zenodo.60550, 2016.
- J. Schindelin, C. T. Rueden, M. C. Hiner and K. W. Eliceiri, Mol. Reprod. Dev., 2015, 82, 518–529 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- T. Ruiz and R. Oldenbourg, Biophys. J., 1988, 54, 17–24 CrossRef CAS PubMed.
- S. M. Clarke, A. Hotta, A. R. Tajbakhsh and E. M. Terentjev, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 64, 061702 CrossRef CAS PubMed.
- S. M. Clarke, A. R. Tajbakhsh, E. M. Terentjev, C. Remillat, G. R. Tomlinson and J. R. House, J. Appl. Phys., 2001, 89, 6530–6535 CrossRef CAS.
- H. Finkelmann, A. Greve and M. Warner, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 5, 281–293 CrossRef CAS.
- Negative order parameters in LCEs were first reported independently by Lagerwall et al. and Mistry et al. at the 2017 European Liquid Crystals conference in Moscow.
- F. Brömmel, D. Kramer and H. Finkelmann, Adv. Polym. Sci., 2012, 250, 1–48 Search PubMed.
- K. Tamashima, T. Takigawa and K. Urayama, Nihon Reoroji Gakkaishi, 2016, 44, 17–27 CrossRef CAS.
- L. Noirez, J. P. Cotton, F. Hardouin, P. Keller, F. Moussa, G. Pepy and C. Strazielle, Macromolecules, 1988, 21, 2889–2891 CrossRef CAS.
- L. Noirez, P. Keller and J. P. Cotton, Liq. Cryst., 1995, 18, 129–148 CrossRef CAS.
- A. R. Tajbakhsh and E. M. Terentjev, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 6, 181–188 CrossRef CAS.
- J. F. D’Allest, P. Maïssa, A. ten Bosch, P. Sixou, A. Blumstein, R. Blumstein, J. Teixeira and L. Noirez, Phys. Rev. Lett., 1988, 61, 2562–2565 CrossRef PubMed.
Footnotes |
| † The full dataset behind this paper is hosted by the University of Leeds' data repository service and can be accessed via the DOI: https://doi.org/10.5518/131. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sm02107k |
| This journal is © The Royal Society of Chemistry 2018 |